


Abstract
The decision to stop smallpox vaccination and the loss of specific immunity in a large proportion of the population could jeopardise world health due to the possibility of a natural or provoked re-emergence of smallpox. Therefore, it is mandatory to improve the current capability to prevent or treat such infections. The DNA repair protein uracil-DNA glycosylase (UNG) is one of the viral enzymes important for poxvirus pathogenesis. Consequently, the inhibition of UNG could be a rational strategy for the treatment of infections with poxviruses. In order to develop inhibitor assays for UNG, as a first step, we have characterised the recombinant vaccinia virus UNG (vUNG) and compared it with the human nuclear form (hUNG2) and catalytic fragment (hUNG) UNG. In contrast to hUNG2, vUNG is strongly inhibited in the presence of 7.5 mM MgCl2. We have shown that highly purified vUNG is not inhibited by a specific uracil-DNA glycosylase inhibitor. Interestingly, both viral and human enzymes preferentially excise uracil when it is opposite to cytosine. The present study provides the basis for the design of specific inhibitors for vUNG.
INTRODUCTION
Poxviruses are large eukaryotic viruses containing a double-stranded DNA (dsDNA) genome with covalently closed hairpin termini (1). The entire life cycle of poxvirus, including DNA replication, occurs exclusively within the cytoplasm of the host cell (2). Therefore, the virus does not depend on cellular nuclear functions, and relies largely on its own gene products for DNA replication and the different phases of gene expression (2–5). Vaccinia virus, a member of the Orthopoxvirus genus and the best characterised member of the poxvirus family, was extensively used as the smallpox vaccine, has gained popularity as a mammalian expression vector, and is being tested as a recombinant vaccine against cancer and infectious diseases (6). Vaccinia virus has a genome of ∼200 kb and can potentially express more than 200 proteins (6). The relative ease of performing genetic and biochemical analyses on vaccinia virus has allowed the identification of numerous virally encoded proteins that participate in DNA replication, including an essential DNA polymerase, a protein kinase, a DNA-independent nucleotide triphosphatase, a topoisomerase, a DNA ligase and several enzymes involved in nucleotide metabolism (5). A viral DNA repair enzyme, uracil-DNA glycosylase (vUNG), encoded by the D4R open-reading frame (ORF), is also involved in vaccinia virus DNA replication (7,8). Poxvirus UNG activity was first identified in Shope fibroma virus (9) and subsequently recognised to be a common feature in all poxviruses, including vaccinia, fowlpox (1,10) and certainly variola viruses. UNGs, which are ubiquitous in nature, catalyse the first step in the base excision repair pathway of uracil residues in DNA by cleaving the glycosidic bond, resulting in an apyrimidinic (AP) site (11). UNG activity is rapidly induced following poxvirus infection, suggesting that the enzyme is required prior to and during DNA synthesis rather than as a post-replicative repair activity. Similar conclusions have been drawn with respect to the nuclear form of the mammalian enzyme, in which an increase in UNG activity slightly precedes the peak of DNA synthesis in normally cycling or stimulated cells (12,13). Thus, UNG removes residues that have been introduced into DNA either through misincorporation of dUTP by DNA polymerase or through the deamination of cytosine (14).
Completion of the base excision repair pathway requires incision of the AP site by AP endonuclease 1 (Ape1) followed by excision of 5′-deoxyribose phosphate and one-nucleotide gap filling by DNA polymerase β, the remaining strand break being sealed by a DNA ligase III/XRCC1 protein complex (15). Herpes simplex virus type-1 (HSV-1) UNG protein is dispensable for HSV-1 replication in cell culture (as with cytomegaloviruses), presumably because the host UNG can supply the activity of the viral enzyme (16,17).
Interestingly, it was initially demonstrated that, unlike all other known UNG, the vaccinia virus protein is essential for virus viability (7,8,18). Several mutations in active-site residues of vUNG were found to prevent virus replication (19). However, a recent study has contradicted these findings and shown that vUNG activity is not essential for virus replication in vitro (20). These authors also reported that vaccinia virus mutants lacking vUNG activity, due to active site mutations, have reduced virulence in a intranasal- infection mouse model suggesting that vUNG plays a role in viral pathogenesis. In fact, animals infected with ≥104 p.f.u. of virus expressing wild-type D4R suffered severe illness and were killed within 6–10 days when their weight loss equalled or exceeded 30%. In contrast, animals infected with 104– 105 p.f.u. of virus with mutated D4R suffered temporary weight loss, and none had to be killed. D4R with a single mutation (H181L) appeared to be slightly less pathogenic than D4R (D68N) (20). Thus, inhibition of vUNG activity should be a rational strategy for the treatment of poxvirus infections (20). The prerequisite being that the enzymatic properties of the vUNG protein are different from the human uracil-DNA glycosylase (hUNG).
In order to test this hypothesis and later on develop an inhibitory assay to screen antiviral agents against vUNG, in the present study we have expressed and characterised the recombinant vUNG. The catalytic fragment of the hUNG and the nuclear form (hUNG2) were used for comparison (21,22). The choice for the hUNG protein was based on the fact that the three-dimensional structure of the hUNG protein complexed to its DNA substrate was resolved, thus providing valuable information for molecular design of specific inhibitors towards vUNG (23–26).
We have determined the kinetic parameters for the excision of uracil residues in single- and double-stranded G·U mismatched substrates for viral and human enzymes. In addition, we have investigated the base-pair specificity of vUNG and its inhibition by several factors. The study of the substrate specificity of vUNG as compared with hUNG and hUNG2 show that they have different properties, thus providing the basis for developing vUNG-specific inhibitors.
MATERIALS AND METHODS
Bacterial strains and plasmids
The Escherichia coli strains MS1021 (ung-1::Spcr) and RZ1032 (ung-1, dut-1) were obtained from laboratory stocks. The strain MS1021 is a derivative of CC102 (27). Isolation of the UNG-deficient mutant of E.coli K12, generated by insertion of a spectinomycin cassette, will be published elsewhere. PCR analysis of genomic DNA from the ung-1:: Spcr mutant confirmed the insertion of the spectinomycin cassette, while crude lysates exhibited no detectable UNG activity (data not shown). The phagemid pBluescript II KS(–) was purchased from Stratagen Cloning Systems (La Jolla, CA). The single-stranded circular pBluescript II KS(–) DNA containing multiple uracil residues was prepared as described (28). The plasmid pTUNGΔ84 coding for the catalytic domain of the hUNG protein was a gift from Dr Hans Krokan (Norwegian University of Science and Technology, Trondheim, Norway).
Enzymes
T4 polynucleotide kinase and uracil-DNA glycosylase inhibitor (UGI) were purchased from New England BioLabs (Saint Quentin Yvelines, France). Purification of the E.coli Fpg protein was performed as described (29). The hUNG recombinant protein lacking the N-terminal 84 amino acids (27 kDa, 230 amino acids) was over-expressed in E.coli MS1021 cells and purified as described (21). The NdeI–HindIII fragment containing the full-length UNG2 cDNA (30) was subcloned into the pET28a vector (Novagen). The pET28a-UNG2 construct was expressed in BL21(DE3)RIL (Stratagene) in 1 l of 3× LB medium. Bacterial pellets were sonicated and extracts were purified on a Ni-NTA Superflow column, which was washed and eluated as recommended (Qiagen). The protein was further purified using a phenyl-Sepharose column (Pharmacia) and finally a Mono-Q column (Pharmacia) (22).
Preparation of radioactively labelled oligonucleotides
All oligonucleotides were purchased from Eurogentec (Angers, France). The sequences of the various oligonucleotides used are shown in Table 1. Aliquots of 50 pmol of single-stranded oligonucleotide were 5′-end labelled with [γ-32P]ATP (4500 Ci mmol–1; ICN Pharmaceuticals, Inc., Costa Mesa, CA) using T4 polynucleotide kinase (New England Biolabs, Beverly, MA). Oligonucleotide duplexes were prepared by annealing in a mixture of non-radioactive and complementary radioactively labelled oligonucleotides in a molar ratio of 2:1 in 0.5× SSC buffer (75 mM NaCl, 7.5 mM citric acid trisodium salt, pH 7.0). The mixture was heated at 95°C for 3 min and then slowly cooled to room temperature. The 5′ 32P-labelled PS-U was annealed to various complements (PHY-G, PHY-C, PHY-T and PHY-A) to generate the following mismatches: U·G, U·C, U·T and U·A. The RT oligonucleotide was annealed to RT-G complement and the εA40 oligonucleotide was annealed to T40 complement (Table 1). The formation of double-stranded oligonucleotides was assessed by 15% polyacrylamide gel electrophoresis (PAGE) under non-denaturing conditions (31).
Table 1. Sequence of the oligonucleotides to measure DNA glycosylase activity.
| Name | Oligonucleotide sequence |
|---|---|
| PS-U | 5′-AGC TAC CAT GCC TGC ACG AAU TAA GCA ATT CGT AAT CAT GGT CAT |
| PHY-G | 5′-ATG ACC ATG ATT ACG AAT TGC TTA GTT CGT GCA GGC ATG GTA GCT |
| PHY-A | 5′-ATG ACC ATG ATT ACG AAT TGC TTA ATT CGT GCA GGC ATG GTA GCT |
| PHY-T | 5′-ATG ACC ATG ATT ACG AAT TGC TTA TTT CGT GCA GGC ATG GTA GCT |
| PHY-C | 5′-ATG ACC ATG ATT ACG AAT TGC TTA CTT CGT GCA GGC ATG GTA GCT |
The bold letters indicate the base targeted by UNG in each oligonucleotide sequence.
DNA glycosylase assays
Single strand plasmid DNA substrate. Circular single-stranded pBluescript II KS(–) DNA (0.25 µg) was incubated at 37°C for 10 min with each purified UNG at various concentrations in reaction buffer containing 100 mM KCl, 5 mM 2-mercaptoethanol, 1 mM EDTA, 60 mM HEPES, pH 8.0. To incise DNA at abasic sites the plasmid was treated with either 20 nM Fpg or piperidine (10% piperidine at 37°C for 15 min) unless otherwise stated. Reaction products were separated by electrophoresis on a 0.8% agarose gel and stained with ethidium bromide.
Single- and double-stranded oligonucleotide substrates. 5′ 32P-labelled single-stranded PS-U or duplex UNG oligonucleotide (10 nM) were incubated with either hUNG or vUNG at 37°C for various periods of time. The standard assay mixture for DNA glycosylase activity (20 µl final volume) contained 0.2 pmol of the labelled single-stranded or duplex oligonucleotide, 60 mM NaCl, 1 mM EDTA, 1 mM DTT, 20 mM Tris–HCl (pH 7.5), 100 µg/ml bovine serum albumin and a limiting amount of enzyme. UNG activity was stopped with 1.5% SDS and 0.3 mg/ml proteinase K at 37°C for 15 min. Abasic sites were incised after glycosylase action by light piperidine treatment [10% (v/v) piperidine at 37°C for 15 min]. Reaction products were analysed by electrophoresis through denaturing 20% (w/v) polyacrylamide gels (7 M urea, 0.5× TBE), visualised using a PhosphorImager Storm 840 (Molecular Dynamics, Sunnyvale, CA), and quantified using ImageQuant software.
Uracil-DNA glycosylase inhibitor (UGI) assay. Assays were performed with concentrations of UGI ranging from 0.002 to 0.1 U for 1 nM hUNG and 0.1 to 5 U of UGI for 50 nM vUNG, at 37°C for 10 min. The reaction contained 10 nM PS-U or U·G.
Expression of vUNG
pRSET-D4R (18) has a T7 promoter with a histidine tag attached to the ORF for purification. Escherichia coli strain BL-21 DE3 transformed with pRSET-D4R was grown at 37°C until the absorbance (600 nm) reached the value of 1. vUNG was induced with 1 mM IPTG and overnight culture at 15°C. The culture was centrifuged (8740 g, 10 min, 4°C) and the pellet was resuspended with buffer A (1 mM 2-mercaptoethanol, 50 mM NaCl, 50 mM Tris–HCl, pH 8.0). Cells were lysed by sonication at 2.4 kV and 4 kΩ (6 × 10 s bursts) on ice and the lysate was centrifuged at 49 000 g for 30 min at 4°C.
Purification of vUNG
A chelating Sepharose fast flow column (Amersham-Pharmacia, Orsay, France) pre-equilibrated with buffer A was loaded with 74 ml of cells and then first washed with buffer A containing 20 mM imidazole and 300 mM NaCl (60× bed volume). The column was then washed with solution B (buffer A containing 60 mM imidazole and 300 mM NaCl) (20× bed volume). Proteins were eluted from the column using a gradient (60–500 mM imidazole in buffer A containing 300 mM NaCl, 30× bed volume). The elution profile was monitored by its absorption at 280 nm. Aliquots from the peak fractions were analysed by 12.5% SDS–PAGE. The peak fractions showing the highest concentration of vUNG were then pooled, subjected to Seperdex G-75 gel filtration chromatography (Amersham-Pharmacia) using a 1 × 48 cm column which was equilibrated with buffer A containing 300 mM NaCl. The elution profile was continuously monitored and aliquots from the peak fractions were subjected to 12.5% SDS–PAGE. The fluorescence emission and the spectrophotometry, measured at 240–340 nm, were measured. The mass of purified vUNG was determined by mass spectrometry with the ion electrospray technique (API III+; Applied Biosystems, Courtaboeuf, France).
RESULTS
Expression and purification of vUNG
In order to over-express vUNG, the plasmid pRSET-D4R was transfected into the E.coli BL21 DE3 strain and grown as described in Materials and Methods. Analysis by PAGE showed that >60% of vUNG was found in the soluble fraction after induction at 15°C overnight. The vUNG protein was purified in one step by affinity chromatography on chelating Sepharose. We obtained 2.5 mg of vUNG protein with molecular weight of 29 kDa (Fig. 1) and with a purity higher than 95% from 1 l of culture. When analysed by gel filtration, vUNG eluted as a single peak (data not shown) showing that the vUNG protein was in a monomeric form and not aggregated. The fluorometric spectrum showed a single peak of emission at 332 nm indicating that the majority of the seven tryptophan residues present in vUNG were not exposed to H2O. This suggests that the protein was correctly folded. The spectrophotometric analysis of the protein in the 240–340 nm range showed a peak at 280 nm and an absorption close to zero from 320 to 340 nm, revealing that vUNG was not aggregated even after 2 months of storage at 4°C. The molecular weight of vUNG measured by mass spectrometry is 30 308 Da, confirming the molecular weight found by SDS–PAGE (data not shown).
Figure 1.

SDS–PAGE analysis of the purified recombinant hUNG and vUNG. Details of the purification are described in Materials and Methods. Lane 1, vUNG (1 µg); lane 2, hUNG (1 µg). The molecular weight standards were rabbit muscle glyceraldehyde-3-phosphate dehydrogenase (36 kDa), bovine erythrocytes carbonic anhydrase (29 kDa), bovine pancreas trypsinogen (24 kDa), soybean trypsin inhibitor (20 kDa) and bovine milk α-lactalbumin (14.2 kDa). Proteins were stained with Coomassie blue R250.
Human and vaccinia virus uracil-DNA glycosylases are highly specific to uracil residues and do not excise other modified bases
As shown in Figure 2A and B, the single-stranded plasmid DNA containing multiple uracil residues migrated as a single species (lane 1). When the plasmid DNA was treated with either Fpg (Fig. 2A, lane 2) or piperidine (Fig. 2B, lane 2), it was not incised, showing that it did not contain the AP sites. When the plasmid was treated with either hUNG or vUNG (Fig. 2A, lanes 8 and 14) or hUNG2 proteins following Fpg and piperidine treatment (Fig. 2B, lane 8), it was degraded, indicating that the UNG proteins generated AP sites by excising the uracil residues. This assay was used to compare the efficiency of the human and viral enzymes. Increasing amounts of each UNG protein were used for the reaction. As shown in Figure 2A, in 10 min, 10 nM vUNG only partially incised the plasmid DNA (lane 13) whereas only 0.1 nM hUNG (lane 5) and 1 nM hUNG2 (Fig. 2B, lane 6) achieved the same degree of incision. This experiment indicated that the human proteins were ∼10–100-fold more active than vUNG towards uracil residues when present in single-stranded DNA (ssDNA). However, 100 nM UNG from human or vaccinia virus was able to completely degrade 0.25 µg of single-stranded plasmid DNA in the presence of Fpg or following piperidine treatment (Fig. 2A, lanes 8, 14 and B, lane 8).
Figure 2.
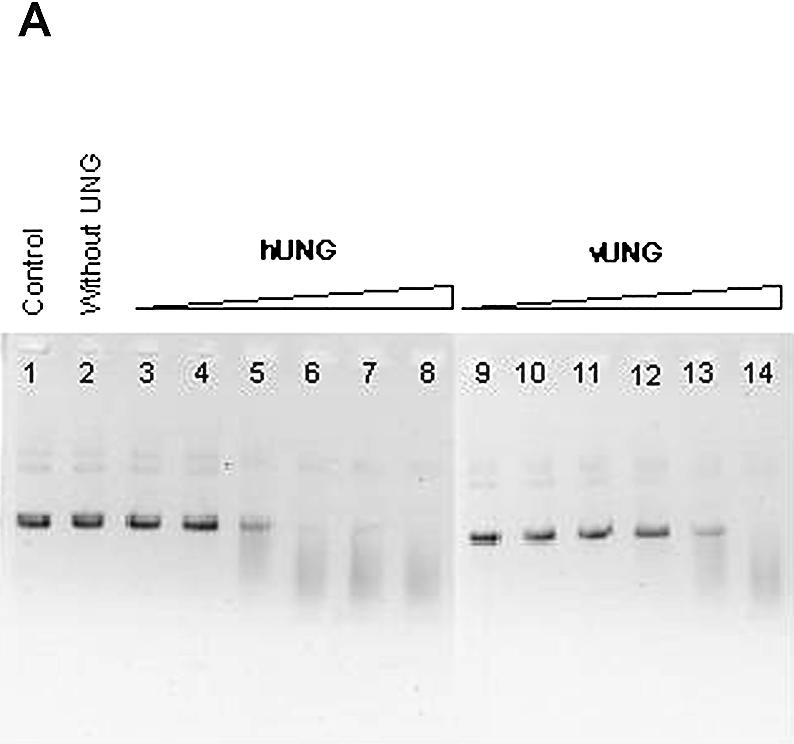
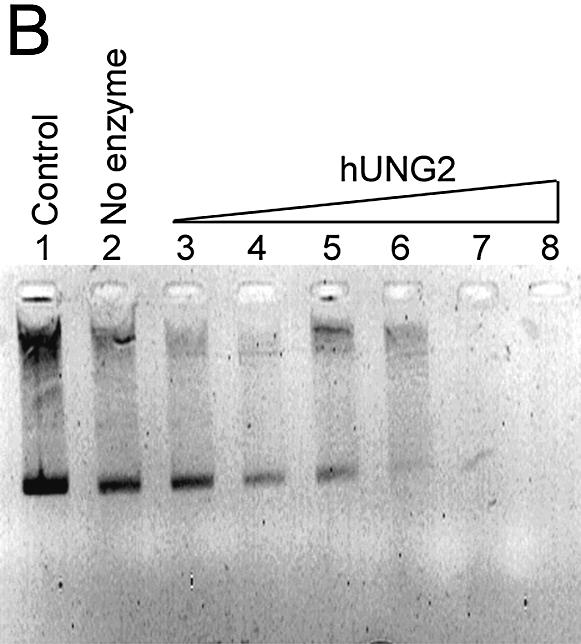
Activity of vUNG, hUNG and hUNG2 on single-stranded plasmid DNA containing uracil residues [pBluescript II KS(–)]. pBluescript II KS(–) (0.25 µg) was incubated with various concentrations of vUNG, hUNG and hUNG2 as described in Materials and Methods. Reaction products were separated by electrophoresis in a 0.8% agarose gel and stained with ethidium bromide. (A) Lane 1, pBluescript II KS(–) control; lane 2, as lane 1 but with 20 nM Fpg; lanes 3–8, as lane 2 but treated with increasing amounts of hUNG; lanes 9–14, as lane 2 but treated with increasing amounts of vUNG. The UNG protein concentrations were 0.001 nM in lanes 3 and 9; 0.01 nM in lanes 4 and 10; 0.1 nM in lanes 5 and 11; 1 nM in lanes 6 and 12; 10 nM in lanes 7 and 13; 100 nM in lanes 7 and 14. (B) Lane 1, pBluescript II KS(–) control; lane 2, as lane 1 but treated with piperidine; lanes 3–8, as lane 2 but treated with increasing amounts of hUNG2; lane 3, 0.001 nM; lane 4, 0.01 nM; lane 5, 0.1 nM; lane 6, 1 nM; lane 7, 10 nM; lane 8, 100 nM.
We then investigated whether a single-stranded oligonucleotide containing a single uracil residue was a substrate for the vUNG protein. 5′ 32P-labelled PS-U was incubated with increasing concentrations of either UNG protein. Light piperidine treatment was used to reveal abasic sites generated by UNG. As shown in Figure 3, piperidine treatment in the absence of the enzyme did not cut oligonucleotides (lanes 1 and 8) showing that there were no detectable AP sites in the substrate. However, piperidine treatment, after reaction with hUNG and vUNG proteins, generated a band migrating at the position of the 20mer fragment (lanes 2–7 for hUNG and lanes 9–14 for vUNG). Similar results were obtained when using double-stranded oligonucleotide as a substrate, showing again that the human protein was ∼100-fold more active as compared with vUNG.
Figure 3.

Activity of vUNG and hUNG on single-stranded oligonucleotide containing a single uracil residue (PS-U). 5′ 32P-labelled PS-U (10 nM) was incubated with various amounts of UNG at 37°C for 5 min. Reaction products were subjected to light piperidine treatment to reveal abasic sites generated by DNA glycosylase action. They were then separated by electrophoresis on a 20% polyacrylamide gel containing 7 M urea and visualised using a PhosphorImager Storm 840. Lanes 1–7, PS-U treated with increasing amounts of hUNG; lanes 8–14, PS-U treated with increasing amounts of vUNG. Lanes 1 and 8, PS-U control. UNG protein concentrations were 0.001 nM in lanes 2 and 9; 0.01 nM in lanes 3 and 10; 0.1 nM in lanes 4 and 11; 1 nM in lanes 5 and 12; 10 nM in lanes 6 and 13; and 100 nM in lanes 7 and 14. a, 45mer PS-U; b, 20mer cleaved products.
Duplex oligonucleotides (Table 1) containing various modified bases, generated by the oxidative stress in cellular DNA, were used to test substrate specificity of vUNG and hUNG proteins. No detectable activity was observed towards mismatched-thymine, 3,N4-ethenocytosine, 5,6-dihydrouracil, 7,8-dihydro-8-oxoguanine and 1,N6-ethenoguanine residues (data not shown).
Characterisation of the susbstrate specificity of vUNG on ssDNA and dsDNA
In order to further characterise the substrate specificities of the vUNG we measured the kinetics of uracil excision in PS-U oligonucleotide and the double-stranded oligonucleotide containing a U·G mismatch. The vUNG protein preferentially excises uracil when present in ssDNA as compared with dsDNA, since the specificity constant (kcat/Km) on the PS-U oligonucleotide was three times higher as compared with U·G duplex oligonucleotide (Table 2). For comparison, the kinetic constants of hUNG acting on the same oligonucleotides were measured (Table 2). Interestingly, the apparent Km for vUNG on ssDNA substrate, calculated from the Michaels–Menten fit (0.5 µM), suggests that vUNG has stronger affinity as compared with the human enzyme (2.9 µM). Furthermore, the excision efficiency of hUNG protein was several orders of magnitude higher than vUNG (Table 2).
Table 2. Kinetic constants of the hUNG and vUNG proteins for excision of uracil when present in single-stranded and duplex oligonucleotides (U·G).
| Substrate | Km (µM)a | kcat (min–1) | kcat/Km (min–1 µM–1) | |||
|---|---|---|---|---|---|---|
| dsDNA | ssDNA | dsDNA | ssDNA | dsDNA | ssDNA | |
| hUNG | 1.6 ± 0.7 | 2.9 ± 0.6 | 11 860 ± 130 | 11 800 ± 80 | 7000 | 4000 |
| vUNG | 2.6 ± 0.7 | 0.5 ± 0.2 | 49 ± 0.9 | 28 ± 0.6 | 19 | 56 |
aThe Km of the UNG from different origins was determined by incubating the respective protein (1 nM hUNG or 50 nM vUNG) with increasing concentrations of oligonucleotide. The PS-U concentrations ranged from 0 to 8 µM for the hUNG protein and 0 to 5 µM for the vUNG protein. The U·G duplex oligonucleotide concentrations ranged from 0 to 8 µM for all proteins tested. The kinetic constants were calculated using the Michaelis–Menten fit.
Effect of the nature of the base opposite to the uracil residue and effect of MgCl2 on the activity of the human and viral UNGs
We next investigated the specificity of vUNG for various oligonucleotides containing each of the four naturally occurring deoxynucleotides opposite to uracil. The hUNG protein was chosen as a control enzyme. Figure 4 presents the initial velocity of the two enzymes when acting upon the four different base pairs. The activity profile was similar for both the vUNG and hUNG proteins. They preferentially excised uracil when it was opposite to a dC and U·A was a less preferred substrate. The relative order of uracil excision by the two UNG proteins was dC > dT > dG >> dA.
Figure 4.

Cleavage of duplex oligonucleotides containing different bases opposite to uracil by hUNG and vUNG. The 5′ 32P-labelled PS-U oligonucleotide was annealed to various complements to generate the following mismatches: U·G, U·C, U·T and U·A. Each duplex oligonucleotide (10 nM) was incubated with hUNG (1 nM) (A) and vUNG (50 nM) (B) at 37°C for various periods of time (1–20 min). Reaction products were subjected to light piperidine treatment to reveal abasic sites generated by UNG and analysed as described in Materials and Methods.
Figure 5 shows the effect of 7.5 mM MgCl2 on the activity of vUNG, hUNG and hUNG2 proteins when acting upon a U·G oligonucleotide. A drastic loss of activity was observed in the presence of MgCl2 for hUNG and vUNG. As expected, hUNG2 was highly stimulated in the presence of 7.5 mM MgCl2 in contrast to hUNG and vUNG. Interestingly, in the presence of MgCl2, hUNG2 is more active than the hUNG.
Figure 5.

Effect of MgCl2 on the activity of the vUNG, hUNG and hUNG2 proteins. The U·G duplex oligonucleotide (250 nM) was incubated with hUNG (1 nM) and vUNG (50 nM) at 37°C for 1–20 min in the presence or absence of 7.5 mM MgCl2. Reaction products were subjected to light piperidine treatment to reveal abasic sites generated by UNG and analysed as described in Materials and Methods. The radioactivity of the reaction products was quantified and plotted against time.
The UGI protein inhibits hUNG activity but not vUNG activity
Ellison et al. previously reported that the UGI from the Bacillus subtilis bacteriophages PBS1 and PBS2 inactivates hUNG but not vUNG (19). Since cell-free extracts were used, the presence of other human proteins cannot be excluded. We have found that purified homogeneous vUNG resists UGI inhibition when either dsDNA or ssDNA are used as substrates, whereas hUNG is highly sensitive to inhibition under both conditions and at very low UGI concentrations (Table 3).
Table 3. Inhibition of the vUNG and hUNG protein activities by UGI protein.
| UGI/UNG (molar ratio) | Inhibition (%)a | |||
|---|---|---|---|---|
| ssDNA | dsDNA | |||
| hUNG | vUNG | hUNG | vUNG | |
| 1 | 95 | 0 | 91 | 0 |
| 10 | 98 | 0 | 98 | 0 |
| 50 | 100 | 0 | 100 | 0 |
aThe extent of inhibition of the UNG from different origins was determined by incubating the respective protein with increasing amounts of UGI. The 10 nM ssDNA (PS-U) and dsDNA (U·G) were incubated with hUNG (1 nM) and vUNG (50 nM) at 37°C for 10 min in the presence of varying concentrations of UGI. Reaction products were subjected to light piperidine treatment and analysed as described in Materials and Methods.
DISCUSSION
Smallpox is a devastating disease with a high case-fatality rate. The causative agent of smallpox, variola virus, is thus a potential biological weapon that could be used by terrorists (31–33). The vaccine is so far the only treatment against smallpox and is active only if administrated in the first 4 days post-exposure (34). If used as a biological weapon (32–34), variola represents a serious threat to civilian populations because of its mortality rate of 30% among unvaccinated individuals and the absence of specific therapy (35–37). Should such an event occur there would be a need for a new therapy for poxvirus infection as the level of immunity against smallpox has declined considerably after discontinuation of vaccination. Furthermore, new antivirals may be required to control the adverse reactions that are sometimes associated with smallpox vaccination.
The D4R ORF encodes UNG (7,8) which is an essential gene for vaccinia virus, as demonstrated by the lethality of a D4R deletion mutant (19). However, recent work has demonstrated that the enzymatic activity of D4R is not required for virus viability and it has become clear that the D4R protein plays a role in virus DNA replication that is independent of UNG activity (20). Nevertheless, vUNG activity may be an important factor for viral pathogenesis; therefore, inhibition of this enzyme could be a rational strategy for the treatment of poxvirus infections.
The purpose of this study was to investigate the substrate specificity of the homogeneous UNGs from vaccinia virus. Expression and purification of the recombinant vUNG in E.coli yielded pure protein which allowed its biochemical characterisation. We over-expressed vUNG using the plasmid carrying the vaccinia virus D4R ORF in E.coli (18). A pure monomeric soluble enzyme (no refolding step was necessary) in high quantity was obtained (Fig. 1). The human truncated and full-length UNGs were prepared as described in Materials and Methods.
Slupphaug et al. (21) showed that hUNG was 3-fold more active on a ssDNA as compared with a dsDNA substrate. However, for hUNG2 in the absence of Mg2+ ions, the difference between ssDNA and dsDNA substrates was not significant (22). In the present study using hUNG, we do not observe significant differences between the two substrates. It should be stressed that, in previous work, calf thymus DNA containing [3H]dUMP residues incorporated by nick-translation resulting in U·A base pairs was used as a substrate. Therefore, the former comparison of relative rates of uracil excision was made using U (ssDNA) and U·A (dsDNA), whereas in the present work we compared U and U·G. It is well established that the activity of various UNG proteins differs according to the sequence context (38). In addition, it has been demonstrated that the relative UNG activity in nuclear extracts strongly depends on the type of DNA substrate used (39). Furthermore, it was shown that excision rates of uracil by hUNG strongly depend on the DNA sequence context (21). Therefore, we expect that both the type of DNA substrate as well as the particular sequence context of the uracil-containing oligonucleotide used in this work may account for the difference between ssDNA and dsDNA substrates (Table 2).
Interestingly, both hUNG and viral enzymes preferentially excise uracil when it is opposite cytosine and thymine (Fig. 4). The base pair specificity of uracil excision was similar for both enzymes and the relative order of excision was U·C > U·T > U·G >> U·A (Fig. 4).
The substrate specificity and kinetic constants of the recombinant vUNG appeared to be similar to that of hUNG2, in the absence of magnesium (22), but not to hUNG, since the kcat/Km values are similar (Table 2). Furthermore, we have found that magnesium drastically inhibits the DNA glycosylase activity of vUNG and hUNG. In agreement with previous observations, we demonstrated that hUNG2 is more active in the presence of physiological concentrations of magnesium (Fig. 4) (22). Importantly, the difference in response to magnesium might provide an efficient way to specifically inhibit the viral enzyme in vivo.
vUNG was highly specific for uracil and did not act on other modified bases that may be generated in cells by oxidative stress (data not shown). Comparison of the kinetic constants for the excision of uracil by vUNG, hUNG and hUNG2 indicated that the efficiency of repair by the human nuclear form UNG in the absence of Mg2+ and the viral enzyme are extremely poor (Table 2). We suggest that the low efficiency of the viral enzyme might be due to the lack of proteins which associate with vUNG in vivo. Indeed, the D4R (vUNG) and D5R (a DNA-dependant ATPase) proteins interact with A20R protein (with unknown function) (40) and it was suggested that the A20R, D4R and D5R proteins are components of a multiprotein DNA replication complex (41–43). Therefore, it is possible that vUNG operates in vivo as a complex and that specific protein–protein interactions are necessary for efficient removal of uracil residues by vUNG. It has been observed that the hUNG2 co-localises in replication foci and physically interacts with the proliferating nuclear antigen (PCNA) (44), the replication protein A (RPA) (44) and DNA polymerase α (45). Altogether, these observations strongly suggest that the UNG proteins are active in DNA replication complexes. Based on the observation that the poxvirus UNGs have limited homology with the hUNG, it has been suggested that the viral proteins may have acquired novel functions allowing them to interact with other proteins (8).
Ellison et al. (19) showed that the UGI proteins of the B.subtilis bacteriophages PBS1 and PBS2 were unable to inhibit vUNG. However, since cell-free extracts were used, the presence of other human enzymes resistant to UGI was not excluded. In the present study, for the first time, we have demonstrated that the pure recombinant vUNG protein is not inhibited by UGI (Table 3). Thus, this supports the notion that the poxvirus proteins may differ significantly in their structure from the other members of the UNG family, including the human enzyme, which are inhibited by UGI (Table 3).
In conclusion, the results presented in this paper will permit the development of a reliable screening method for inhibitors which are specific against vUNG. Enzymatic properties of the viral protein differ substantially from the human enzymes; this could facilitate the search for specific vUNG inhibitors. The considerable sequence homology observed among poxvirus UNGs suggests that drugs active against vUNG should also be able to inhibit variola virus and thus may be attractive candidates for chemotherapy.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Protein’Expert S.A. for their technical advice, Dr F. G. Falkner (Baxter Vaccine AG) for plasmids and for critically reading the manuscript, Dr Hans Krokan (Norwegian University of Science and Technology, Trondheim, Norway) for the plasmid, the technical assistance of Danielle Gratier, Josette Guimet, Corinne Rothlisberger and Henri Blancquaert, and Bernard Souberbielle, King’s College, London for reviewing the manuscript. This work was supported by grants from the Service de Santé des Armées (SSA) and the Délégation Générale pour l’Armement (DGA) to D.G. and Association pour la Recherche sur le Cancer (ARC) to M.S.
REFERENCES
- 1.Goebel S.J., Johnson,G.P., Perkus,M.E., Davis,S.W., Winslow,J.P. and Paoletti,E. (1990) The complete DNA sequence of vaccinia virus. Virology, 179, 247–266, 517–263. [DOI] [PubMed] [Google Scholar]
- 2.Moss B. (2001) Poxviridae: the virus and their replication. In Knipe,D.M. and Howley,P.M. (eds), Fields Virology. Raven Press, New York, Vol. 2, pp. 2849–2884. [Google Scholar]
- 3.Traktman P. (1990) Poxviruses: an emerging portrait of biological strategy. Cell, 62, 621–626. [DOI] [PubMed] [Google Scholar]
- 4.Moss B., Ahn,B.Y., Amegadzie,B., Gershon,P.D. and Keck,J.G. (1991) Cytoplasmic transcription system encoded by vaccinia virus. J. Biol. Chem., 266, 1355–1358. [PubMed] [Google Scholar]
- 5.Traktman P. (1996) Poxvirus DNA replication. In DePamphilis,M.L. (ed.), DNA Replication in Eucaryotic Cells. Cold Spring Harbor Laboratory Press, New York, pp. 775–793. [Google Scholar]
- 6.Moss B. (1996) Genetically engineered poxviruses for recombinant gene expression, vaccination and safety. Proc. Natl Acad. Sci. USA, 93, 11341–11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stuart D.T., Upton,C., Higman,M.A., Niles,E.G. and McFadden,G. (1993) A poxvirus-encoded uracil DNA glycosylase is essential for virus viability. J. Virol., 67, 2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Millns A.K., Carpenter,M.S. and DeLange,A.M. (1994) The vaccinia virus-encoded uracil DNA glycosylase has an essential role in viral DNA replication. Virology, 198, 504–513. [DOI] [PubMed] [Google Scholar]
- 9.Upton C., Stuart,D.T. and McFadden,G. (1993) Identification of a poxvirus gene encoding a uracil DNA glycosylase. Proc. Natl Acad. Sci. USA, 90, 4518–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tartaglia J., Winslow,J., Goebel,S., Johnson,G.P., Taylor,J. and Paoletti,E. (1990) Nucleotide sequence analysis of a 10.5 kbp HindIII fragment of fowlpox virus: relatedness to the central portion of the vaccinia virus HindIII D region. J. Gen. Virol., 71, 1517–1524. [DOI] [PubMed] [Google Scholar]
- 11.Lindahl T. (2001) Keynote: past, present and future aspects of base excision repair. Prog. Nucleic Acid Res. Mol. Biol., 68, xvii–xxx. [DOI] [PubMed] [Google Scholar]
- 12.Gupta P.K. and Sirover,M.A. (1981) Stimulation of the nuclear uracil DNA glycosylase in proliferating human fibroblasts. Cancer Res., 41, 3133–3136. [PubMed] [Google Scholar]
- 13.Vollberg T.M., Siegler,K.M., Cool,B.L. and Sirover,M.A. (1989) Isolation and characterization of the human uracil DNA glycosylase gene. Proc. Natl Acad. Sci. USA, 86, 8693–8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindahl T. and Nyberg,B. (1974) Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry, 13, 3405–3410. [DOI] [PubMed] [Google Scholar]
- 15.Kubota Y., Nash,R.A., Klungland,A., Schar,P., Barnes,D.E. and Lindahl,T. (1996) Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J., 15, 6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 16.Mullaney J., Moss,H.W. and McGeoch,D.J. (1989) Gene UL2 of herpes simplex virus type 1 encodes a uracil-DNA glycosylase. J. Gen. Virol., 70, 449–454. [DOI] [PubMed] [Google Scholar]
- 17.Reddy S.M., Williams,M. and Cohen,J.I. (1998) Expression of a uracil DNA glycosylase (UNG) inhibitor in mammalian cells: varicella-zoster virus can replicate in vitro in the absence of detectable UNG activity. Virology, 251, 393–401. [DOI] [PubMed] [Google Scholar]
- 18.Holzer G.W. and Falkner,F.G. (1997) Construction of a vaccinia virus deficient in the essential DNA repair enzyme uracil DNA glycosylase by a complementing cell line. J. Virol., 71, 4997–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellison K.S., Peng,W. and McFadden,G. (1996) Mutations in active-site residues of the uracil-DNA glycosylase encoded by vaccinia virus are incompatible with virus viability. J. Virol., 70, 7965–7973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Silva F.S. and Moss,B. (2003) Vaccinia virus uracil DNA glycosylase has an essential role in DNA synthesis that is independent of its glycosylase activity: catalytic site mutations reduce virulence but not virus replication in cultured cells. J. Virol., 77, 159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slupphaug G., Eftedal,I., Kavli,B., Bharati,S., Helle,N.M., Haug,T., Levine,D.W. and Krokan,H.E. (1995) Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry, 34, 128–138. [DOI] [PubMed] [Google Scholar]
- 22.Kavli B., Sundheim,O., Akbari,M., Otterlei,M., Nilsen,H., Skorpen,F., Aas,P.A., Hagen,L., Krokan,H.E. and Slupphaug,G. (2002) hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J. Biol. Chem., 277, 39926–39936. [DOI] [PubMed] [Google Scholar]
- 23.Slupphaug G., Mol,C.D., Kavli,B., Arvai,A.S., Krokan,H.E. and Tainer,J.A. (1996) A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature, 384, 87–92. [DOI] [PubMed] [Google Scholar]
- 24.Parikh S.S., Mol,C.D., Slupphaug,G., Bharati,S., Krokan,H.E. and Tainer,J.A. (1998) Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J., 17, 5214–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mol C.D., Arvai,A.S., Sanderson,R.J., Slupphaug,G., Kavli,B., Krokan,H.E., Mosbaugh,D.W. and Tainer,J.A. (1995) Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: protein mimicry of DNA. Cell, 82, 701–708. [DOI] [PubMed] [Google Scholar]
- 26.Mol C.D., Arvai,A.S., Slupphaug,G., Kavli,B., Alseth,I., Krokan,H.E. and Tainer,J.A. (1995) Crystal structure and mutational analysis of human uracil-DNA glycosylase: structural basis for specificity and catalysis. Cell, 80, 869–878. [DOI] [PubMed] [Google Scholar]
- 27.Cupples C.G. and Miller,J.H. (1989) A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc. Natl Acad. Sci. USA, 86, 5345–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kunkel T.A., Bebenek,K. and McClary,J. (1991) Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol., 204, 125–139. [DOI] [PubMed] [Google Scholar]
- 29.Boiteux S., O’Connor,T.R., Lederer,F., Gouyette,A. and Laval,J. (1990) Homogeneous Escherichia coli FPG protein. A DNA glycosylase which excises imidazole ring-opened purines and nicks DNA at apurinic/apyrimidinic sites. J. Biol. Chem., 265, 3916–3922. [PubMed] [Google Scholar]
- 30.Nilsen H., Otterlei,M., Haug,T., Solum,K., Nagelhus,T.A., Skorpen,F. and Krokan,H.E. (1997) Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res., 25, 750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castaing B., Geiger,A., Seliger,H., Nehls,P., Laval,J., Zelwer,C. and Boiteux,S. (1993) Cleavage and binding of a DNA fragment containing a single 8-oxoguanine by wild type and mutant FPG proteins. Nucleic Acids Res., 21, 2899–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henderson D.A., Inglesby,T.V., Bartlett,J.G., Ascher,M.S., Eitzen,E., Jahrling,P.B., Hauer,J., Layton,M., McDade,J., Osterholm,M.T. et al. (1999) Smallpox as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. J. Am. Med. Assoc., 281, 2127–2137. [DOI] [PubMed] [Google Scholar]
- 33.Klietmann W.F. and Ruoff,K.L. (2001) Bioterrorism: implications for the clinical microbiologist. Clin. Microbiol. Rev., 14, 364–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berche P. (2001) The threat of smallpox and bioterrorism. Trends Microbiol., 9, 15–18. [DOI] [PubMed] [Google Scholar]
- 35.Lane J.M., Ruben,F.L., Neff,J.M. and Millar,J.D. (1969) Complications of smallpox vaccination, 1968. N. Engl. J. Med., 281, 1201–1208. [DOI] [PubMed] [Google Scholar]
- 36.Kaplan E.H., Craft,D.L. and Wein,L.M. (2002) Emergency response to a smallpox attack: the case for mass vaccination. Proc. Natl Acad. Sci. USA, 99, 10935–10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Halloran M.E., Longini,I.M.,Jr, Nizam,A. and Yang,Y. (2002) Containing bioterrorist smallpox. Science, 298, 1428–1432. [DOI] [PubMed] [Google Scholar]
- 38.Delort A.M., Duplaa,A.M., Molko,D., Teoule,R., Leblanc,J.P. and Laval,J. (1985) Excision of uracil residues in DNA: mechanism of action of Escherichia coli and Micrococcus luteus uracil-DNA glycosylases. Nucleic Acids Res., 13, 319–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nilsen H., Haushalter,K.A., Robins,P., Barnes,D.E., Verdine,G.L. and Lindahl,T. (2001) Excision of deaminated cytosine from the vertebrate genome: role of the SMUG1 uracil-DNA glycosylase. EMBO J., 20, 4278–4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ishii K. and Moss,B. (2001) Role of vaccinia virus A20R protein in DNA replication: construction and characterization of temperature-sensitive mutants. J. Virol., 75, 1656–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCraith S., Holtzman,T., Moss,B. and Fields,S. (2000) Genome-wide analysis of vaccinia virus protein–protein interactions. Proc. Natl Acad. Sci. USA, 97, 4879–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klemperer N., McDonald,W., Boyle,K., Unger,B. and Traktman,P. (2001) The A20R protein is a stoichiometric component of the processive form of vaccinia virus DNA polymerase. J. Virol., 75, 12298–12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Punjabi A., Boyle,K., DeMasi,J., Grubisha,O., Unger,B., Khanna,M. and Traktman,P. (2001) Clustered charge-to-alanine mutagenesis of the vaccinia virus A20 gene: temperature-sensitive mutants have a DNA-minus phenotype and are defective in the production of processive DNA polymerase activity. J. Virol., 75, 12308–12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Otterlei M., Warbrick,E., Nagelhus,T.A., Haug,T., Slupphaug,G., Akbari,M., Aas,P.A., Steinsbekk,K., Bakke,O. and Krokan,H.E. (1999) Post-replicative base excision repair in replication foci. EMBO J., 18, 3834–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seal G. and Sirover,M.A. (1986) Physical association of the human base-excision repair enzyme uracil DNA glycosylase with the 70,000-dalton catalytic subunit of DNA polymerase alpha. Proc. Natl Acad. Sci. USA, 83, 7608–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]