


Abstract
Insertion of the T3 DNA polymerase thioredoxin binding domain (TBD) into the distantly related thermostable Taq DNA polymerase at an analogous position in the thumb domain, converts the Taq DNA polymerase from a low processive to a highly processive enzyme. Processivity is dependent on the presence of thioredoxin. The enhancement in processivity is 20–50-fold when compared with the wild-type Taq DNA polymerase or to the recombinant polymerase in the absence of thioredoxin. The recombinant Taq DNA pol/TBD is thermostable, PCR competent and able to copy repetitive deoxynucleotide sequences six to seven times more faithfully than Taq DNA polymerase and makes 2–3-fold fewer AT→GC transition mutations.
INTRODUCTION
An enzyme is processive if, upon binding to its substrate, it carries out multiple catalytic cycles before dissociation. When referring to polymerases, processivity relates to the number of nucleotides added to the nascent strand during one round of binding and dissociation from the primer template. The processivity of DNA polymerases is intimately related to their biological role. Enzymes that copy genomes exhibit high processivity, repair of long gaps during DNA repair may proceed with lower processivity while a distributive mode of catalysis appears adequate for repair of small gaps or for bypass polymerases that synthesize past blocking lesions at the expense of fidelity.
Replication slippage occurs during synthesis of microsatellite repeat sequences and is characterized by an expansion or deletion in the number of repeat units. One proposed mechanism for polymerase slippage involves dissociation of the growing strand and formation of a loop consisting of one or more repeat units either in the template or nascent strand, which is stabilized by hydrogen bonding with adjacent repeat elements (1,2). It has been previously established that the processivity of T7 DNA polymerase enzyme is related to reduction of slippage during copying of repetitive DNA sequences, presumably because highly processive replication results in a lowered probability of enzyme dissociation during replication of the repetitive nucleotide sequence.
T7 bacteriophage replication requires formation of a 1:1 complex of Escherichia coli thioredoxin with T7 DNA polymerase. Thioredoxin binds the polymerase at a 76 amino acids domain that is absent from other members of the Pol I family of enzymes (3). Binding of thioredoxin converts the enzyme from a processivity of <15 nt to a processivity >2000 nt; it increases the affinity to the primer template by 80-fold and enhances the rate of elongation (4,5). T7 DNA polymerase shares extensive structural homologies to other members of the Pol I class of enzyme including E.coli Pol I and Taq DNA polymerase.
The T7 DNA polymerase thioredoxin binding domain (TBD) is located in the thumb of the polymerase between helices H and H1. Thioredoxin has no reported affinity for DNA, so presumably upon binding to the polymerase the thioredoxin acts as a clamp, tethering the polymerase onto the DNA. Bedford et al. (6), demonstrated that insertion of the TBD into E.coli Klenow Pol I confers thioredoxin-dependent processivity, suggesting that the TBD alone is able to accomplish the processivity functions (6). By comparing the co-crystal structures of T7 DNA polymerase bound to DNA and nucleotide (PDB:1T7P), with the homologous ternary structure of the Taq DNA polymerase (PDB:3KTQ), we generated hybrid Taq DNA polymerase variants containing the TBD of the homologous T3 bacteriophage DNA polymerase. The T3 DNA polymerase is 96% identical to the T7 DNA polymerase (7). The amino acid sequence of the TBD of T3 DNA polymerase differs from the T7 DNA polymerase TBD only at position 291, where isoleucine is found instead of threonine. Our goal was to confer thioredoxin-specific processivity on Taq DNA polymerase and investigate the effect of processivity on fidelity of replication during PCR.
MATERIALS AND METHODS
Bacterial strains, bacteriophage and plasmids
T3 bacteriophage were obtained from ATCC. Plasmid pTacTaq, containing the entire Taq DNA polymerase gene in pHSG576 was described previously (8). Site-directed mutagenesis was performed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Escherichia coli strain GI724 [F-, lacIq, lacPL8, ampC::Ptrp cI, mcrA, mcrB, INV(rnnD-rnnE)] was obtained from ATCC and JS200 polAts is temperature sensitive, can be complemented by Taq DNA polymerase and was obtained from the laboratory collection. Circular single-stranded M13mp18 (Panvera Corp, Madison, WI) was hybridized to M13–40 primer in 50 mM KCl by heating to 95°C and slowly cooling to room temperature. Escherichia coli thioredoxin was used for all experiments requiring thioredoxin.
Generation of hybrid Taq DNA pol/TBD
Comparisons of the ternary crystal structures for the large fragment of Taq DNA polymerase (PDB: 3KTQ) and T7 DNA polymerase (PDB: 1T7P) were preformed using the VAST structure comparison software and Cn3D viewer from NCBI. The TBD of bacteriophage T3 gene5 (DNA polymerase) consisting of nucleotides 780–1011, was amplified by PCR and cloned into Taq DNA polymerase using silent XbaI and MluI restriction sites introduced into the Taq DNA polymerase gene at positions 1572 and 1638, respectively. Nucleotides encoding the amino acids 480–485 of Taq DNA polymerase were replaced with nucleotides encoding the 76 amino acid residues 262–337 of the T3 DNA polymerase, to generate the hybrid protein. Sequencing of the entire hybrid gene confirmed that the inserted DNA corresponded to the TBD of bacteriophage T3. The amino acid sequence of the TBD of T3 DNA polymerase differs from the T7 DNA polymerase only at position 291, where isoleucine is found instead of threonine (7). The hybrid protein was designated Taq DNA pol/TBD. A low copy plasmid pHSG576 was initially used to clone Taq DNA pol/TBD and enabled the activity of the polymerase to be assessed by in vivo complementation of the E.coli strain JS200 polAts as previously reported (8).
Protein purification
Six histidine codons were introduced onto the N-terminus of the Taq DNA polymerase and Taq DNA pol/TBD gene and the recombinant genes were cloned in frame into the NdeI site of the pLEX vector (Invitrogen). 5′–3′ Exonuclease-deficient mutants which lack the first 235 amino acids of the N-terminal were likewise generated. These truncated exonuclease- deficient proteins were designated Taq DNA polymerase (exo–) and Taq DNA pol/TBD(exo–). Growth and induction of protein expression was performed in GI724 E.coli that were cotransfected with pRARE (Novagen, Madison, WI, USA). Crude lysates were obtained by incubation with lysozyme (100 µg/ml) for 20 min at 25°C in heparin binding/wash buffer [50 mM Tris pH 7.5, 50 mM NaCl, 5% glycerol (v/v)], 5 ml/g wet cell weight, followed by three cycles of freezing and thawing. Protease inhibitors without EDTA (Cocktail set V; Calbiochem, San Diego, CA) were included at a 1/100 dilution in the crude lysate and at 1/1000 dilution in all other buffers. The lysate was centrifuged at 16 000 g for 20 min at 4°C and the supernatant adsorbed onto a 5 ml heparin column (Amersham, Piscataway, NJ). The column was washed with 30 ml of heparin binding/wash buffer and eluted with a 25 ml linear gradient from 50 mM to 2 M NaCl in 50 mM Tris pH 7.5, 5% glycerol (v/v), and 1 ml fractions were collected. Fractions were assessed by DNA polymerase activity assay and peak activities were pooled. Pooled fractions were incubated with 1 ml of bed volume Ni2+-NTA Sepharose slurry (Novagen His Bind Kit) for 20 min at 4°C before chromatography. Fractions were eluted after washing the columns in 10 bed volumes of Ni2+-NTA binding buffer [5 mM imidazole, 500 mM NaCl, 40 mM Tris pH 7.9, 20% glycerol (v/v)] with a 12 ml linear gradient from 50 to 800 mM imidazole in 40 mM Tris pH 7.9, 20% glycerol (v/v). Peak fractions contained a single polypeptide corresponding to the molecular weight for each of the Taq DNA polymerase (94 kDa), Taq/TDB (103 kDa) and Taq DNA polymerase (exo–) (68 kDa) and Taq/TDB(exo–) (76 kDa) variants, as evident by SDS–PAGE (10%) with Coomassie blue staining using molecular weight markers (Kaleidoscope Prestained Standards; Bio-Rad). The proteins were dialyzed into a final storage buffer of 50% glycerol (v/v), 100 mM KCl, 1 mM DTT, 50 mM Tris pH 8.0, 0.5% IGEPAL and 0.5% Tween-20.
Polymerase activity assay
Activity assays were performed using as a template primer, 10 nM M13mp18 hybridized to M13–40 primer (IDT). The reaction mixture (20 µl) contained 50 mM Tris pH 8.8, 16 mM (NH4)2SO4, 0.1% Tween-20, 5.0 mM DTT, 5.0 mM MgCl2, 200 µM each of dATP, dTTP, dCTP and 5 µM dGTP, 250 nCi 32P-dGTP and 250 µg/ml BSA. The amount of Taq DNA polymerase and Taq DNA pol/TBD used in each reaction was 230 ng, while 60 ng was used for the Taq(exo–) and Taq DNA pol/TBD(exo–) enzymes as determined by the Bradford protein quantification assay (Bio-Rad) using a standard curve derived from BSA. Reactions were incubated for 20 min at 60°C, and stopped by the addition of 40 mM EDTA. The reaction proceeds with linear kinetics for at least 30 min (data not shown). The DNA product of the reaction was bound to 96 well DNA binding filter plates (Silent screen; Nalgene). The spots were visualized by phosphorimaging and quantified by densitometry using Imagequant software. Escherichia coli thioredoxin (Promega), when present was at a final concentration of 100 µM.
Processivity
Processivity was measured as described by Bambara et al. (9), using a M13mp18 single-stranded template hybridized to primer labeled at the 5′ end with [γ-32P]ATP by T4 polynucleotide kinase (New England Biolabs) (9). The primer was labeled by incubation for 30 min at 37°C using T4 polynucleotide kinase. After annealing, the M13mp18/primer template substrate was isolated using a G25 spin column (Amersham Pharmacia). The reaction mixture (10 µl) contained 50 mM Tris pH 8.8, 16 mM (NH4)2SO4, 0.1% Tween-20, 5.0 mM DTT, 5.0 mM MgCl2, 200 µM each of dATP, dTTP, dCTP and dGTP, 3.75 nM of primer/template (1:1) and enzyme concentrations as stated in the caption to Figure 3. This corresponded to a ratio of 470 and 67 primer/template to enzyme molecules, respectively; the excess primer/template ensures that the size of the products observed result from only one binding event. The final thioredoxin concentration was 100 µM for data in Figure 3A and 0.2, 2 and 20 µM for data in Figure 3B. Thioredoxin was preincubated with polymerase in the presence of 5 mM DTT for 10 min at 25°C prior to the addition of the reaction mixture, primer and template. Reactions were incubated for 5 min at 60°C and were stopped by the addition of 40 mM EDTA. Products were separated by 14% PAGE containing 8 M urea and visualized by exposure to a phosphor screen.
Figure 3.

The effect of thioredoxin on processivity of the hybrid Taq DNA pol/TBD. (A) Extension assays were performed with a molar excess of template corresponding to a primer/template ratio of 470 for Taq DNA polymerase and Taq DNA polymerase (exo–) and 67 for Taq DNA pol/TBD and Taq DNA pol/TBD(exo–). Different ratios for the enzymes were used to ensure equal activity was loaded on the gel. (+) 100 µM thioredoxin, (–) no thioredoxin. No enzyme control shows the labeled primer alone. (B) Extension assay showing the effect of increasing concentrations of thioredoxin and enzyme dilution for Taq DNA pol/TBD(exo–). For each thioredoxin concentration (0.2, 2 and 20 µM), three enzyme concentrations were used (56, 28 and 5.6 pM) corresponding to a primer/template ratio of 67, 134 and 670.
Streptavidin processivity assay
A 2 kb DNA template was generated by PCR amplification of the luciferase gene from the positive control plasmid obtained from the Promega in vitro transcription/translation kit, using a 5′ biotinylated forward primer (Biotin-luctaq3, B, 5′-GTT TTCCCAGTCACGACGTT-3′ and primer luctaq4, 5′-TTA CACTTTATGCTTCCGGCTCGTA-3′). The PCR product was purified from a 0.8% agarose gel using a gel purification kit (Qiagen). Two micrograms of DNA was bound to 10 µl of streptavidin-coated magnetic beads in kilobase binding buffer (Dynal) by incubation for 3 h at room temperature. The experiment was performed in a 96 well plate, one reaction per well. The beads were magnetically separated, washed in binding buffer and the DNA was denatured in a solution containing 0.125 M NaOH, 1 mM EDTA for 20 min at room temperature. Following strand separation, the single-stranded biotinylated DNA–bead complexes were washed three times in TE buffer (10 mM Tris pH 8.0, 1 mM EDTA) and once in H2O prior to extension. The initial 30 µl reaction contained 400 µM primer luctaq4, 10 nM polymerase, 100 µM thioredoxin, 5 mM DTT, in the same reaction mixture used for polymerase activity assays without dNTPs. Reactions were preincubated for 10 min at 60°C to allow binding of polymerase to the primer terminus. DNA synthesis was initiated by the addition of dNTPs, including [α-32P]dGTP and a large excess of activated calf thymus trap DNA (final concentration of 0.8 mg/ml). The excess of trap DNA is to ensure that each primer template is extended only once so that the distribution of extension products reflects the processivity of the polymerase. Reactions were quenched after 10 min by the addition of 40 mM EDTA and placed on ice. Bead-immobilized extension products were washed three times in TE and once in H2O. Incubation of individual reactions with the restriction enzymes BsrBI, KpnI, ClaI, XcmI and HindIII that cleave double-stranded DNA at 18, 96, 492, 1122 and 1898 nt, respectively, from the primer terminus. Only double-stranded radiolabeled fragments generated during single binding and extension events, were released into solution for quantitation. Untrapped fully extended control reactions were incubated with 5 U Taq DNA polymerase (Promega) and used to quantitate the maximum signal obtained for each restriction cleavage. Data values from pretrapped negative controls in which trap was added prior to polymerase were subtracted from the background. The results are expressed as a percentage of the maximum signal for each restriction enzyme.
PCR conditions
The reaction consisted of 50 mM Tris pH 8.8, 16 mM (NH4)2SO4, 0.1% Tween-20, 5.0 mM DTT, 1.8 mM MgSO4, 100 µM thioredoxin, 1 ng of plasmid or λ DNA and 200 µM of each dNTP. PCR amplification was performed for 25 cycles of 94°C for 30 s, 50°C for 30 s, 60°C for 60 s, preceded by a 2 min denaturation at 94°C and completed with a final 5 min extension step at 60°C.
Amplification of λ DNA was performed with the forward primer 5′lambda2 (5′-GATTCTGGATACGTCTGAAC-3′) and the reverse primers for 1kLambda3 (5′-GGATTTTCACCACATCAAT-3′), 5kLambda2 (5′-AGGTC TTTTTCTGCTCTGA-3′), 7kLambda2 (5′-ACAGCGGAACTTATGAATC-3′) and 10kLambda2 (5′-GTCTGCAGTGACTTCTGC-3′) for 1, 5, 7 and 10 kb products, respectively.
Base substitution fidelity
PCR products were obtained by nested amplification of 10 ng of human genomic DNA at the D2S123 loci using reaction mixtures as above with either Taq DNA polymerase or Taq DNA pol/TBD. Two rounds of PCR was performed, each with 35 cycles using primers D2S123F (5′-CAAAAATTGACTCAAGAAGAA-3′), D2S123R (5′-TTAGGAGCTCTTTTGAATTG-3′) and nested primers, D2S123nestF (3′-AAATCTGAACAAACCTATGC-5′) and D2S123nestR (5′-GGGACTGTGTCATCTACAAT-3′). PCR products were cloned using the TopoTA cloning strategy (Invitrogen). Single read sequences were obtained from 88 individual clones totaling 24 531 and 19 631 bases sequenced for Taq DNA polymerase and Taq DNA pol/TBD, respectively.
PCR slippage fidelity assay
In order to determine the effect of processivity on slippage during PCR, we utilized a frame shift reporter assay described previously (10). PCR was performed as above using plasmid templates pAJ32, pAJ33 or pAJ47, that contain a stretch of either 10 A, 10 G or 11 CA repeats, respectively, at the beginning of the β-lactamase gene, rendering it out of frame by +1 (A10, G10) or +2 (CA11) nucleotides. The vector also contains a tetracycline resistance marker allowing for selection of cells with non-revertant plasmids. Primers used were MSI2-F (5′-GAGTAAGTAGTTCGCCAGTT-3′) and MSI2-R (5′-CGTAGAGGATCCACAGGA-3′).
The 1275 bp fragment contained the repeat sequence in the center of the PCR fragment flanked by NheI and Bglll sites. A 508 bp fragment obtained by digestion with NheI and Bglll was ligated into the reporter vector (pAJ49) at the NheI and Bglll sites and transformed into DH10B electro-competent cells. Cells were plated on LB agar plates containing tetracycline or both tetracycline and carbenicillin. Deletion of one nucleotide or the addition of two nucleotides during DNA synthesis restores the frame of the β-lactamase gene and allows the frequency of frame shift mutations to be determined by dividing the number of Tet+ Carb+ revertants by the total number of Tet+ colonies. The regions containing the repetitive nucleotide stretches were sequenced from five revertant colonies to verify the location and nature of the frame shift.
Separation and quantitation of slippage at repeat sequences
PCR was performed using plasmid pAJ32 as the template (A10). PCR conditions were performed as above except 0.1 ng of template DNA was amplified for 35 cycles and primer pMSI4-R (5′-TGTGATAAACTACCGCATTA-3′), labeled at the 5′ end with the fluorophore 6-FAM (IDT) was used. The 1035 bp fragment obtained was digested with EcoRI to produce a 350 bp fragment labeled with 6-FAM. The DNA was gel purified and slippage was detected using an automated DNA sequencer (model 377, Applied Biosystems; Perkin Elmer, Foster City, CA, USA), and GENESCAN 672 software (Applied Biosystems; Perkin Elmer) for slippage peak analyses. Length determination of DNA fragments was achieved by running in-lane molecular weight markers.
RESULTS
We first measured the ability of the T3 bacteriophage TBD to confer enhanced processivity on Taq DNA polymerase. The processive polymerase was created by introducing the TBD domain of the T3 bacteriophage DNA polymerase into the Taq DNA polymerase at an analogous position in the thumb domain. We first compared the structure of T7 DNA polymerase bound to nucleotide and primer template, with the ternary crystal structure of Taq DNA polymerase. A site for insertion of the TBD was determined by analysis of the superimposed structures at the thumb domain (Fig. 1). Insertion of 76 residues encompassing the T3 bacteriophage gene5 TBD into the Taq DNA polymerase at the analogous position in the thumb domain, resulted in a hybrid enzyme that when expressed from a single copy plasmid, fully complemented the PolA temperature-sensitive mutation of E.coli strain JS200 at the restrictive temperature (data not shown). Purification of both Taq/TDB hybrid and Taq DNA polymerase full-length and N-terminal truncated 5′–3′ exonuclease-deficient enzymes were achieved using heparin and immobilized metal affinity chromatography.
Figure 1.

Superimposition of the thumb domains of Taq DNA polymerase (blue) with T7 DNA polymerase (pink). The arrows indicate the site of insertion of the T3 TBD (yellow). The primary amino acid sequence of Taq DNA polymerase from residue 470–507 is indicated below (blue) with the sequence of T3 TBD in yellow and the deleted region in red.
DNA polymerase activities of the hybrid Taq DNA pol/TBD and 5′–3′ exo minus Taq DNA pol/TBD enzymes
Polymerase activity was compared between Taq DNA polymerase, Taq DNA polymerase (exo–) enzyme and the cognate Taq DNA pol/TBD hybrid proteins using primed M13mp18 DNA in the presence and absence of thioredoxin (Table 1). In the absence of thioredoxin, both Taq DNA pol/TBD and Taq DNA pol/TBD(exo–) display ∼15% activity compared with the unmodified wild-type Taq DNA polymerase. Thioredoxin stimulates the activity of Taq DNA pol/TBD by ∼4-fold and Taq DNA pol/TBD(exo–) enzymatic activities by >30-fold but had no effect on Taq DNA polymerase. Both Taq DNA pol/TBD and Taq DNA pol/TBD(exo–) are as thermostable as the Taq DNA polymerase and Taq DNA polymerase (exo–) enzymes, respectively, following exposure to 94°C for 30 min (Fig. 2). The exonuclease minus variants show elevated thermostability as previously reported (11).
Table 1. Effect of thioredoxin on DNA polymerase activity.
| Polymerase | Polymerase activity (units/mg protein) | |
|---|---|---|
| – Thioredoxin | + Thioredoxin | |
| Taq DNA polymerase | 2218 | 2224 |
| Taq DNA pol/TBD | 302 | 1291 |
| Taq DNA polymerase (exo–) | 1308 | 1630 |
| Taq DNA pol/TBD(exo–) | 201 | 6521 |
DNA polymerase activity was determined using singularly primed single-stranded M13 DNA as described in the Materials and Methods. One unit of polymerase corresponds to the incorporation of 10 nmoles of all four nucleotides in 20 min at 60°C. The amount of Taq DNA polymerase and Taq DNA polymerase (exo–) used in each reaction was 60 ng, while 230 ng was used for the Taq DNA pol/TBD and Taq DNA pol/TBD(exo–) enzymes. Thioredoxin when added was at a final concentration of 100 µM.
Figure 2.

Polymerase activity was measured as described in the Materials and Methods following exposure to 94°C for 0, 2, 10 and 30 min. The amount of Taq DNA polymerase and Taq DNA polymerase (exo–) used in each reaction was 230 ng, while 60 ng was used for the Taq DNA pol/TBD and Taq DNA pol/TBD(exo–) enzymes. Thioredoxin was included in all reactions at 100 µM.
Thioredoxin increases processivity of the hybrid proteins
Two protocols were employed to quantify the processivity of the hybrid and wild-type polymerases. In Figure 3A, we show the 32P-labeled primer extension products following synthesis after a single binding event. The length of the products of the reaction can be visualized on a polyacrylamide gel and demonstrate that the full-length Taq DNA polymerase protein displays a maximal processivity of 50–80 nt that was unaffected by thioredoxin addition. The hybrid Taq DNA pol/TBD protein exhibits a slightly reduced processivity in the absence of thioredoxin; however, in the presence of a 1.7 × 106-fold molar excess of thioredoxin the majority of the products are >300 nt in length. In the absence of thioredoxin, the Taq DNA polymerase (exo–) displays a range of products indicating a lower processivity of 1–25 nt compared with that of the full-length Taq DNA polymerase and processivity is not increased upon addition of thioredoxin. The processivity of Taq DNA pol/TBD(exo–) is the lowest, with a range from 1 to 10 nt. The addition of thioredoxin stimulated both the Taq DNA pol/TBD and Taq DNA pol/TBD(exo–) hybrid proteins to produce a population of slowly migrating extension products, many of which were too large to enter the gel. Serial dilution of the polymerase enzyme resulted in fewer larger products rather than shorter products, indicating that the increased shift in product size observed between the hybrid enzymes was not an increase in copying of the DNA template caused by the addition of thioredoxin (Fig. 3B).
To further quantify the processivity of the polymerases we developed a novel assay to establish the maximum length of extension of products produced during a single binding event. Primed single-stranded DNA immobilized to a streptavidin bead was incubated with the polymerase in the presence and absence of a 10 000-fold molar excess of thioredoxin. Extension was commenced by addition of dNTPs and Mg2+ in the presence of a large excess of competitor DNA. Under these conditions the enzyme extends the immobilized DNA until dissociation, thereafter it binds onto the excess of trap DNA. After washing, the extension products are cleaved at designated distances from the primer terminus using a series of restriction enzymes. As only double-stranded DNA is cleaved, the fragments released into solution allow the length of the extension product sizes to be determined. The result, when expressed as a percentage of the total signal obtained when all primers are fully extended, is shown for each restriction digest in Figure 4. For the Taq DNA polymerase (exo–) enzyme, >60% of the primers were extended up to 18 nt, 25% reached 96 nt and <1% of primers were extended by 492 nt or greater. The Taq DNA pol/TBD(exo–) hybrid protein in the absence of thioredoxin resulted in only 10% of the primers becoming extended to 18 nt, and ∼3% reached 96 nt. Upon addition of thioredoxin, the Taq DNA pol/TBD(exo–) enzyme increased to 40% of primers becoming extended to 18 and 96 nt, in concordance with the increased activity observed in Table 1. Moreover, significant extension to longer products was observed: 26, 11 and 7% of the reaction products obtained lengths >492, >1122 and >1898 nt, respectively. DNA products of this length were not detected using the Taq DNA polymerase (exo–) enzyme which has an average processivity of ∼1–25 nt (Fig. 3). This represents an increase of 20–50-fold in processivity.
Figure 4.

Streptavidin processivity assay. An immobilized single-stranded DNA molecule of 2000 nt in length was incubated in a reaction containing a primer hybridized to the 5′ end, and polymerase. Extension was initiated by the addition of dNTPs including [α-32P]dGTP, Mg2+ and 0.8 mg/ml activated calf thymus DNA as described in Materials and Methods. Cleavage with restriction enzymes located 18, 96, 492, 1122 and 1898 nt, respectively, from the primer terminus only occurs if primer extension results in a double-stranded DNA substrate. Full extension with 5 U Promega Taq DNA polymerase in the absence of trap DNA allowed the percentage of primers extended to be determined.
PCRs with Taq DNA pol/TBD
PCR was performed using λ phage as the template DNA and the primers listed in Materials and Methods with equivalent units of both Taq DNA polymerase and Taq DNA pol/TBD + thioredoxin activity (2.5 U per reaction). The Taq DNA pol/TBD polymerase was as able as Taq DNA polymerase to perform PCR when thioredoxin was included in the reaction mix (Fig. 5) producing up to a 5 kb fragment. In the absence of the thioredoxin cofactor, the Taq DNA pol/TBD enzyme is unable to amplify even the 1 kb fragment and is incapable of amplification of any fragments larger than 350 bp (data not shown). Interestingly, there were fewer non-specific amplification products present in the Taq DNA pol/TBD + thioredoxin amplified DNA compared with the Taq DNA polymerase amplified DNA.
Figure 5.
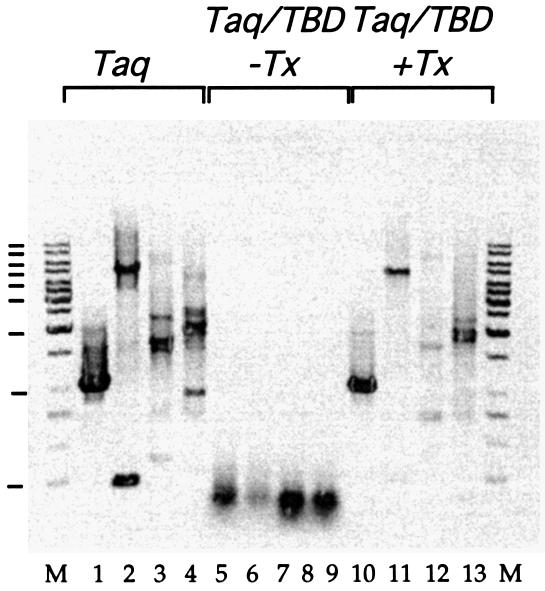
PCR using 1 ng of Lambda phage DNA as template. Ethidium bromide stained 0.8% agarose gel (image inverted). Lane M, 1 kb marker (GeneRuler); lanes 1–4, Taq DNA polymerase amplified 1, 5, 7 and 10 kb; lanes 5–8, Taq DNA pol/TBD no thioredoxin (–Tx) 1, 5, 7 and 10 kb; lanes 9–12, Taq DNA pol/TBD + thioredoxin (+Tx) 1, 5, 7 and 10 kb. Molecular weight marker sizes are indicated by the dark bars on the left of the gel (from top to bottom: 10, 8, 6, 5, 4, 3, 2, 1 and 0.25 kb).
Base substitution errors during PCR
Incorporated errors made during PCR were measured by cloning PCR products and sequencing. The overall fidelity of replication was not significantly different between Taq DNA pol/TBD and Taq DNA polymerase enzyme. However, the processive Taq DNA pol/TBD enzyme showed 2–3-fold fewer AT→GC transitions (P = 0.0014 measured by chi-square) compared with Taq DNA polymerase (Table 2). The other classes of base substitution were not statistically significantly different as the number of observed events were too few.
Table 2. Base substitution errors during PCR.
| Taq DNA polymerase | Taq/TBD | |||
|---|---|---|---|---|
| Mutations | Frequency (×10–4) | Mutations | Frequency (×10–4) | |
| AT→TA | 5 | 2.0 | 9 | 4.60 |
| AT→CG | 1 | 0.40 | 3 | 1.50 |
| AT→GC | 40 | 16.3 | 14a | 7.10 |
| CG→TA | 1 | 0.40 | 3 | 1.50 |
| Total | 47 (24 531) | 19.2 | 29 (19 631) | 14.8 |
Base substitution frequency was determined by sequencing cloned PCR products amplified by either Taq DNA polymerase or Taq DNA pol/TBD. Total base pairs sequenced for each enzyme are shown in brackets.
aP < 0.005 (chi-squared).
Fidelity of replication of repetitive DNA during PCR
Two methods were employed to determine the slippage frequency obtained during PCR with the highly processive Taq DNA pol/TBD versus Taq DNA polymerase. PCR fragments containing A10, G10 or CA11 repeats generated with Taq DNA polymerase or Taq DNA pol/TBD in the presence of thioredoxin were made. Cloning into the frame shift reporter vector enabled determination of frame shifting frequencies in the PCR samples. The frequency of frame shift mutations in the A10 or CA11 microsatellites were both decreased by 6- and 7-fold, respectively, when the Taq DNA pol/TBD enzyme was used compared with Taq DNA polymerase, while the frequency of G10 frame shift mutations was unchanged (Fig. 6). Sequencing of five revertant clones from each miscrosatellite showed that deletion mutations only occurred within the microsatellite regions, as expected. Slippage alleles were separated and quantitated from DNA amplified from pAJ32 containing the A10 microsatellite. Total slippage was determined by dividing the area of each slippage peak by the total area of all peaks. A decrease of 4-fold in slippage peak was observed in DNA obtained from processive amplification using Taq DNA pol/TBD compared with Taq DNA polymerase. Slippage chromatograms are shown in Figure 7.
Figure 6.

Slippage frequency was determined by the number of frame shifted revertant colonies (Tet+ Carb+) divided by the total number of cells plated (Tet+). The result is representative of one experiment. Similar results were obtained in two similar separate experiments. The background frequency of frame shift molecules for template and vector DNA was determined to be <1 revertant per 105.
Figure 7.

Slippage chromatograms were obtained from PCR products amplified with either Taq DNA polymerase or Taq DNA pol/TBD. One primer was labeled with 6-FAM fluorophore and the PCR product was digested with EcoRI. The DNA was gel purified and slippage polymorphisms detected using an automated DNA sequencer (model 377; Applied Biosystems) and GENESCAN 672 software. The result is one representative of three experiments.
DISCUSSION
In general, DNA polymerases possess a remarkably similar three-dimensional structure even amongst enzymes with considerable sequence diversity (12). The generalized polymerase structure looks vaguely like a right hand, with fingers, palm and thumb subdomains (12,13). The catalytic domain is located in the palm, the fingers bind incoming nucleotides and make contacts with the single-stranded template, while the thumb grips the newly synthesized double-stranded DNA (12). It is these contacts made by the thumb domain that largely determine processivity in Pol I family enzymes. For example, deletion of eight amino acids from the thumb in the Klenow fragment reduces the DNA binding affinity by >100-fold and greatly reduces the enzyme processivity (14).
We have shown that insertion of the T3 bacteriophage TBD into the Taq DNA polymerase at the analogous position in the thumb domain, resulted in the formation of a hybrid protein that becomes stimulated by thioredoxin to produce highly processive DNA synthesis at elevated temperature. Insertion of the T3 thioredoxin loop resulted in both full-length and N-terminally truncated hybrid Taq DNA pol/TBD having ∼15% of the activity of their wild-type counterparts at 60°C in the absence of thioredoxin. Prolonged exposure to high temperature (94°C) did not result in less thermostability for either Taq DNA pol/TBD or Taq DNA pol/TBD(exo–) protein compared with the Taq DNA polymerase enzymes. In addition, the truncated Taq DNA pol/TBD(exo–) hybrid polymerase retained the enhanced thermostability previously observed for Taq DNA polymerase (exo–) polymerase (11). The decreased activity of the hybrid proteins may be due to imperfect folding, or perhaps interference of DNA binding or catalysis by the added TBD. Alternatively, the slight reduction in processivity observed for the hybrid enzymes (Fig. 3) could contribute to the decreased activity observed. Thioredoxin increased the activity of both hybrid proteins with Taq DNA pol/TBD(exo–) having 30-fold more activity compared with Taq DNA pol/TBD(exo–) in the absence of thioredoxin cofactor. As Taq DNA polymerase has only 25% amino acid identity with T3 DNA polymerase, this lack of similarity between the proteins at the primary sequence supports the notion that the TBD of T3 polymerase is by itself sufficient to confer processivity.
Thioredoxin binds the T7 DNA polymerase with high affinity, having a Kd of 15 nM at 37°C (5). The Taq DNA pol/TBD hybrids requires high micromolar concentrations of thioredoxin in order to convert the majority of polymerase molecules from low to high processive replication at 60°C (Fig. 3). Thioredoxin itself is remarkably thermostable, having a melting temperature of 88°C (15), suggesting that denaturation of thioredoxin is not the reason for the requirement for high concentrations of thioredoxin in order to confer processive replication by the Taq DNA pol/TBD enzymes at 60°C. Addition of thioredoxin to the PCR stimulated the Taq DNA pol/TBD enzyme sufficiently to enable amplification of upwards of 5 kb in length, although pieces larger than that produced by Taq DNA polymerase polymerase were not observed (Fig. 5). This suggests that maximum amplicon size is not limited by processivity, but rather frequency of misincorporations resulting in formation of poorly extended mismatched 3′ termini (16). Addition of a proof-reading exonuclease-proficient thermostable polymerase to the Taq DNA pol/TBD reaction might alleviate this restriction and allow for synthesis of larger amplicons than the current capability of Taq DNA polymerase.
The Taq DNA pol/TBD hybrid enzyme showed slightly lower overall misinsertion frequencies, which were not significantly different to that of Taq DNA polymerase. However, when the spectrum of base substitutions was analyzed the rate of AT→GC transitions were 2–3-fold lower that that observed for Taq DNA polymerase (Table 2). This finding is contrary to what was observed for both T7 DNA polymerase and Pol γ in the absence of their respective processivity cofactors (17,18). Unlike these polymerases, Taq DNA polymerase does not have a proof-reading 3′–5′ exonuclease activity, relying instead on an extremely low mismatch extension efficiency (19). That fewer C or G nucleotides were misincorporated across from template A or T positions may be due to a higher stringency against mismatched T-G or C-A extensions or alternatively an increased discrimination at the active site for misinsertion of C or G opposite A or T.
In addition to the more accurate replication of non-iterated DNA, processive synthesis of microsatellite DNA is also reflected in the reduced frequency of slippage during PCR by 6–7-fold for the poly(A/T) and poly(CA/TG). Slippage in copying poly(G/C) DNA was not reduced. It is interesting to speculate that the lack of slippage in copying poly(G/C) may be related to the increased base substitution fidelity we observed, perhaps by an alteration of active site conformation by the thioredoxin complex resulting in changes in the kinetics of nucleotide incorporation. Alternatively, the processivity across different microsatellite regions may differ due to formation of alternative DNA structures that impede polymerase progression allowing dissociation and slippage to occur.
Mutation in mismatch repair results in enhanced mutagenesis and is specifically characterized by microsatellite instability in vivo (20). Defective mismatch repair is principally associated with hereditary non-polyposis colorectal carcinoma (HNPCC), which is typified by microsatellite instability (21). Slippage rates are particularly high following amplification of microsatellite DNA in vitro (22) and is a limitation on the use of PCR-based technology to measure endogenous rates of microsatellite instability (23). This is important since microsatellite instability is a common occurrence in many tumors (24). We have generated a PCR-competent polymerase with high processivity and enhanced fidelity, which should be useful in strategies that require more accurate amplification of microsatellite DNA.
Acknowledgments
ACKNOWLEDGEMENTS
Support for this work was received from NCI grants CA80993 and CA78885 as well as the NIH molecular training program in cancer research 5 T32 CA09437.
REFERENCES
- 1.Sia E.A., Jinks-Robertson,S. and Petes,T.D. (1997) Genetic control of microsatellite stability. Mutat. Res., 383, 61–70. [DOI] [PubMed] [Google Scholar]
- 2.Kunkel T.A. and Alexander,P.S. (1986) The base substitution fidelity of eucaryotic DNA polymerases. Mispairing frequencies, site preferences, insertion preferences and base substitution by dislocation. J. Biol. Chem., 261, 160–166. [PubMed] [Google Scholar]
- 3.Himawan J.S. and Richardson,C.C. (1996) Amino acid residues critical for the interaction between bacteriophage T7 DNA polymerase and Escherichia coli thioredoxin. J. Biol. Chem., 271, 19999–20008. [DOI] [PubMed] [Google Scholar]
- 4.Tabor S., Huber,H.E. and Richardson,C.C. (1987) Escherichia coli thioredoxin confers processivity on the DNA polymerase activity of the gene 5 protein of bacteriophage T7. J. Biol. Chem., 262, 16212–16223. [PubMed] [Google Scholar]
- 5.Huber H.E., Tabor,S. and Richardson,C.C. (1987) Escherichia coli thioredoxin stabilizes complexes of bacteriophage T7 DNA polymerase and primed templates. J. Biol. Chem., 262, 16224–16232. [PubMed] [Google Scholar]
- 6.Bedford E., Tabor,S. and Richardson,C.C. (1997) The thioredoxin binding domain of bacteriophage T7 DNA polymerase confers processivity on Escherichia coli DNA polymerase I. Proc. Natl Acad. Sci. USA, 94, 479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pajunen M.I., Elizondo,M.R., Skurnik,M., Kieleczawa,J. and Molineux,I.J. (2002) Complete nucleotide sequence and likely recombinatorial origin of bacteriophage T3. J. Mol. Biol., 319, 1115–1132. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki M., Baskin,D., Hood,L. and Loeb,L.A. (1996) Random mutagenesis of Thermus aquaticus DNA polymerase I: concordance of immutable sites in vivo with the crystal structure. Proc. Natl Acad. Sci. USA, 93, 9670–9675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bambara R.A., Fay,P.J. and Mallaber,L.M. (1995) Methods of analyzing processivity. Methods Enzymol., 262, 270–280. [DOI] [PubMed] [Google Scholar]
- 10.Jackson A.L., Chen,R. and Loeb,L.A. (1998) Induction of microsatellite instability by oxidative DNA damage. Proc. Natl Acad. Sci. USA, 95, 12468–12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawyer F.C., Stoffel,S., Saiki,R.K., Chang,S.Y., Landre,P.A., Abramson,R.D. and Gelfand,D.H. (1993) High-level expression, purification and enzymatic characterization of full-length Thermus aquaticus DNA polymerase and a truncated form deficient in 5′ to 3′ exonuclease activity. PCR Methods Appl., 2, 275–287. [DOI] [PubMed] [Google Scholar]
- 12.Patel P.H., Suzuki,M., Adman,E., Shinkai,A. and Loeb,L.A. (2001) Prokaryotic DNA polymerase I: evolution, structure and ‘base flipping’ mechanism for nucleotide selection. J. Mol. Biol., 308, 823–837. [DOI] [PubMed] [Google Scholar]
- 13.Joyce C.M. and Steitz,T.A. (1994) Function and structure relationships in DNA polymerases. Annu. Rev. Biochem., 63, 777–822. [DOI] [PubMed] [Google Scholar]
- 14.Minnick D.T., Astatke,M., Joyce,C.M. and Kunkel,T.A. (1996) A thumb subdomain mutant of the large fragment of Escherichia coli DNA polymerase I with reduced DNA binding affinity, processivity and frameshift fidelity. J. Biol. Chem., 271, 24954–24961. [DOI] [PubMed] [Google Scholar]
- 15.Lu Z., DiBlasio-Smith,E.A., Grant,K.L., Warne,N.W., LaVallie,E.R., Collins-Racie,L.A., Follettie,M.T., Williamson,M.J. and McCoy,J.M. (1996) Histidine patch thioredoxins. Mutant forms of thioredoxin with metal chelating affinity that provide for convenient purifications of thioredoxin fusion proteins. J. Biol. Chem., 271, 5059–5065. [PubMed] [Google Scholar]
- 16.Cheng S., Fockler,C., Barnes,W.M. and Higuchi,R. (1994) Effective amplification of long targets from cloned inserts and human genomic DNA. Proc. Natl Acad. Sci. USA, 91, 5695–5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Longley M.J., Nguyen,D., Kunkel,T.A. and Copeland,W.C. (2001) The fidelity of human DNA polymerase gamma with and without exonucleolytic proofreading and the p55 accessory subunit. J. Biol. Chem., 276, 38555–38562. [DOI] [PubMed] [Google Scholar]
- 18.Kunkel T.A., Patel,S.S. and Johnson,K.A. (1994) Error-prone replication of repeated DNA sequences by T7 DNA polymerase in the absence of its processivity subunit. Proc. Natl Acad. Sci. USA, 91, 6830–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang M.M., Arnheim,N. and Goodman,M.F. (1992) Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res., 20, 4567–4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vaish M. and Mittal,B. (2002) DNA mismatch repair, microsatellite instability and cancer. Indian J. Exp. Biol., 40, 989–994. [PubMed] [Google Scholar]
- 21.Perucho M. (1996) Cancer of the microsatellite mutator phenotype. Biol. Chem., 377, 675–684. [PubMed] [Google Scholar]
- 22.Shinde D., Lai,Y., Sun,F. and Arnheim,N. (2003) Taq DNA polymerase slippage mutation rates measured by PCR and quasi-likelihood analysis: (CA/GT)n and (A/T)n microsatellites. Nucleic Acids Res., 31, 974–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oda S., Maehara,Y., Sumiyoshi,Y. and Sugimachi,K. (2002) Microsatellite instability in cancer: what problems remain unanswered? Surgery, 131, S55–S62. [DOI] [PubMed] [Google Scholar]
- 24.Eshleman J.R. and Markowitz,S.D. (1995) Microsatellite instability in inherited and sporadic neoplasms. Curr. Opin. Oncol., 7, 83–89. [PubMed] [Google Scholar]