


Abstract
We identified a type II topoisomerase enzyme from Leishmania infantum, a parasite protozoon causing disease in humans. This protein, named Li topo II, which displays a variable C-terminal end, is located in the kinetoplast. The cloned gene encoding Li-TOP2 compensates for the slow growth of topo II-deficient mutants of Saccharomyces cerevisiae, resulting in a catalytically active DNA topoisomerase in yeast. Analysis of the specific mRNA levels of the Li-TOP2 gene showed variations throughout the parasite cell cycle in synchronized cells as well as between the distinct forms of the parasite. Thus, the enzyme had higher levels of mRNA expression in the highly infective intracellular form of the parasite, the amastigote, than in the extracellular promastigote form, suggesting a relation with the distinct developmental and infectious phases of the protozoon. In addition, western blot analysis showed differences in protein expression between the proliferative and non-proliferative forms of L.infantum promastigotes, which displayed similar levels of mRNA. This indicated possible post-transcriptional regulation mechanisms. The data suggest that Li topo II has a part in DNA decatenation and probably at the initial stages of proliferation in the intracellular form of L.infantum, a parasite that has to proliferate into the host macrophage to survive its hostile environment in its first moments of intracellular infection.
INTRODUCTION
The parasite protozoa of the genus Leishmania cause disease in more than 15 million people. Leishmaniasis is endemic in the tropical and subtropical regions of Africa and America, the Indian subcontinent and South Asia. It is associated in many cases with AIDS patients as an opportunistic disease (1,2). The parasite has two forms, the extracellular promastigotes, which increase its infectivity before entering the host macrophage, and the intracellular amastigotes (3). Its developmental process, related to parasite infectivity, can be mimicked by promastigotes in liquid medium at the laboratory (4).
The genus Leishmania is among the most primitive eukaryotes. It belongs to the order of Kinetoplastida, which bear single mitochondria called kinetoplasts (5). Kinetoplasts contain an unusual structure formed by a disc-shaped network of catenated DNA circles (6,7). These circles are divided into two categories: minicircles (0.5–3 kb), up to several thousand of which are present, and maxicircles (30–40 kb) of which there are only a few dozen (8). Minicircles encode the small guide RNAs that regulate RNA editing, whereas most maxicircles encode ribosomal RNAs and proteins involved in energy transduction (9). The replication of kinetoplast DNA is another unusual feature because it occurs in synchrony with the replication of nuclear DNA (10), unlike mitochondrial DNA from higher eukaryotes which replicates through the whole cell cycle (11). During replication, minicircles interlocked with several neighboring molecules are released from the interior of the network towards the external part of the structure. The daughter chains are then re-attached to the network periphery (12). These complex topological problems are solved by the action of type II topoisomerases (13,14). These enzymes use ATP to transport one DNA double-helix backbone through a transient double-strand break that they create in another (15,16). This mechanism allows type II topoisomerases to relax, catenate–decatenate and knot–unknot DNA molecules. These enzymes are essential in DNA replication and chromosome segregation, and are also involved in the maintenance of chromosome structure (17,18). Eukaryotic type II topoisomerases are homodimeric enzymes comprising three functional domains. The highly conserved N-terminal domain, which contains the ATP binding pocket; the core domain, with the active site for DNA cleavage-rejoining; and the less conserved C-terminal domain, where the regulatory elements such as phosphorylation sites and localization signals are located.
Type II DNA topoisomerase activity is specifically targeted by various agents, most of which interfere in the DNA rejoining reaction catalyzed by the enzyme. These compounds lead to DNA breaks, which induce subsequent genome fragmentation and cell death (15). Therefore, eukaryotic type II topoisomerases have emerged as an ideal target for antitumoral or antiparasite drugs (19–21). In the case of Trypanosomatidae, these enzymes have an additional specificity because they function in the kinetoplast, a structure that is absent in higher eukaryotes. Nevertheless, the use of type II DNA topoisomerases as targets for screening of specific inhibitors is not an easy task due to difficulties in preserving the stability of the enzyme, mainly its regulatory domain (15). To our knowledge, no purification of any recombinant type II topoisomerase from Trypanosomatidae has been reported (22–24).
We describe here the cloning and functional analysis of a gene encoding a type II DNA topoisomerase from Leishmania infantum, the parasite protozoon responsible for visceral leishmaniasis in the Mediterranean basin. The gene was identified by sequence comparison and through complementation experiments in Saccharomyces cerevisiae. Immuno fluorescence experiments revealed that the gene product is a kinetoplast-located enzyme, which achieves higher levels of expression in the infective intracellular form of the parasite following post-transcriptional regulation. The relevance of these data for the different phases of the parasite cycle is discussed.
MATERIALS AND METHODS
Materials
The parasite strain (MHOM/FR/80/LEM75) was kindly provided by Dr J. Alvar (Reference Parasitology Service, Majadahonda, Madrid, Spain). Leishmania infantum promastigotes were grown at 26°C in RPMI-1640 (Gibco BRL, Scotland, UK). They were supplemented with 10% heat-inactivated fetal calf serum (Gibco BRL, Scotland, UK), penicillin (50 U/ml) and streptomycin (50 µg/ml). Promastigote cultures were initiated at 1 × 106 cells/ml and harvested at logarithmic and stationary phases of growth, as defined by morphology and cell concentration (25).
The amastigote phase of the parasite was obtained by infecting an X-ray-irradiated (100 rads in a single exposition) monolayer of J774 cells grown on 75-cm2 tissue culture flasks (Costar, Cambridge, MA, USA) with stationary phase promastigotes (107 cells/ml) in culture medium (20:1, parasite/cell ratio). The infected cells were incubated at 37°C in a 5% CO2 atmosphere for 6 days to allow development of the L.infantum amastigotes into macrophages (26). This process was followed up by phase-contrast microscopy (ELWD 0.3; Nikon, Japan).
Synchronous cultures
Leishmania infantum promastigotes (7 × 106 cell/ml) were incubated for 6 h in medium containing 200 µg/ml hydroxyurea (27). Synchronous cells were harvested by centrifugation at 2000 g for 10 min at room temperature and suspended in the same volume of fresh medium lacking hydroxyurea. Leishmania infantum promastigotes remained synchronous for ∼6 h. Samples of 10 µg of RNA were taken every 30 min for further northern blot analysis (see below).
Oligonucleotides and DNA probes
Oligonucleotides LiF1 (AAGCTGACGAACATTCTTTCCACTAA) and LiR2 (GAGGCCTCGCCGTGGTGAAAC), designed according to the conserved regions of the sequences of the DNA TOP2 family, were used for reverse transcription–polymerase chain reaction (RT–PCR) amplification under the following conditions: with 1 min at 95°C, 1 min at 55°C and 2 min at 72°C, a fragment of 1985 bp was obtained. The 5′-terminal end was amplified with the spliced leader oligonucleotide SL (5′-ATCAGTTTCTGTACTTTATTG) and the LiR3 oligonucleotide (5′-AGACGACGGAGAACTTAGTGGAAAG). The 3′-terminal end was amplified (3′-RACE) with a specific oligonucleotide from the known sequence LiF2 (5′-CGGTGCGGGCCATGATCAAC) and the oligo(dT) primer [5′-GCGCCAGGAATTCGC (dT17)]. The DNA probes for the encoding sequence of Li-TOP2 were labeled with [α-32P]dCTP, using the Random Primed DNA Labeling Kit (Boehringer Mannheim, Germany).
Expression of L.infantum TOP2 in Escherichia coli
The open-reading frame containing the DNA TOP2 from L.infantum was cloned into a pQE/.REP4 bacterial expression vector, which allows the expression of recombinant proteins as fusions with a multifunctional leader peptide containing a hexahystidyl sequence for purification on Ni++-affinity resins (28). The open-reading frame of Li-TOP2 was PCR-amplified using oligonucleotides containing SphI and HindIII restriction sites, 5′TOP SphI (5′-GCGCGCATGCATGACAGACGCTTCCA-3′) and 3′TOP HindIII (5′-GCGCAAGCTTTTACATCAAAACATGTGGC-3′). The resulting 3.7 kb PCR product was cloned at the SphI/HindIII sites of vector pQE30 (Qiagen, Germany). The construction of the recombinant expression plasmid, named pQ30-Li TOP2, was confirmed by DNA sequencing. Expression of the His6-tagged protein was carried out in the M15 E.coli strain, which contains the pREP4 repressor plasmid and the T5 promoter under the control of isopropyl-β-d-thiogalactopyranoside (IPTG)-regulated lac I gene (29). Cells were transformed with the pQE30 plasmid and grown in 37°C Luria–Bertani medium until the optical density (OD) reached 0.6 (∼6 h). Then, IPTG was added to a final concentration of 1 mM and the incubation was continued for 4 h at 26°C to reduce the inclusion bodies. Cells were collected by centrifugation at 4000 g for 20 min and lysed in buffer A (6 M Gu-HCl, 0.1 M sodium phosphate, 10 mM Tris–HCl, pH 8.0). The suspension was cleared by centrifugation at 10 000 g for 20 min and the supernatant containing the recombinant protein was used for the purification process. A 1.1 kb fragment from the N-terminal domain was amplified using a sense primer with a SphI site 5′-GCGCGCATGCATGACAGACGCTTCCA-3′ and an antisense primer with a HindIII site 5′-GCGCAAGCTTTTAGAGCTGGTCGTCCAT-3′. The amplification product was cloned at the SphI/HindIII sites of vector pQ30 (Qiagen) to obtain a pQ30-LiTOP2-1.1 kb construct encoding for a 40 kDa topo II fragment used to obtain specific antiserum. Induction, overproduction and solubility of the recombinant proteins were analyzed by 10% polyacrylamide gel electrophoresis (PAGE) in the presence of SDS and viewed by Coomassie blue staining. Western blot analysis was carried out as described (30,31). In all cases, unless otherwise indicated, 10 µg of protein were loaded per well.
Purification of Li topo II and Li topo II-1.1
Ni++-NTA agarose beads (Qiagen) previously equilibrated with buffer A (see above), were incubated for 30 min at room temperature with the soluble fraction containing the recombinant protein, as described above. The resin was loaded into a column and washed with 10 ml of buffers B (8 M urea, 0.1 M sodium phosphate, 10 mM Tris–HCl, pH 8.0) and C (8 M urea, 0.1 M sodium phosphate, 10 mM Tris–HCl, pH 6.3). The recombinant protein was eluted in 1 ml fractions with 5 ml of buffers D (8 M urea, 0.1 sodium phosphate, 10 mM Tris–HCl, pH 5.9) and E (8 M urea, 0.1 M sodium phosphate, 10 mM Tris–HCl, pH 4.5). The fractions were analyzed by SDS–PAGE. The amount of protein was quantified by the method of Bradford (32).
Southern and northern blot analysis
Genomic DNA was isolated from L.infantum promastigotes following standard procedures, digested with different restriction endonucleases, subjected to electrophoresis in 0.8% agarose gels and transferred to nylon membranes (33). Blots were hybridized at 65°C for 16 h in hybridization buffer (Amersham Pharmacia Biotech, UK), containing 100 µg/ml denatured salmon sperm DNA. Filters were washed once at room temperature for 10 min in 2× SSC followed by two washes for 15 min at 65°C, 2% SDS and two additional washes for 10 min at 65°C in 1× SSC containing 1% SDS. Leishmania infantum total RNA was obtained from the distinct parasite forms and prepared as described (31). Total RNA was isolated from the synchronous culture every 30 min using TRIzol (Gibco BRL, NY). For analysis, 10 µg of total RNA were fractionated in formaldehyde agarose gels and transferred onto nylon membranes following standard procedures (33). The blots were hybridized with the PCR probe described for the Li-TOP2 coding region (see above) and washed as described above. Blots were analyzed by autoradiography and bands were scanned and quantified by using a Molecular Dynamics Computer Densitometer and Image Quant Software 3.3 program. To avoid sampling errors, densitometry scanning of ethidium bromide-stained gels was carried out.
RNA extraction and semi-quantitative RT–PCR
Total RNA was isolated from each life cycle form using the TRIzol Reagent (Gibco BRL, NY) according to the manufacturer’s protocol. RNA from the intracellular amastigotes was obtained directly from the infected macrophages as previously described (34). Before reverse transcription, genomic DNA was removed by incubating with 10 U of DNase I-RNase free (Boehringer Mannheim). Two micrograms of RNA were reverse-transcribed using the First Strand cDNA Synthesis Kit for RT–PCR (Boehringer Mannheim) with random hexamers in a 20 µl reaction. Semi-quantitative RT–PCR conditions were established in our laboratory to allow comparisons between the expression of Li-TOP2 mRNAs and the rRNA S24α transcript. The number of cycles, 26, used in this experiment was kept to a minimum. The relative expression of Li-TOP2 mRNA was determined by using 1 µl of cDNA of the different parasite life cycle forms, 10 µl of a 10× Amplitaq Gold buffer, 2 mM MgCl2, 200 µM of each dNTP, 5% DMSO, 250 mM of forward and reverse primer for the Li-TOP2 gene (TOP2 F33, 5′-TTATTACAGAGGGTGACTCTGCGAA-3′ and TOP2 R35, 5′-AGACCTTTGTAGTACTTGGCCGTG-3′) and for the ribosomal control (rRNA F, 5′-CCCGGGAAACCCAGGTGGAGGCC-3′ and rRNA R, 5′-CCGGTGGATTCGGTTGGTGAGTTGTT-3′), 2.5 U of AmpliTaq Gold DNA polymerase and water to 100 µl. PCR conditions were as follows: denaturation for 30 s at 94°C; annealing for 30 s at 60°C; extension for 30 s at 72°C, for 35 cycles for Li TOP 2 and 25 cycles for rRNA S24α amplification. As a negative control, RT–PCRs were performed in the absence of reverse transcriptase. The size of the Li-TOP2 and rRNA S24α amplicons were 460 and 470 bp, respectively. These were the optimal conditions for the semi-quantitative analysis of Li-TOP2 mRNAs, which was limited to the products generated during the exponential phase of amplification. The PCR products were analyzed on 1.5% TAE agarose gels. Southern transfer and hybridization were performed under the same stringent conditions indicated previously. Blots were hybridized with the probes for the Li-TOP2 gene and the S24α ribosomal control, and analyzed by autoradiography. The bands were scanned and quantified using a Molecular Dynamics Computer Densitometer and the Quant Software 3.3 program. The experiments were carried out independently three times and all points were determined in duplicate.
Enzyme activity determination throughout the parasite life cycle
Cell extracts were prepared from 1 × 107 parasites in each life cycle form. Parasite pellets were washed once with PBS and lysed by the addition of 12 µl of DNA topo II assay buffer (50 mM Tris–HCl pH 8.0, 120 mM KCl, 10 mM MgCl2, 0.5 mM each of ATP and DTT), to which 100 µg/ml PMSF and 2 µg/ml leupeptin had been previously added. The lysate was centrifuged at 12 000 g at 4°C to remove the insoluble cell debris.
Decatenation assays containing 200 ng of kinetoplast DNA (35) were carried out in a final volume of 20 µl of lysis buffer. The reactions were performed at 30°C for 30 min and were stopped by the addition of 6× loading buffer (100 mM EDTA pH 8.0, 60% sucrose, 0.1% bromphenol blue, 0.1% xylene cyanol). The products of the reaction were fractionated by size on a 1% agarose gel containing 0.5 µg/ml ethidium bromide.
Phylogenic comparisons
Three different segments of the Li topo II were used to generate the data matrix to infer the phyletic relationships of the members of the type II topoisomerase family: (i) the complete sequence of the enzyme; (ii) the N-terminal segment including the first 800 amino acids spanning the most conserved portion of the topo II proteins; and (iii) the C-terminal segment spanning from residue 800 to the end of the polypeptide. The proteins aligned are in the SWISS-PROT, GenBank, EMBL and dbEST databases, and the corresponding identifiers are: Homo sapiens α isoform (NP_001058); Homo sapiens β isoform (NP_001059); Mus musculus (NP_035753); Drosophila melanogaster (P15348); S.cerevisiae (P06786); L.infantum (AF86355); Crithidia fasciculata (P27570); Chlorella (NP048939); Arabidopsis thaliana (NP189031). The multiple alignments were carried out using the CLUSTALW program. The phylogenetic trees derived from the multiple alignments were viewed with the Tree View program developed by Rod Page.
Specific antibodies and immunofluorescence localization
New Zealand rabbit specific polyclonal antiserum was obtained with 800 µg of purified Li topo II 40 kDa fragment, administered in four weekly subcutaneous shots: the first one in Complete Freund’s Adjuvant and the following in Incomplete Freund’s Adjuvant. After one additional week, the rabbit was bled, the serum obtained, decomplementarized at 55°C for 1 h and subsequently stored at –20°C. Immunofluorescence was carried out as described (36) with slight modifications. In short, 106/ml promastigotes were washed twice with phosphate-buffered saline (PBS) containing 1% fetal calf serum and 0.1% sodium azide and resuspended in the same PBS solution with identical final cell concentration. Twenty-five microliters were spotted onto 12 mm round cover slips. After 1 h at 37°C, the cells were treated with methanol/acetone (1:1) for 15 min at –20°C. The fixed cells were incubated for 15 min at room temperature in DAPI-methanol (0.1 µg/ml). After an additional washing with methanol, the cells were treated with PBS containing 0.5% Triton X-100 for 5 min and incubated in a humidity chamber for 1 h at room temperature with rabbit polyclonal anti-Li-topoII-40 kDa fragment antibodies, diluted 1:200. After washing in PBS, cells were incubated with anti-rabbit IgG-FITC (Dako, Denmark), diluted 1:2000, for 1 h. After washing with PBS, the slides were mounted with Mowiol (Calbiochem, CA) and observed through a Zeiss Axioplan microscope. The images were captured by a Hamamatsu CCD camera and processed in Adobe Photoshop on a Mackintosh computer.
Complementation of Li-TOP2 gene
Yeast strains. The topoisomerase-deficient strain JCW28 (Δtop1 top2-4) derives from FY251 (TOP1 TOP2 MATα his3-Δ200 leu2-Δl trp1-Δ63 ura3-52). The thermo-sensitive mutation top2-4 was obtained by one-step gene replacement (37), and the null mutation Δtop1 was obtained by the hit-and-run method of gene replacement (38). Conditionally deficient yeast cells were transformed by the lithium acetate method and cultures grown in synthetic selective or rich media (39).
Plasmids. Construction of pEMBL-LiTOP2: the L.infantum topoisomerase II gene was PCR-amplified with the primers LiT2Bam and LiT2Hind. Primer LiT2Bam, 5′-GCGCGGATCCCGTAACCATGTCAATCGCTTCCAAGTATAAGCTC-3′, created a BamHI site (in bold) upstream of the coding sequences (underlined). Primer LiT2Hind, 5′-GCGCAAGCTTTTAGTGATGGTGATGCACAAAACATGTGG-3′ created a HindIII site (in bold) at the end of the coding sequences (underlined). The PCR products were inserted in regular cloning vectors and the BamHI-HindIII DNA fragment containing the topoisomerase II coding sequence was then inserted downstream of the yeast GAL1 promoter in pEMBLyex4 plasmid.
DNA relaxation activity of L.infantum topoisomerase II in Δtop1 top2-4 yeast cells
YeptopA-PGPD (LEU) is a derivative of Yep13, which carries the E.coli topA gene under the yeast GPD promoter. This vector provides constitutive expression of E.coli topoisomerase I in yeast (40). Yeast DNA isolation and blot hybridization DNA from yeast cells was prepared from yeast spheroplasts (41). Blot hybridization (42) used 32P-labeled DNA probes obtained by random priming of gel-purified DNA sequences.
Cultures containing 2% glucose or 2% galactose of JCW28 cells harboring YEptopA-PGPD(LEU) plus pEMBLyex4 or pEMBL-LiTOP2 were set as described (see above). Cultures were shifted to 35°C for 2 h 30 min before cell harvesting. Extracted DNA samples were examined by two-dimensional electrophoresis in a 0.6% agarose gel, run in TBE buffer plus 0.6 and 3 µg/ml chloroquine in the first (top to bottom, 12 h at 60 V) and second (left to right, 6 h at 60 V) dimensions, respectively. The gel was blot-hybridized with a 32P-labeled probe to reveal the 2µ yeast plasmid (6.3 kb).
RESULTS
Identification and cloning of L.infantum TOP2 gene
The complete cDNA encoding sequence of the Li topo II polypeptide was obtained by RT–PCR. First, specific oligonucleotides were designed according to conserved sequences of type II topoisomerases from different species to obtain a central region of the gene of 1985 bp. In a subsequent step, the 5′ and 3′ ends were obtained by the same procedure using specific oligonucleotides from the known sequence and oligonucleotides containing specific sequences for the ‘spliced leader’ and the mRNA poly(A) tail (5′-RACE and 3′-RACE, respectively). Additionally, a 5′ flanking sequence of 520 bp was obtained. The open-reading frame 3711 bp long (GenBank accession no. AF86355), with a high percentage (60.5%) of its codons ending in G or C as described for the genes of the genus Leishmania (43), encodes a 1237 amino acid protein for an estimated molecular mass of 136 kDa, predicted to be a type II topoisomerase.
Leishmania infantum topo II belongs to the type II topoisomerase family
The hypothesis that Li topo II was a member of the type II topoisomerase family was confirmed by multiple amino acid sequence alignment of Li topo II with H.sapiens, D.melanogaster, C.fasciculata and S.cerevisiae topo II proteins, as well as with other Trypanosomatidae. The 1237 amino acid residues of Li topo II were 91% identical with other closely related species, Leishmania chagasi (AAC05295) and Leishmania donovani (AAD34021). In the case of other Trypanosomatidae, there was 78% identity with the topo II of C.fasciculata, and 64 and 61% with Trypanosoma cruzi and Trypanosoma brucei. For topo II enzymes from humans, Drosophila and yeast, identities were 28, 34 and 32%, respectively. Li topo II contained the residues that are invariant in all type II topoisomerases, including the tyrosine involved in enzyme transesterification with the DNA phosphate group (residue 775). Interestingly, Li topo II contained two of the three leading nuclear sequences described (44) at residues 998 (KRRR) and 1165 (PPSKRR) (see below). The alignment also showed that the Li topo II enzyme is shorter than the polypeptides from other eukaryotes, as it has deletions at the N-terminal and C-terminal ends. The similarities between the Li topo II and the rest of the enzymes considered were higher at the N-terminal end and decreased after the active site tyrosine towards the C-terminal end, as shown in Figure 1A, which displays the alignment of the C-terminal ends of the enzymes.
Figure 1.



(A) Multiple amino acid alignment of C-terminal ends of topo II enzymes from different species. Black columns represent identical residues and gray columns represent homologous residues. (B) Phylogenetic trees showing the evolutionary distances between members of the topo II family. The species employed were: H.sapiens α isoform (NP001058); H.sapiens β isoform (NP_001059); M.musculus (NP035753); D.melanogaster (P15348); S.cerevisiae (P06786); L.infantum (AF86355); C.fasciculata (P27570); Chlorella (NP048939); and A.thaliana (NP189031). The multiple alignments were carried out using the CLUSTALW program. The phyletic trees derived from the multiple alignments were viewed by the Tree View program developed by Rod Page. For further experimental details, see Materials and Methods.
Figure 1B shows the phylogenetic trees deduced from the proteins belonging to the type II topoisomerase family from lower and higher eukaryotes and one virus protein. The trees were obtained with the full proteins, with the N-terminal end sequences (Fig. 1B.1) or the more variable C-terminal ends (from residue 948 to the end, Fig. 1B.2). The tree for the complete polypeptides fits with the one for the N-terminal polypeptides. In this tree, Li topo II has an intermediate position close to yeast and between the higher eukaryotes (from human to Drosophila) and the Chlorella and Arabidopsis enzymes which are furthest away from mammals’. In relation to the humans, L.infantum polypeptide was closer to the beta isoform enzyme than to the alpha one. This phylogenetic distance is clearly modified when the tree includes only the protein domains containing the more variable C-terminal ends. In this case, the topo II proteins appear to be grouped not according to their eukaryotic or prokaryotic affiliation, as expected, but to the type of organism considered, uni- or pluricellular. The polypeptide for Leishmania appears at one of the extremes of the tree, close to the Chlorella virus and closer to human than to Drosophila among the higher eukaryotes. In all cases, A.thaliana is at one extreme of the tree. The C-terminal end sequence in Trypanosomatidae and in Leishmania seems to be especially divergent and confers a higher degree of variability on the type II topoisomerase than on the other eukaryotes.
Li topo II over-expression in E.coli
Li topo II was expressed in the E.coli strain M15 (Fig. 2A) (see above). The IPTG induced new polypeptides to migrate to the expected position. Based on the His6 tag present at its N-terminal end, the recombinant protein can be purified in a Ni2+-NTA affinity chromatography column. However, the protein had a transient life as a complete polypeptide and is quickly degraded into minor polypeptides, mainly of 80 and 40 kDa, even in the presence of protease inhibitors (data not shown). Although the purified fraction showed topoisomerase activity (see Materials and Methods), this activity cannot be used as a reference for purification in this case, due to the existence in the recombinant cells of an accompanying activity coming from E.coli, topoisomerase IV, a potent decatenase (45). This topo IV activity may be retained by unspecific binding during the purification process, would not be detectable as a band by PAGE (see below) and might be responsible for the topoisomerase activity of the purified fraction. The expression of Li topo II was attempted in S.cerevisiae, but the recombinant protein expressed was not enough to allow its purification (data not shown).
Figure 2.
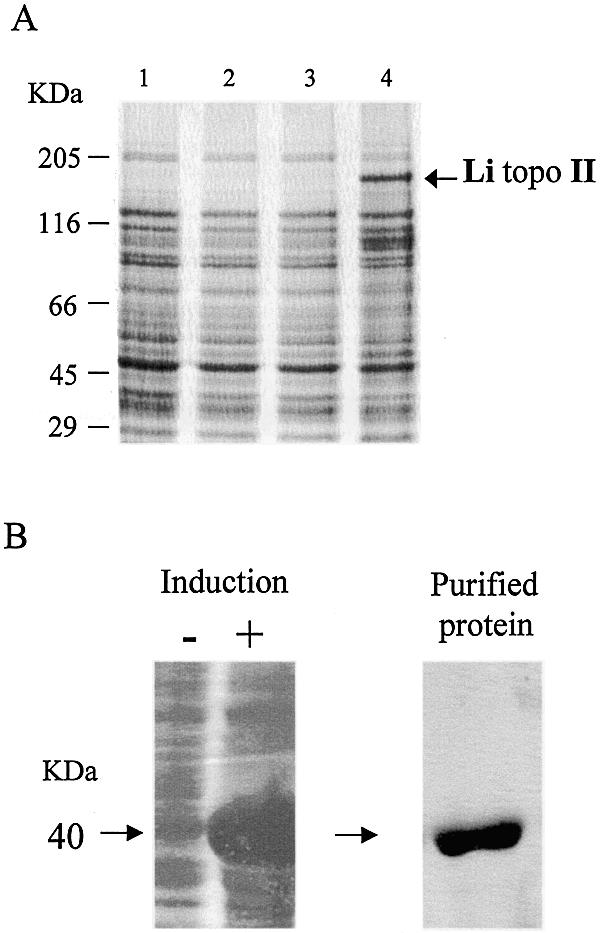
Li topo II over-expression and Li topo II-1.1 purification. (A) Coomassie blue staining of Li topo II over-expression. Lane 1, E.coli M15 strain; lane 2, E.coli M 15 strain harbored with plasmid pQE30; lane 3, E.coli M15 strain harbored with the plasmid pQE30-Li-topo II without IPTG; lane 4, E.coli M15 strain harbored with the plasmid pQE30-Li-topo II after 4 h of addition of IPTG. (B) Expression of Li topo II-1.1 polypeptide fragment (40 kDa) by an E.coli M15 strain harbored with the pQE30-Li topo II-1.1 plasmid before (not induced) and after (induced) the addition of IPTG and protein purification on Ni++-affinity resin. For further experimental details, see Materials and Methods.
A stable 40 kDa fragment of the Li topo II protein was specifically cloned, expressed and purified using an identical procedure (Fig. 2B). This region was chosen because it corresponds to the most conserved region among different type II topoisomerases (see above). This fragment was used to obtain the specific antibody against Li topo II that was fully reactive against the whole protein in western blot and immunofluorescence experiments (see below).
Li topo II is encoded by a single gene and is differentially expressed throughout the cell cycle
The molecular analysis of the TOP2 gene by Southern blotting showed a clear pattern. The enzymes HindIII and XhoI, which showed no target in the encoding sequence, gave a single hybridization band, whereas BclI, ScaI, SmaI and ClaI, which showed a single target within the sequence, gave two hybridization bands (Fig. 3). This analysis and the presence of a single RNA band in the corresponding northern blot analysis (Fig. 4A) indicated the existence of a single encoding gene.
Figure 3.
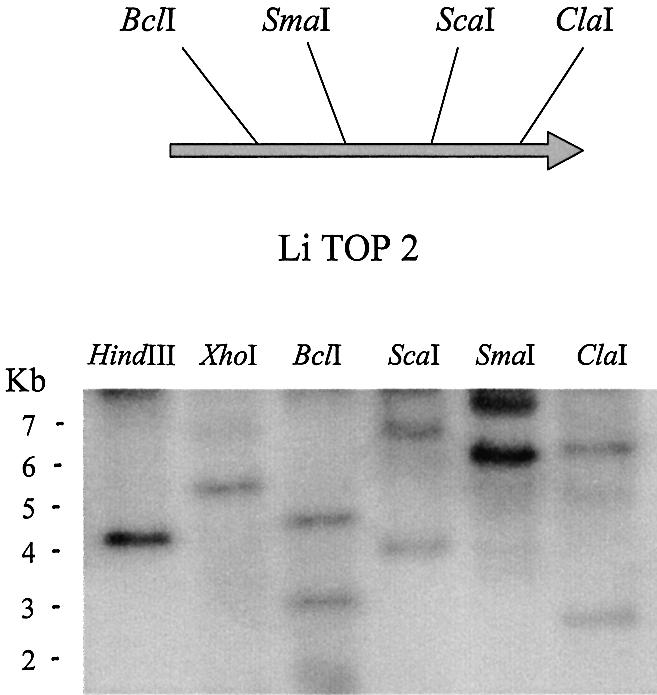
Molecular gene analysis of Li-TOP2 gene. Southern blot analysis and physical map of Li-TOP2 gene deduced from its nucleotide sequence with the location of the restriction enzymes used. Leishmania infantum genomic DNA was subjected to total digestion with the indicated restriction enzymes. For further experimental details, see Materials and Methods.
Figure 4.

Differential expression of Li topo II throughout the cell and parasite life cycle. (A) Northern blot analysis using 5 µg of total RNA extracted from L.infantum promastigotes taken from hydroxyurea-synchronous cell culture at different times (indicated in minutes). (B) Levels of expression by RT–PCR of S24α L.infantum ribosomal protein mRNA (control) and Li topo II mRNA from logarithmic (Log) and stationary (Sta) promastigotes as well as in intracellular amastigotes (Amas). (C) Relative levels of expression of Li topo II mRNA transcripts in the different L.infantum phases: logarithmic (Log), stationary (Sta) promastigotes and amastigotes (Amas). (D) Western blot analysis of the levels of Li topo II at the logarithmic and stationary phases of the parasite. (E) Decatenation activity assay of the Li topo II enzyme in cell extracts from the logarithmic (Log) and stationary (Sta) phases of L.infantum promastigotes. The positions of open circular (OC) and covalently closed circular (CCC) DNA products are indicated. For further experimental details, see Materials and Methods.
In the 5′UTR sequence there is a hexameric ATAGAA sequence that has been considered responsible for mRNA cyclic expression during the cell cycle in C.fasciculata (46). Thus, the expression of Li-TOP2 was analyzed along the distinct cell cycle phases of the parasite. The use of hydroxyurea, which is lethal for the cells in S phase, allows synchronization of the protozoa culture. Once removed from the culture medium, the synthesis of the nuclear and kinetoplast DNA starts, and the synchronous cell cycle lasts at least for two generations. The northern blot analysis of the samples obtained at 30 min periods showed that the levels of Li-TOP2 mRNA varied throughout the cell cycle. A single band for a molecular mass of 4.8 kb with a maximum expression at 60 and 120 min was observed (Fig. 4A).
Semi-quantitative RT–PCR provided a more precise evaluation of the differences in the abundance of Li-TOP2 gene mRNA transcripts during the different life cycle stages of L.infantum (34). Figure 4B shows the curves for the relative levels of expression of Li TOP2 mRNA in logarithmic and stationary promastigote forms, as well as in amastigote forms. As a control, we used the ribosomal transcript S24α, which is expressed at a constant level in all three stages of the life cycle of L.infantum (47). Figure 4C shows the relative ratio (34) resulting from three independent experiments of the Li-TOP2 transcript expression in the distinct forms of the parasite, relative to the constant expression of the ribosomal transcript S24α. The data indicate that there are differences between the distinct forms of L.infantum, with a more than 20% increase in mRNA Li-TOP2 expression in the amastigote intracellular form. Interestingly, the extracellular promastigote form showed no differences in mRNA expression between the proliferative logarithmic phase and the non-proliferative stationary phase.
Li topo II protein expression was measured by western blotting of L.infantum cell extracts obtained from samples taken at the different growth phases. The expression of the protein was clear in promastigotes at the proliferative logarithmic phase, whereas the protein was absent at the infective stationary phase, although here there was a high level of mRNA expression (see Fig. 4C and D). The DNA type II topoisomerase activity was examined by using a decatenation activity assay (see Materials and Methods). The presence of topo II activity in the cell extracts corresponding to logarithmic and stationary promastigotes was measured by decatenation assays in agarose gels (35). Figure 4E shows the presence of bands corresponding to free minicircles in addition to the kDNA band in the samples treated with logarithmic cell extracts, whereas the samples treated with parasite extracts from the stationary phase did not show these minicircle bands, which indicated the absence of topo II activity. Thus, the enzyme is present and functionally active in the logarithmic growth phase of L.infantum and is absent in the non-proliferative stationary phase, regardless of the existence in this phase of mRNA transcripts at similar levels to those in the active logarithmic phase.
Leishmania infantum TOP2 compensates for the slow growth of Δtop1 top2-4 yeast mutants
To verify the functionality of L.infantum TOP2, its compensatory effects in topoisomerase-deficient yeast cells were examined. The Li-TOP2 gene was inserted downstream of the galactose-inducible GAL1 promoter in the yeast multicopy vector pEMBLyex4. Transformants of the topoisomerase-deficient strain JCW28 (Δtop1 top2-4), harboring either pEMBLyex4 or pEMBL-LiTOP2, were grown at 26°C in liquid media containing different amounts of glucose and galactose, and its duplication times were monitored. As summarized in Figure 5, cells carrying pEMBLyex4 duplicated approximately every 8 h in media containing 2% glucose, and approximately every 9 h in media containing 2% galactose. These values are four times slower than those of the parental strain FY251 (TOP1+, TOP2+). However, when JCW28 cells carried pEMBL-LiTOP2, they grew faster as the galactose/glucose ratio steadily grew. Its duplication time was reduced to ∼5 h in media containing 2% galactose. This growth rate compensation is comparable with that achieved when S.cerevisiae topoisomerase I or II is expressed from plasmid-borne genes in the JCW28 strain (48,49). Yet, when a similar experiment was done at 35°C, cells were unable to grow due to the temperature-dependent lethality of the top2-4 mutation. This indicated that the expressed Li topo II was not enough to substitute all essential functions of the yeast topoisomerase II.
Figure 5.

Growth rate of Δtop1 top2-4 yeast cells expressing Li topo II. Transformants of JCW28 cells, transformed either with pEMBLyex4 or pEMBL-LiTopoII, were grown in selective media SD (–ura) containing different amounts (w/v) of galactose and glucose as indicated. Duplication times (min) of each culture were measured during exponential growth.
The product of the Li-TOP2 gene is a catalytically active topoisomerase II
Further evidence that the product of the Li-TOP2 gene is catalytically active was obtained by examination of its effects on DNA topology in yeast cells. In S.cerevisiae, DNA supercoils are relaxed by yeast topoisomerases I and II. Unbalanced relaxation of positive and negative supercoils occurs when yeast topoisomerases are inactivated, and the E.coli topoisomerase I is expressed from a plasmid-borne gene. Because this bacterial enzyme only removes negative supercoils, yeast DNA will rapidly accumulate positive supercoils unless other topoisomerase activity removes them (49,50). The experiment described below shows that the positive supercoiling generated by the unbalanced activity of E.coli topoisomerase I does not accumulate when L.infantum topoisomerase II is expressed.
JCW28 (Δtop1 top2-4) cells carrying YEptopA-PGPD (LEU), a plasmid that provides constitutive expression of E.coli topoisomerase I, were transformed with either pEMBLyex4 or pEMBL-LiTOP2. Single colonies of these transformants were grown at 26°C in selective media (–ura, –leu) containing either 2% glucose or 2% galactose. When cells reached exponential growth (OD ∼0.7), cultures were shifted to 35°C to inactivate yeast topoisomerase II. After 2 h 30 min, cells were harvested and DNA was extracted to examine the topology of the yeast 2µ circle by two-dimensional gel electrophoresis (Fig. 6). When the cells harbored pEMBLyex4 and grew in either glucose (lane 1) or galactose (lane 2), the 2µ circles migrated as a single spot (S+), which corresponds to positively supercoiled DNA in these gel conditions. However, when the cells harbored pEMBL-LiTopoII (lanes 3 and 4), the topology of the 2µ was different. In the cells that grew in glucose (lane 3), positive supercoils accumulated but to a lesser extent than in lanes 1 and 2. In the cells that grew in galactose (lane 4), positive supercoils did not accumulate and most of the 2µ circles migrated as a characteristic arch of topoisomers (R). Therefore, an activity able to remove DNA supercoils was present in the cells harboring pEMBL-LiTopoII that grew in galactose.
Figure 6.
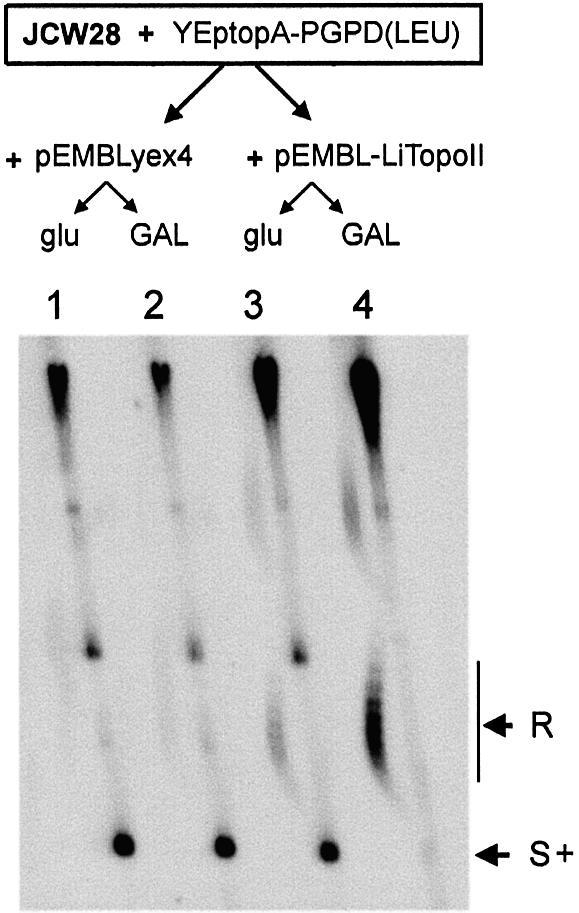
DNA relaxation activity of L.infantum topoisomerase II in Δtop1 top2-4 yeast cells. Cultures containing 2% glucose or 2% galactose of JCW28 cells harboring YeptopA-PGPD(LEU) plus pEMBLyex4 (lanes 1 and 2) or pEMBL-LiTopoII (lanes 3 and 4) were set as described in the text. Cultures were shifted to 35°C for 2 h 30 min before cell harvesting. DNA extracts were examined by two-dimensional electrophoresis, carried out in a 0.6% agarose gel, that was run in TBE buffer plus 0.6 and 3 µg/ml chloroquine in the first (top to bottom, 12 h at 60 V) and second (left to right, 6 h at 60 V) dimensions, respectively. The gel was blot-hybridized with a 32P-labeled probe to reveal the 2µ yeast plasmid (6.3 kb). The positions of the arched distribution of topoisomerse and of the positively supercoiled forms (S+) of the 2µ are indicated. Bands appearing on the upper half of the gel mostly correspond to multimeric forms of the 2µ plasmid, chromosomal DNA fragments and less abundant DNA plasmids.
Localization of Li topo II detected by immunofluorescence
Immunofluorescence detection of Li topo II in Leishmania parasites used specific polyclonal antibodies raised against a 1143 bp (40 kDa) recombinant fragment (see above). Simultaneous DAPI staining enabled the DNA of the nucleus and kinetoplast to be traced. The specific immunofluorescence of the protein appeared as two spots located at the kinetoplast disc (Fig. 7B), coinciding with the mitochondrial topo II at the related species C.fasciculata but different from T.cruzi in which it is located in the nucleus (51,52). This immunofluorescence pattern corresponds to the promastigote form in the logarithmic phase. At the stationary phase, there was no fluorescent signal (data not shown), as expected from the previous data.
Figure 7.

Kinetoplast localization of Li topo II by immunofluorescence. Leishmania infantum promastigotes were recovered from asynchronous cultures at the logarithmic phase. (A) Phase contrast. (B) DNA staining with DAPI shows the nucleus (n) and kinetoplast (k) localization at the promastigotes. (C) Li topo II at logarithmic phase promastigotes. Negative immunofluorescence in stationary phase promastigotes is not shown. Bar 2 µm. For further experimental details, see Materials and Methods.
DISCUSSION
DNA type II topoisomerases evolved to solve the topological problems of DNA, mainly due to its double-helix structure (18). Thus, these enzymes seem to be indispensable for chromosome condensation and segregation (17,53,54), so that the absence of type II topoisomerases coincides with the loss or breakage of chromosomes (54). The structural problems created during DNA replication and transcription (55,56) also require the action of these enzymes to eliminate the resulting torsional stress (18,57,58). The absence of the type II beta enzyme affects neuronal differentiation in mammals (59). Type II alpha is absolutely required for chromosome segregation (60) in higher eukaryotes. In addition, its absence dramatically increases gene recombination in yeasts (48).
A few topo II enzymes with different cell locations have been described in Trypanosmatidae. Thus, whereas in C.fasciculata topo II is located at the kDNA, in T.cruzi the enzyme is located at the nucleus (44,61). In L.donovani it is found at both the nucleus and the kinetoplast (24). Functional inhibitory studies with RNA interference in T.brucei showed a striking loss of kinetoplast DNA after interference of TOP2 transcripts. Free minicircle intermediates accumulated: these coincided with growth inhibition and cell death in many cases (62,63). Thus, whether it is one or two enzymes that act on the nucleus and kinetoplast is not yet resolved.
The Li topo II enzyme described here is located at two antipodal sites flanking the kDNA and not in the nucleus as in T.brucei (62) and the related L.donovani (24). This happens regardless of the existence in the enzyme polypeptide of two out of the three nuclear leading sequences described and a high percentage of identity in the amino acid residues. These differences in location may be explained by the distinct epitopes recognized by the different antisera; although the possibility that Trypanosomatidae may have a different topo II in the kinetoplast than that in the nucleus cannot be excluded. Therefore, it is tempting to speculate about the existence of two closely related enzymes that play a similar role, but are targeted to different subcellular compartments.
This is, to our knowledge, the first TOP2 gene that has been unequivocally identified in Trypanosomatidae by functional studies such as partial gene complementation and measurement of the enzyme activity in yeast. Thus, Li-TOP2 is shown to compensate for the slow growth of functionally active Δtop1-top2-4 yeast cells, as measured by yeast DNA relaxation. The enzyme had a high degree of variability in the C-terminal end domain of the polypeptide, as shown by the phyletic comparisons. The fact that a putative role in chromosome segregation was assigned to this domain through specific interactions with other proteins required for this key cell function (58) makes it especially interesting in a parasite like Leishmania, which does not condense definite chromosomes during mitosis (3).
Li-TOP2 gene is differentially expressed throughout the cell cycle and the different phases of the parasite. Thus, Li topo II is cell cycle regulated, present in the replicative logarithmic phase of the parasite and absent in the non-replicating, infectious metacyclic promastigotes. However, the detected level of mRNA transcripts is similar in both cell stages. Therefore, it seems to be post-transcriptionally regulated. This could be done either by keeping this mRNA pool in a special subcellular organelle unavailable to the translation system or by the action of a specific degradation mechanism of the translated enzyme. The existence of high levels of mRNA at the infective, non-dividing, stationary phase parasites may be useful for the protozoon in the macrophage, once transformed into the amastigote phase, to initiate the rapid division necessary for the survival of the parasite. The existence of a pool of molecules ready to be used could be one adaptive defense mechanism to counteract the macrophage attack. The amount of mRNA even increases in the highly replicative intracellular amastigote but the crucial first hours in the mammal cell may be covered by the Li-TOP2 transcripts coming from the extracellular form of Leishmania.
The cloning and functional characterization of the Li topo II are the first steps towards a more detailed biochemical characterization including in vitro analysis, which together with a model of Li topo II deficiency will serve to determine the relevance of the enzyme to the infectivity and development of L.infantum. This will allow its use as a specific target for anti-parasite drugs, avoiding the treatment failures and subsequent disease relapses that are now spreading visceral leishmaniasis. This has become a health hazard with important economic and social consequences not only in traditionally affected areas but also in the Northern Mediterranean basin.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Dr P. Hernandez for a helpful discussion about the manuscript. This study was partially supported by grants BIO1999-0853 and BIO2000-0149-P4-04 from the Ministry of Science and Technology. T.H. thanks CAM for a fellowship. M.J.R. thanks the Spanish Research Council for a fellowship.
REFERENCES
- 1.Neva F. and Sacks,S. (1995) Leishmaniasis. In Warren,K.S. and Mahmoud,A.A.S. (eds), Leishmaniasis in Tropical and Geographical Medicine. McGraw-Hill, New York, pp. 296–308. [Google Scholar]
- 2.Jimenez M.I., Gutierrez-Solar,B., Benito,A., Aguiar,A., García,E., Carcenado,E. and Alvar,J. (1991) Cutaneous Leishmania infantum zymodems isolated from bone marrow in AIDS patients. Res. Rev. Parasitol., 51, 91–95. [Google Scholar]
- 3.Sacks D.L. and Perkins,P.V. (1984) Identification of an infective stage of Leishmania promastigotes. Science, 223, 1417–1419. [DOI] [PubMed] [Google Scholar]
- 4.Da Silva R. and Sacks,D.L. (1987) Metacyclogenesis is a major determinant of Leishmania promastigote virulence and attenuation. Infect. Immun., 55, 2802–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soguin M.L., Elwood,H.J. and Gunderson,J.H. (1986) Evolutionary diversity of eukaryotic small-subunit rRNA genes. Proc. Natl Acad. Sci. USA, 83, 1383–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donelson J.E., Gardner,M.J. and El-Sayed,N.M. (1999) More surprises from kinetoplastida. Proc. Natl Acad. Sci. USA, 96, 2579–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alexeieff A. (1917) Sur la fonction plycoplastique du kinetoplaste((kinetonucleus) chez les flagellés. C. R. Soc. Biol., 80, 512–514. [Google Scholar]
- 8.Shapiro T.A. and Englund,P.T. (1995) The structure and replication of kinetoplast DNA. Annu. Rev. Microbiol., 49, 117–143. [DOI] [PubMed] [Google Scholar]
- 9.Sturm N.R and Simpson,L. (1991) Leishmania tarentolae minicircles of different sequence classes encode single guide RNAs located in the variable region approximately 150 bp from the conserved region. Nucleic Acids Res., 19, 6277–6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cosgrove W.B. and Skeen,M.J. (1970) The cell cycle in Crithidia fasciculata. Temporal relationship between synthesis of deoxyribonucleic acid in the nucleus and the kinetoplast. J. Protozool., 17, 172–177. [DOI] [PubMed] [Google Scholar]
- 11.Clayton D.A. (1991) Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell. Biol., 7, 453–478. [DOI] [PubMed] [Google Scholar]
- 12.Englund P.T. (1978) The replication of DNA networks in Crithidia fasciculata. Cell, 14, 157–168. [DOI] [PubMed] [Google Scholar]
- 13.Englund P.T. (1979) Free minicircles of kinetoplast DNA in Crithidia fasciculata. J. Biol. Chem., 254, 4895–4900. [PubMed] [Google Scholar]
- 14.Melendy T., Sheline,C. and Ray,D.S. (1988) Localization of type II DNA topoisomerase to two sites at the periphery of the kinetoplast DNA of Crithidia fasciculata. Cell, 55, 1083–1088. [DOI] [PubMed] [Google Scholar]
- 15.Cheesman S.J. (2000) The topisomerases of protozoan parasites. Parasitol. Today, 16, 277–281. [DOI] [PubMed] [Google Scholar]
- 16.Roca J. (1995) The mechanism of DNA topoisomerases. Trends Biochem. Sci., 20, 156–160. [DOI] [PubMed] [Google Scholar]
- 17.Wang J.C. (1996) DNA topoisomerases. Annu. Rev. Biochem., 65, 635–692. [DOI] [PubMed] [Google Scholar]
- 18.Wang J.C. (2002) Cellular roles of DNA topoisomerases: a molecular perspective. Nature Rev., 3, 430–440. [DOI] [PubMed] [Google Scholar]
- 19.Staruss P.R. and Wang,J.C. (1990) The TOP2 gene of Trypanosoma brucei—a single copy gene that shares extensive homology with other TOP2 genes encoding eukaryotic DNA topoisomerase II. Mol. Biochem. Parasitol., 38, 141–150. [DOI] [PubMed] [Google Scholar]
- 20.Schneider E., Hsiang,Y.H. and Liu,L.F. (1991) DNA topoisomerase as anticancer drug targets. Adv. Pharmacol., 21, 149–183. [DOI] [PubMed] [Google Scholar]
- 21.Shapiro T.A. (1994) Drugs affecting Trypanosome topoisomerases. Adv. Pharmacol., 29, 187–199. [PubMed] [Google Scholar]
- 22.Fragoso S.P. and Goldenberg,S. (1992) Cloning and characterization of the gene encoding Trypanosoma cruzi DNA topoisomerase II. Mol. Biochem. Parasitol., 55, 127–134. [DOI] [PubMed] [Google Scholar]
- 23.Cheesman S., Horrocks,P., Tosh,K. and Kilbey,B. (1998) Intraerythrocytic expression of topoisomerase II from Plasmodium falciparum is developmentally regulated. Mol. Biochem. Parasitol., 42, 39–46. [DOI] [PubMed] [Google Scholar]
- 24.Das A., Dasgupta,A., Sharma,S., Ghosh,M., Sengupta,T., Bandopadhyay,S. and Majumber,H.K. (2001) Characterisation of the gene encoding type II topoisomerase from L.donovani: a key molecular target in anti leishmanial therapy. Nucleic Acids Res., 29, 1844–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zarley J.H., Britigan,B.E. and Wilson,M.E. (1991) Hydrogen peroxide-mediated toxicity for Leishmania donovani chagasi promastigotes. Role of hydroxyl radical and protection by heat shock. J. Clin. Invest., 88, 1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.González-Aseguinolaza G., Taladriz,S., Marquet,A. and Larraga,V. (1999) Molecular cloning, cell localization and binding affinity to DNA replication proteins of the p36/LACK protective antigen from Leishmania infantum. Eur. J. Biochem., 259, 909–916. [DOI] [PubMed] [Google Scholar]
- 27.Pasion S.G., Brown,G.W., Brown,L.M. and Ray,D.S. (1994). Periodic expression of nuclear and mitochondrial DNA during the trypanosomatidae cell cycle. J. Cell Sci., 107, 3515–3520. [DOI] [PubMed] [Google Scholar]
- 28.Kroll D.J., Abedel-MalekAbdel Hafiz,H., Marcell,T., Simpson,S., Chen,C.Y., Gutierrez-Hartman,A., Lustbader,J.W. and Hoeffler,J.P. (1993) A multifunctional prokaryotic protein expression system: overproduction, affinity purification and selective detection. DNA Cell Biol., 12, 441–453. [DOI] [PubMed] [Google Scholar]
- 29.Farabaugh P.J. (1978) Sequence of the lac I gene. Nature, 274, 765–767. [DOI] [PubMed] [Google Scholar]
- 30.Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 31.González-Seguinolaza G., Almazan,F., Rodriguez,F.J., Marquet,A. and Larraga,V. (1997) Cloning of the gp63 surface protease of Leishmania infantum. Differential post-translational modifications correlated with different infective forms. Biochim. Biophys. Acta, 1361, 92–102. [PubMed] [Google Scholar]
- 32.Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 33.Sambrook J., Fristch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 34.Ramiro M.J., Hanke,T., Taladriz,S. and Larraga,V. (2002) DNA polymerase beta mRNA determination by relative quantitative RT–PCR from Leishmania infantum intracellular amastigotes. Parasitol. Res., 88, 760–767. [DOI] [PubMed] [Google Scholar]
- 35.Haldane A. and Sullivan,D. (2001) DNA topoisomerase II-catalyzed DNA decatenation. In Osheroff,N. and Bjornsti,M.A. (eds), DNA Topoisomerase Protocols. Human Press, Totowa, NJ, pp. 13–23. [Google Scholar]
- 36.Taladriz S., Hanke,T., Ramiro,M.J., García-Díaz,M., García de Lacoba,M., Blanco,L. and Larraga,V. (2001) Nuclear DNA polymerase beta from Leishmania infantum. Cloning, molecular analysis and developmental regulation. Nucleic Acids Res., 29, 3822–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rothstein R.J. (1983) One-step gene disruption in yeast. Methods Enzymol., 101, 202–211. [DOI] [PubMed] [Google Scholar]
- 38.Roca J., Gartenberg,M.R., Oshima,Y. and Wang,J.C. (1992) A hit-and-run system for targeted genetic manipulations in yeast. Nucleic Acids Res., 20, 4671–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ito H., Fukuda,Y., Murata,K. and Kimura,A. (1983) Transformation of intact yeast cells treated with alkali cations. J. Bacteriol., 153, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trigueros S. and Roca,J. (2002) A GyrB-GyrA fusion protein expressed in yeast cells is able to remove DNA supercoils but cannot substitute eukaryotic topoisomerase II. Genes Cells, 7, 249–257. [DOI] [PubMed] [Google Scholar]
- 41.Sherman F. (1983) Methods in Yeast Genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 42.Southern E.M. (1975) Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol., 98, 503–517. [DOI] [PubMed] [Google Scholar]
- 43.Broccoli S., Marquis,J.F., Papadopoulou,B., Olivier,M. and Drolet,M. (1999) Characterization of Leishmania donovani gene encoding a protein that closely resembles a type IB topoisomerase. Nucleic Acids Res., 27, 2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pasion S.G., Hines,J.C., Aebershold,R. and Ray,D.S. (1992) Molecular cloning and expression of the gene encoding the kinetoplast associated type II DNA topoisomerase of C.fasciculata. Mol. Biochem. Parasitol., 50, 57–67. [DOI] [PubMed] [Google Scholar]
- 45.Zechiedrich E.L., Khodursky,A.B. and Cozzarelli,N.R. (1997) Topoisomerase IV, not gyrase, decatenates products of site-specific recombination in E.coli.Genes Dev., 11, 2580–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mahmood R., Hines,J.C. and Ray,D.S. (1999) Identification of cis and trans elements involved in the cell cycle regulation of multiple genes in Crithidia fasciculata. Mol. Cell. Biol., 19, 6174–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berberich C., Machado,G., Morales,G., Carrillo,G., Jimenez-Ruiz,A. and Alonso,C. (1998) The expression of the Leishmania infantum KMP-11 protein is developmentally regulated and stage specific. Biochim. Biophys. Acta, 1442, 230–237. [DOI] [PubMed] [Google Scholar]
- 48.Trigueros S. and Roca,J. (2002) Failure to relax negative supercoiling of DNA is a primary cause of mitotic hyper-recombination in topoisomerase-deficient yeast cells. J. Biol. Chem., 40, 37207–37221. [DOI] [PubMed] [Google Scholar]
- 49.Giaever G.N. and Wang,J.C. (1988) Supercoiling of intracellular DNA can occur in eukaryotic cells. Cell, 55, 849–856. [DOI] [PubMed] [Google Scholar]
- 50.Wang J.C. (1991) DNA topoisomerases: why so many? J. Biol. Chem., 266, 6659–6662. [PubMed] [Google Scholar]
- 51.Melendy T., Sheline,C. and Ray,D.S. (1998) Localization of a type II DNA topoisomerase to two sites of the periphery of the kinetoplast DNA of C.fasciculata. Cell, 70, 1083–1086. [DOI] [PubMed] [Google Scholar]
- 52.Fragoso .P., Mattei,D., Hines,J.C., Ray,D.S. and Goldenberg,S. (1998) Expression and cellular localization of Trypanosoma cruzi type II DNA topoisomerase. Mol. Biochem. Parasitol., 94, 197–204. [DOI] [PubMed] [Google Scholar]
- 53.Nitiss J.L. (1998) Investigating the biological functions of DNA topoisomerases in eukaryotic cells. Biochim. Biophys. Acta, 1400, 63–81. [DOI] [PubMed] [Google Scholar]
- 54.Uemura T., Ohkura,H., Adachi,Y., Morino,K., Shiozaki,K. and Yanagida,M. (1987) DNA topoisomerase II is required for condensation and separation of mitotic chromosomes in S.pombe. Cell, 50, 917–925. [DOI] [PubMed] [Google Scholar]
- 55.Spell R.M. and Holm,C. (1994) Nature and distribution of chromosomal intertwinings in Saccharomyces cerevisiae. Mol. Cell. Biol., 14, 1465–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crookie E., Hwang,D.S., Skanstad,K., Thony,B. and Kornberg,A. (1991) E.coli minichromosome replication: regulation of initiation at oriC. Res. Microbiol., 142, 127–130. [DOI] [PubMed] [Google Scholar]
- 57.Liu L. and Wang,J.C. (1987) Supercoiling of the DNA template during transcription. Proc. Natl Acad. Sci. USA, 84, 7024–7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jensen S., Andersen,A.H., Kjeldensen,E., Biersack,H., Olsen,E.H.N., Andersen,T.B., Westergaard,O. and Jakobsen,B.T. (1996) Analysis of functional domain organization in DNA Topoisomerase II from humans and Saccharomyces cerevisiae. Mol. Cell. Biol., 16, 3866–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsutsui K., Tsutsui,K., Sano,K., Kikuchi,A. and Tokunaga,A. (2001) Involvement of DNA topoisomerase II β in neuronal differentiation. J. Biol. Chem., 276, 5769–5778. [DOI] [PubMed] [Google Scholar]
- 60.Watt P.M., Louis,E.J., Borts,R.H. and Hickson,I.D. (1995) Sgs1: a eukaryotic homolog of E. coli RecQ that interacts with topoisomerase II in vivo and is required for faithful chromosome segregation. Cell, 81, 253–260. [DOI] [PubMed] [Google Scholar]
- 61.Fragoso S., Mattei,D., Hines,J.C., Ray,D. and Goldenberg,S. (1998) Expression and cellular localization of Trypanosoma cruzi type II topoisomerase. Mol. Biochem. Parasitol., 94, 197–204. [DOI] [PubMed] [Google Scholar]
- 62.Wang Z., Morris,J.C., Drew,M.E. and Englund,P.T. (2000) Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J. Biol. Chem., 275, 40174–40179. [DOI] [PubMed] [Google Scholar]
- 63.Wang Z. and Englund,P.T. (2001) RNA interference of a trypanosome topoisomerase II causes progressive loss of mitochondrial DNA. EMBO J., 20, 4674–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]