


Abstract
Phospholipid hydroperoxide glutathione peroxidase (phGPx) is a member of the seleno glutathione peroxidase family that is comprised of five selenoproteins capable of reducing hydroperoxy lipids to the corresponding alcohols. The enzyme has been implicated in antioxidative defense, but its high expression level in testicular tissue suggests a more specific function during sperm maturation. The phGPx is encoded for by a joint sperm nucleus/phGPx gene (sn/phGPx) and can be expressed as a mitochondrial or cytosolic isoform. Although sn/phGPx genes have been cloned from various mammalian species expression regulation of the enzyme has not been studied in detail. We investigated the 5′-flanking region of the murine sn/phGPx gene and observed basic promoter activity in a 200 bp region localized immediately upstream of the translational initiation site of the cytosolic isoform (3′-ATG). DNase protection assays indicated the presence of five distinct protein-binding regions and electrophoretic mobility shift assays and supershift experiments revealed binding of stimulating protein 1 (SP1), nuclear factor Y (NF-Y) and members of the SMAD family. Site-directed mutagenesis of the consensus binding sequences abolished in vitro transcription factor binding. Expression of reporter genes was most effectively impaired when SP1/SP3 and NF-Y binding site-deficient constructs were tested. Chromatin immunoprecipitation suggested the in vivo relevance of these transcription factors. Our data indicate that the basic phGPx promoter constitutes a 200 bp oligonucleotide, which is localized immediately upstream of the 3′-ATG and involves functional SP1/SP3, NF-Y and SMAD binding sites. The corresponding trans-regulatory proteins may contribute to differential expression regulation of the mitochondrial and cytosolic phGPx isoforms.
INTRODUCTION
Selenium-containing glutathione peroxidases (GPx) constitute a family of anti-oxidative enzymes that are capable of reducing organic and inorganic hydroperoxides to the corresponding hydroxy compounds utilizing glutathione or other hydrogen donors as reducing equivalents (1,2). Five different selenium GPx sub-types have been identified (3,4), but there are only four seleno GPx genes. Among the seleno GPx isoforms the phospholipid hydroperoxide glutathione peroxidase (phGPx) is unique (5,6) because of its capability of reducing ester lipid hydroperoxides even if they are incorporated in biomembranes (7,8) or lipoproteins (9,10). The phGPx cDNAs of various species contain two in-frame translational initiation sites (5′-ATG and 3′-ATG, Scheme 1), both of which are functional under certain conditions. In vitro translation of the rat full-length phGPx mRNA is initiated predominantly at the 5′-ATG. However, translation did start at the 3′-ATG when the 5′-ATG was deleted (11). The sequence between the two translational initiation sites was suggested to encode for a signal peptide required for mitochondrial import (11). In fact, experiments in which this targeting sequence was fused to a green fluorescence protein indicated predominant mitochondrial localization of the fusion protein (12). More detailed investigations of phGPx expression at the mRNA level indicated a high degree of variation of transcriptional initiation. Two major phGPx mRNA populations appear to exist and both of them are heterogeneous (11). (i) One mRNA family lacks the mitochondrial insertion peptide and utilizes the 3′-ATG for translational initiation. These messengers are translated to the cytosolic isoform. (ii) The second mRNA population contains both the 5′- and the 3′-ATG as well as the complete mitochondria targeting sequence. Translation of these transcripts is initiated at the 5′-ATG and leads to the formation of the mitochondrial phGPx (Scheme 1). After mitochondrial import the targeting peptide is cleaved off (13) so that the two phGPx isoforms cannot be distinguished on the basis of their protein chemical properties. The existence of two mRNA populations suggests differential transcription mechanisms of the ph/sperm nucleus (sn) GPx gene utilizing various transcription initiation sites. Alternatively, a joint primary transcript may be processed differentially at post-transcriptional levels. Expression of the ph/snGPx gene is tissue specifically regulated (14). In most cells the enzyme is expressed at relatively low levels (15,16) but in testis it constitutes a major protein (17). Unfortunately, the regulatory mechanisms involved in high-level expression of the enzyme in germinative cells and its repression in somatic tissues are largely unknown. The previous observation that expression of the enzyme is strongly down-regulated in testis of hypophysectomized rats and that this defect can partially be reversed by gonadotropin administration, suggested a role of testosterone in the regulation of phGPx expression (18). However, more recent data suggested that testosterone does not directly activate transcription of the ph/snGPx gene (19).

Scheme 1. Multiple transcription initiation sites in mouse and rat tissues. The location of the murine translational start codons at +145 (5′-ATG) and ATG at +226 (3′-ATG) are shown. Thick arrows indicate different transcription initiation sites identified in mice for the mitochondrial and cytosolic phGPx isoform. The most 5′ transcription start site identified so far in mice (testis) was set +1 (33). The asterisk indicates two additional transcription start sites reported for various murine organs (15). The two windows of transcription start sites identified in rat tissues (11) are indicated by brackets.
The gene encoding for the porcine phGPx was cloned in 1996 (20) and more recently the complete genomic sequences of the human (21) and the murine (22) orthologs became available. Comparison of the 5′-flanking regions suggested the existence of conserved consensus binding sequences for several transcription factors (22). However, the functionality of these binding motifs has never been studied in detail. Only recently (23), experiments on the human ph/snGPx gene suggested the regulatory importance of a conserved CCAAT box that binds transcription nuclear factor Y (NF-Y). This binding motif was localized –156 to –151 bp upstream of the 5′-ATG (Scheme 1). Other cis-regulatory elements, which have been identified structurally in various mammalian phGPx genes, have not been studied for their functionality. Moreover, it remained unclear whether the synthesis of the two phGPx isoforms (mitochondrial and cytosolic) is regulated by a uniform mechanism or whether their expression is under the control of different cis-regulatory elements.
The lack of detailed experimental data on the regulation of phGPx expression and the interesting biological functions of the enzyme prompted us to investigate the functionality of putative transcription factor binding sites present in the 5′-flanking region of the murine ph/snGPx gene. DNase protection assays suggested the existence of at least five protein-binding sites and electrophoretic mobility shift assays (EMSAs), which included supershift studies, identified stimulating protein 1 (SP1), NF-Y and members of the SMAD family as regulatory proteins.
MATERIALS AND METHODS
Materials
The chemicals used were from the following sources: trisodium citrate dihydrate, magnesium sulfate heptahydrate, Triton X-100, bovine serum albumin (BSA), penicillin–streptomycin solution and FBS were from Sigma (Deisenhofen, Germany); sodium hydroxide, sodium chloride and tris(hydroxymethyl)aminomethane (Tris) were from Merck (Darmstadt, Germany); PWO DNA polymerase, agarose, ampicillin, AMV reverse transcriptase and proteinase inhibitor cocktail tablets (complete, Mini) were from Roche Diagnostics (Mannheim, Germany); restriction endonucleases and the DNA molecular weight markers (100 bp, 1 kb) were from New England Biolabs GmbH (Schwalbach, Germany); PanScript DNA Polymerase was from Pan Biotech GmbH (Aidenbach, Germany); Advantage 2 Polymerase was from Clontech (Palo Alto, CA, USA); Bacto yeast extract, Bacto agar and Bacto tryptone were from Difco (Detroit, MI); Dulbecco’s modified Eagle’s medium (DMEM) was from Invitrogen (Karlsruhe, Germany); rabbit polyclonal anti-NF-Y antibody (H209), rabbit anti-SP1 antibody (PEP2) and rabbit anti-SP3 antibody (D-20) X were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The primers were custom-synthesized by TIB MOLBIOL (Berlin, Germany).
Cell lines and culture conditions
Human embryonic kidney cells 293 (HEK293) and murine macrophages (J774) were obtained from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). The cells were maintained in DMEM supplemented with 10% (v/v) fetal calf serum and antibiotics (100 U/ml penicillin and 100 µg/ml streptomycin) at 37°C under 5% CO2.
Functional promoter assays
The pBlueTOPO reporter vector (Invitrogen, Groningen, The Netherlands) containing the β-galctosidase as reporter gene was used for analysis of promoter activity of subcloned promoter fragments. For the assays, putative promoter fragments of phGPx were amplified by PCR and subcloned into the pBlue-TOPO reporter vector following the user’s manual. PCR was carried out with a Biometra TRIO Thermoblock 2.51BB (Biometra, Göttingen, Germany) using purified plasmid DNA (Qiagen, Hilden, Germany) of the subcloned ph/snGPx gene as template and the desired primer combinations: 5′-flanking region of the ph/snGPx gene upstream of the 5′-Cap (constructs A0–A6), 5′-CGC TAA GAC GCG AGG CCC CTG-3′; immediate 5′-flanking region of the first phGPx exon containing the 5′-untranslated region (5′-UTR) of the mRNA (constructs B0–B4), 5′-AGC CAG CGG CTC CTC CCC TCA-3′; region of the ph/snGPx gene immediately upstream of the 3′-ATG containing the sequence encoding for the mitochondrial targeting peptide (constructs C0–C4), 5′-TGG TGC CTG CCA GAC CAG GCG-3′; for the constructs B0 and C0, 5′-GAG GGG CCT CGC GTC TTA GCG-3′; for the constructs A1, B1 and C1, 5′-CGC GTC CCT ATC ACT GGG CAT G-3′; for the constructs A2, B2 and C2, 5′-CCC AAA GCG CCC TGG CAC CCT-3′; for the constructs A3, B3 and C3, 5′-GGC CGA CAC CGA GAT CCC AGC-3′; for the constructs A4, B4 and C4, 5′-AAG CTA CAT CTG GGG TTC GGG AC-3′; for construct A5, 5′-GGC AAT GGC TTT CTC ACC AAG CC-3′; for construct A6, 5′-AGA GTG TGA CCA CTA GGC CCA TC-3′. For transient transfection, HEK293 cells were resuspended at a density of 0.5 × 106 cells/5 ml of medium and plated in 60-mm culture dishes. After a culturing period of 24 h, cells were transfected with a calcium phosphate transfection kit (Invitrogen). We used 10 µg of the β-Gal promoter construct and 5 µg of an internal reference plasmid containing the luciferase cDNA (pGL3; Promega, Mannheim, Germany). After 72 h, cells were harvested and both β-galactosidase and luciferase activities were assayed. The β-galactosidase activities were normalized for transfection efficiency using the data of the luciferase assay. For functional promoter studies with mutated reporter gene constructs, cells were transiently transfected using FuGene 6 as described by the manufacturer (Roche). HEK293 cells were plated in six-well plates at a density of 2.5 × 105 cells/well in DMEM supplemented with 10% (v/v) fetal calf serum 24 h before transfection. For transient transfection we used each of 1 µg of β-gal promoter construct, 1 µg of the luciferase vector pGL3 and 6 µl of FuGene 6. After 72 h, cells were harvested and both β-galactosidase and luciferase activities were assayed as described above.
Site-directed mutagenesis
Site-directed mutagenesis in the plamsid pBlueTOPO was performed using the QuikChangeTM Site-Directed Mutagenesis Kit (Stratagene, Amsterdam, The Netherlands) and the DNA sequences of mutated plasmids were confirmed by sequence analysis. For each mutation the loss of transcription factor binding activity was checked.
DNase I protection assay
A genomic fragment containing the 5′-UTR of the cytosolic phGPx isoform as well as ∼100 bp of the 5′-flanking region of the ph/snGPx gene (–99 to +228 bp) was subcloned in the pBlueTOPO vector (Invitrogen). After excision with HindIII (NcoI) and BamHI the probe was radio-labeled using T4 polynucleotide kinase. The labeled probe was then treated with BamHI or HinDIII to achieve selective labeling of either the sense- or antisense strand. Aliquots of the labeled probe containing 10 000–20 000 c.p.m. of 32P-labeled DNA were added to reaction mixes consisting of a 40 mM HEPES buffer, pH 7.9 containing 100 mM KCl, 12.5 mM MgCl2, 1 mM EDTA, 20% glycerol, 1 mM DTT, 1.5 µg of poly(dI–dC) and ∼100 µg of nuclear proteins in a total volume of 50 µl. The mixture was kept on ice for 30 min before addition of 50 µl of 5 mM CaCl2, 10 mM MgCl2 and 2–10 U DNase I depending on the input of nuclear proteins. Digestion was carried out at 4°C for 5 min and terminated by adding 100 µl of a solution containing 200 mM NaCl, 20 mM EDTA and 1% SDS. DNA was phenol extracted and precipitated with ethanol. The digestion products were analyzed on 6% acrylamide sequencing gel containing 8 M urea. The protected sequences were identified by comparison with the migration of Maxam-Gilbert sequencing products. Gels were dried and exposed to X-ray films overnight at –80°C.
Electrophoretic mobility shift assay and supershifts
EMSAs were carried out with the DIG Gel Shift Kit (Roche Diagnostics) following the user’s manual. In the first step, single-stranded complementary oligonucleotides containing the binding sequences for transcription factors were annealed and end-labeled with digoxygenin. The labeled probes (48 fmol of double-stranded oligonucleotides) were then incubated for 30 min at 4°C with 10 µg of nuclear extract proteins in 40 mM HEPES buffer, pH 7.9 containing 100 mM KCl, 12.5 mM MgCl2, 1 mM EDTA, 20% glycerol, 1 mM DTT, 2 µg of poly(dI–dC), 0.2 µg of poly-(l)-lysine (total assay volume of 15 µl). Then the mixtures were subjected to electrophoresis on a 6% polyacrylamide gel with 0.5-fold TBE running buffer. The DIG-oligonucleotide/protein complexes were transferred to a Hybond-N blotting membrane (Amersham Life Science, Freiburg, Germany) and the shift bands were visualized following the user’s manual. For competition studies, unlabeled probe competitor or consensus oligonucleotides (Santa Cruz Biotechnology) were added at a 200-fold excess over the DIG-labeled probe. When competitor was used, it was pre-incubated with the nuclear extracts at 4°C for 10 min before the addition of the DIG-labeled probe DNA.
For supershift experiments polyclonal antibodies raised against the SP1, NF-Y and SP3 (all from Santa Cruz Biotechnology) were added to the incubation mixture. Labeling, incubation and electrophoresis were carried out as described above.
Chromatin immunopreciptation
107 HEK293 cells were washed twice with phosphate-buffered saline (PBS) and the cellular proteins were cross-linked to DNA by adding formaldehyde to a final concentration of 1% for 15 min at room temperature. Cells were subsequently washed, removed from the culture dishes by scraping and resuspended in 1 ml of PBS. Cells were lysed in 1.5 ml of lysis buffer (10 mM HEPES buffer, pH 7.9 containing 10 mM KCl, 1.5 mM MgCl2, 0.1 mM EDTA, 0.5 mM DTT and the proteinase inhibitor cocktail). After 10 min on ice, the cells were homogenized in a glass Dounce homogenizer and centrifuged at 5200 g. The nuclear pellet was resuspended in 20 mM HEPES buffer, pH 7.9 containing 10% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.1 mM EDTA, 0.5 mM DTT and proteinase inhibitor cocktail and was sonicated on ice to fragment the genomic DNA. The mixture was then centrifuged to remove cell debris and aliquots were stored at –80°C. For chomatin immunoprecipitation, aliquots were diluted in RIPA buffer (50 mM Tris–HCl buffer, pH 7.5 containing 150 mM NaCl, 0.1% sodium desoxycholat, 1% Triton X-100, 0.25 mM EDTA and the proteinase inhibitor cocktail) and samples were incubated overnight at 4°C with 2.5 µl of a rabbit polyclonal anti-NF-Y antibody (H209) or with a rabbit anti-SP1 antibody (PEP2) obtained from Santa Cruz Biotechnology. Immune complexes were precipitated by the addition of 50 µl of protein A-agarose (Santa Cruz Biotechnology) and low speed spinning. Precipitates were washed twice with 1 ml of RIPA buffer, twice with 1 ml of PBS and DNA/protein complexes were eluted from the agarose by 15 min of incubation with 250 µl of elution buffer (1% SDS, 100 mM NaHCO3). Elution was repeated, the eluates were pooled, NaCl was added to reach a final concentration of 0.3 M and the samples were incubated at 65°C for 4 h to reverse formaldehyde-induced cross-linking. The proteins were then digested with 2 µl of proteinase K (20 mg/ml) in 40 mM Tris–HCl buffer, pH 7.5 containing 10 mM EDTA. The samples were incubated for 1 h at 60°C and the DNA was extracted with phenol/chloroform followed by ethanol precipitation. The DNA pellet was resuspended in 50 µl of of sterile H2O and 10 µl of DNA solution was used as a template for PCR. The following primer combinations were designed to amplify the precipitated binding regions of the transcription factors and 35 PCR cycles were run: P1 (5′-CGC CAA CAA GTC CGC ACG-3′) and P2 (5′-AAA GGC GGC CGA GGC TCA TC-3′). The PCR products were separated on 2% agarose gel and visualized by ethidium bromide staining.
Miscellaneous methods
For preparations of nuclear extracts cultured cells were rinsed with ice-cold PBS, scraped from the surface and collected by centrifugation. The cells were washed with 4 vol of hypotonic buffer (10 mM K-HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT). Subsequently, the cells were resuspended in 4 vol of hypotonic buffer containing 0.4% Nonidet P-40, incubated on ice for 10 min and centrifuged. The nuclear pellet was resuspended in 2 vol of high salt buffer (20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 420 mM NaCl, 1 mM DTT, 0.2 mM EDTA, 20% glycerol) and the nuclei were extracted for 60 min on ice. Subsequently, the mixture was centrifuged for 15 min at 7500 g. The supernatant was snap frozen in liquid nitrogen and stored at –80°C. Protein concentrations of the nuclear extracts were measured with the Bio-Rad protein assay system.
RESULTS
Functional promoter studies
Two major populations of phGPx mRNA species have been identified in previous studies (11,13), which differ with respect to their molecular weights (Scheme 1). The long messengers that contain both the 3′- and the 5′-ATG are translated to the mitochondrial phGPx isoform. Here, the 5′-ATG is utilized as a translational initiation site. In contrast, the family of short phGPx messengers, which contain the 3′-ATG only, encodes for the cytosolic phGPx isoform and translation of this messenger is initiated at the 3′-ATG. This interesting genomic constellation (for direct sequence information see Fig. S1 in the Supplementary Material) is of dual consequence for the regulatory mechanisms of transcription: (i) if the cytosolic (short) and the mitochondrial (long) mRNA species are differentially transcribed there must be at least two separate transcriptional initiation sites (Scheme 1) for the two isoforms; (ii) if separate transcriptional initiation sites exist the potential regulatory sequences for transcription are likely to be different and for the cytosolic enzyme the region between the 3′- and the 5′-ATG might contribute to transcriptional regulation. To shed light on the relative contribution to the promoter activity of the different parts of the 5′-flanking region a comprehensive set of reporter genes was constructed (Fig. 1A) covering a 1000 bp region immediately upstream of the translational initiation site of the mitochondrial isoform (5′-ATG). After transient transfection of HEK293 cells with these constructs, the enzymatic activity of the reporter gene (β-galactosidase) was assayed. The data summarized in Figure 1B indicate that the 5′-flanking region of the ph/snGPx gene located immediately upstream of the transcriptional initiation site of the mitochondrial isoform (A-set) exhibits only a weak promoter activity (relative galactosidase activity of ∼0.61). However, it was significantly higher than that of the negative control (relative galactosidase activity of 0.1). The different reporter genes of the A-set did not exhibit major differences in their promoter activities although the shortest construct appeared to be most active. The second set of reporter genes (B-set) contained the 5′-UTR of the mitochondrial messenger and variable sections of the 5′-flanking region of the ph/snGPx gene. The average activity of this set of reporter gene constructs was almost one order of magnitude higher than that of the A-set indicating significant promoter activity in the region of the ph/snGPx gene, which is transcribed as the 5′-UTR of the mitochondrial messenger. This conclusion is also supported by two other observations: (i) if only the 5′-UTR of the mitochondrial messenger was fused to the reporter gene (construct 0 of the B-set) the activity was higher than that for all constructs of the A-set; (ii) the relative promoter activity of each B-set member was almost one order of magnitude higher than the activity of the corresponding A-set counterpart. Interestingly, as in the A-set (A1), construct B1, which contains only a 100 bp fragment of the 5′-flanking region, exhibited the highest promoter activity.
Figure 1.

Functional promoter studies of the 5′-flanking region of the joint ph/snGPx gene. Different sections of the 5′-flanking region of the joint ph/snGPx gene were ligated into a β-galactosidase-based reporter gene and HEK293 cells were transfected with the different constructs using the calcium phosphate transfection method. In order to correct the β-galactosidase activity measured for transfection efficiency, cotransfections with luciferase containing control plasmid (pGL4) were carried out. (A) Length (bp) of the PCR fragments of the 5′-flanking regions used for reporter gene construction. (B) Relative β-galactosidase activity of the different promoter constructs corrected for luciferase activity. Co-negative control (pBlueTopo without insert).
Next, we addressed the question of whether or not there is significant promoter activity in the region of the ph/snGPx gene encoding for the mitochondrial targeting peptide. This region is completely transcribed for both the mitochondrial and cytosolic isoforms, and contributes to the 5′-UTR of the cytosolic phGPx. The average promoter activity of this set of reported genes (C-set) was almost twice as high as that of the B-set suggesting significant promoter activity in this region. As in the other sets, construct C1 containing the complete sequence of exon 1 and a –100 bp fragment of the 5′-flanking region exhibited the highest promoter activity.
Taken together these data indicate that the –100 to –900 bp part of the 5′-flanking region of the ph/snGPx gene may not exhibit major promoter activity. Only the first 100 bp located immediately upstream of the 5′-Cap may contribute to the basic promoter. However, the majority of the promoter activity was mapped to exon 1 of the ph/snGPx gene, which is comprised of the 5′-UTR of the mitochondrial messenger as well as the sequences encoding for the mitochondrial targeting peptide.
DNase protection studies
As indicated above, reporter gene construct C1 exhibited the highest promoter activity. Thus, the 326 bp region immediately upstream of the 3′-ATG (Fig. 1) may constitute the basic phGPx promoter. Interspecies sequence alignments indicated that this fragment contains several conserved transcription factor binding sequences including the recently identified CCAAT box (23). To find out whether there are additional protein-binding sites in this region, DNAse protection assays were carried out. From Figure 2 it can be seen that four distinct protein-binding regions (footprints 1–4, FP-1 to FP-4) have been detected and three of them were observed using sense and anti-sense strands as templates. Two footprints (FP-1, FP-2) were mapped to the 5′-flanking region of the ph/snGPx gene; the others (FP-3, FP-4) were located in the 5′-UTR of the long phGPx messenger. FP-1, FP-2 and FP-3 were of similar size (20–30 bp), but FP-4 was comprised of 50 bp. This size suggested the existence of two or more protein-binding regions. A very similar footprint pattern has recently been reported for the human phGPx (23), suggesting a high interspecies conservation of the regulatory elements.
Figure 2.

DNA footprinting of the 5′-flanking region of the joint ph/snGPx gene. DNase protection assays (footprints) were carried out as described in Materials and Methods. A genomic fragment containing the 5′-UTR of the cytosolic phGPx isoform as well as ∼100 bp of the 5′-flanking region of the phGPx gene (–99 to +228 bp) was used. The areas of quenched signals indicate the protein-binding sites (FP-1 to FP-4). Unspecific quenching was tested using BSA but we did not observe any unspecific effects. The sequences of the footprints were determined with the G+A ladder.
Electrophoretic mobility shift assays
To identify the DNA-binding proteins involved in foot printing we first inspected the corresponding DNA regions for the presence of consensus binding sequences for known transcription factors using the MatInspector program (24). These data (see Table S1 in Supplementary Material) suggested that FP-1, FP-3 and FP-4 contain high core SP1/SP3 binding regions. In FP-2 and FP-4 the consensus sequence for the binding of NF-Y was found. In addition, a binding sequence for the SMAD family member forkhead activin signal transducer (FAST1) was observed in FP-4, although its core value was not particularly impressive. To identify the functionality of these cis-regulatory elements EMSAs were carried out using the nuclear extracts of HEK293 cells and murine macrophages (J774) as a source of nuclear proteins. When a digoxygenin-labeled oligonucleotide of FP-1 was incubated with these nuclear extracts two strong shift signals were observed (Fig. 3, lanes b and d). Both signals were competed off with a 200-fold molar excess of unlabeled competitor (data not shown) and by a SP1/SP3 consensus probe (lanes c and e). In contrast, when an unlabeled probe containing the sequence of FP-4A (contains a NF-Y binding site only; see below) was added, the two shift signals were not influenced (data not shown). The upper shift band migrated in a molecular weight region of an SP1/SP3-oligonucleotide complex, whereas the lower signal exhibited a higher electrophoretic mobility. These data suggest that the upper shift band may be due to SP1/SP3 binding whereas the lower band was caused by an unknown transcription factor.
Figure 3.
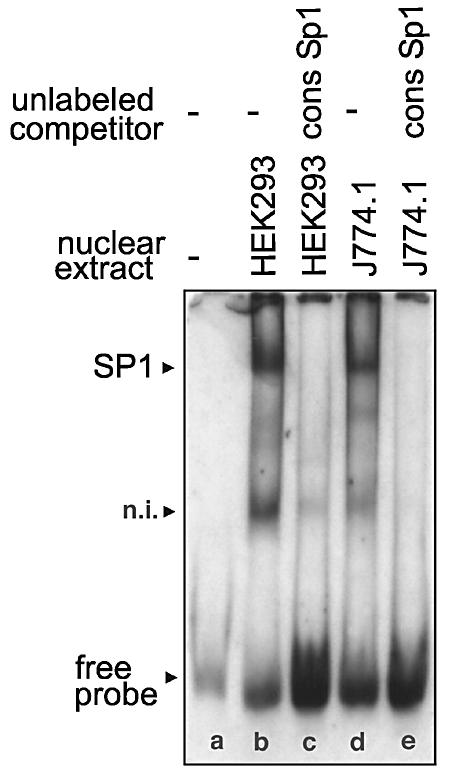
EMSAs using an oligonucletide of FP-1 as labeled probe. The labeled oligonucleotide dimer (GTA AGC CCA GCC CCG CCC AAG CCG TCC CTT CAT TCA) containing the binding sequence for SP1 was incubated with nuclear extracts of HEK293 cells (10 µg of protein) or J774.1 cells (10 µg of protein) in the presence or absence of unlabeled SP1 consensus oligonucleotides (9.6 pmol) as described in Materials and Methods. After 30 min of incubation at 4°C the entire binding sample was applied to polyacrylamide gel electrophoresis. The arrows indicate the shift bands. n.i., not identified.
Sequencing of FP-2 indicated the presence of a NF-Y binding site and EMSA studies with nuclear extracts of HEK293 (lane b) and J774 cells (lane f) indicated single shift signals (Fig. 4), which were competed off by unlabeled probes (lanes c and g) and by a NF-Y consensus oligonucleotide (lanes e and h). Mutagenesis (see Table S2 in the Supplementary Material) of the NF-Y consensus sequence abolished transcription factor binding and competition with an unlabeled FP-4A probe also eliminated the shift signal (lane d). In contrast, when a mutated FP-4 oligonucleotide was used the signal remained unaltered (data not shown). Taken together, these data indicate that FP-2, but also FP-4A (see below), contain functional NF-Y binding sites. The FP-2 sequence indicated a CCAAT box, which is conserved among all mammalian species.
Figure 4.
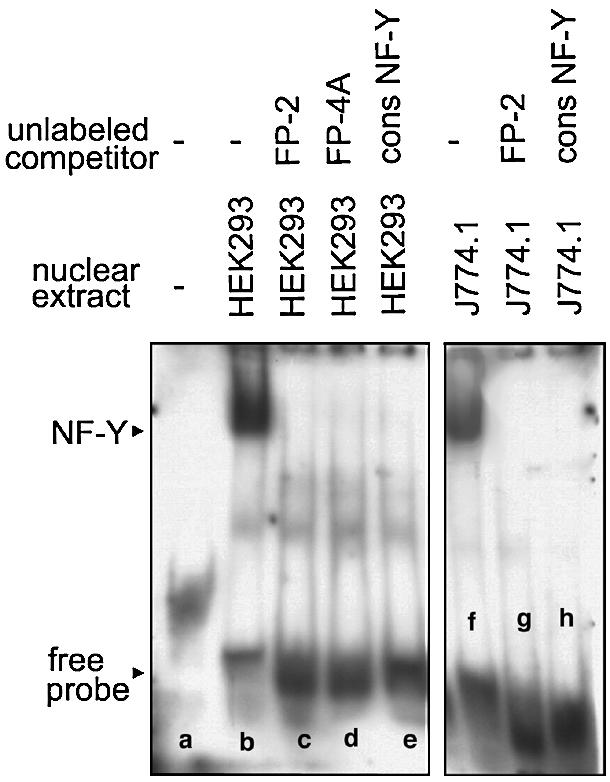
EMSAs using an oligonucletide of FP-2 as labeled probe. The labeled oligonucleotide dimer (ATT CAG GCT TCC CAT TGG CTG CAG GGG CCT CGC GTC) containing the binding sequence for NF-Y was incubated with nuclear extracts of HEK293 cells (10 µg of protein) or J774.1 cells (10 µg of protein) in the presence or absence of unlabeled competitors or NF-Y consensus oligonucleotides (9.6 pmol) as described in Materials and Methods. After 30 min of incubation at 4°C the entire binding sample was applied to polyacrylamide gel electrophoresis. The arrows indicate the shift bands.
In silico analysis of FP-3 suggested the existence of a single SP1/SP3 binding site (see Table S1 in the Supplementary Material). Incubation of nuclear extracts of HEK293 and J774 cells with a labeled FP-3 oligonucleotide indicated strong shift signals (Fig. 5A, lanes b and d), which were competed off with an unlabeled SP1/SP3 consensus oligonucleotide (lanes c and e). To confirm the specificity of SP1/SP3 binding we used unlabeled oligonucleotides of FP-1, FP-2, FP-3, FP-4A and FP-4B as competitors (Fig. 5B) and found that the shift band disappeared with FP-1, FP-3 and FP-4B oligonucleotides (lanes d, b and f). In contrast, the signal remained unchanged when the experiment was carried out with unlabeled probes of FP-2 and FP-4A, which only contain NF-Y binding sequences (lanes c and e).
Figure 5.

EMSAs using an oligonucletide of FP-3 as labeled probe. The labeled oligonucleotide dimer (AAT AAG AGA CGT CAG TGG GCG TGC CCG AGG GCG GGC) containing the binding sequence for SP1 was incubated with nuclear extracts of HEK293 cells (10 µg of protein) or J774.1 cells (10 µg of protein) in the presence or absence of unlabeled (A) SP1 consensus oligonucleotides or (B) competitors (9.6 pmol) as described in Materials and Methods. After 30 min of incubation at 4°C the entire binding sample was applied to polyacrylamide gel electrophoresis. The arrow indicates the shift bands.
FP-4 is comprised of 50 nt and because of this size it was divided into two overlapping parts (FP-4A and FP-4B) for functional promoter studies. In silico analysis of FP-4A suggested the presence of single NF-Y and SMAD binding sites. EMSA with nuclear extracts of HEK293 cells (Fig. 6A) revealed two shift signals (lane b), which were quenched when unlabeled FP-4A competitor was added (lane c). The upper shift band disappeared when NF-Y consensus oligonucleotide or unlabeled FP-2 oligonucleotide (contains the NF-Y consensus sequence) were added (lanes f and d). In contrast, a FP-2 oligonucleotide, in which the NF-Y consensus sequence was mutated (see Table S2 in the Supplementary Material), did not influence NF-Y binding but abolished the lower shift band (lane e). More detailed in silico promoter analysis suggested the additional occurrence of a binding site for SMAD transcription factors. In fact, after addition of a SMAD3/4 consensus oligonucleotide (Fig. 6B, lane b) the lower shift signal disappeared. After combined addition of unlabeled SMAD and NF-Y competitors no shift bands were observed at all (data not shown). These data suggested that the upper shift signal was caused by NF-Y and the lower one by a SMAD protein.
Figure 6.

EMSAs using an oligonucletide of FP-4 as labeled probe. The labeled oligonucleotide dimer (CCA TAC TCG GCC TCG CGC GTC CAT TGG TCG GCT GCG) containing the overlapping binding sequences for SMAD and NF-Y (A and B) or the labeled oligonucleotide (TCG GCT GCG TGA GGG GAG GAG CCG CTG GCT CCG GCC) containing the binding sequence for SP1 (C) was incubated with nuclear extracts of HEK293 cells (10 µg of protein) or J774.1 cells (10 µg of protein) in the presence or absence of unlabeled competitors or consensus oligonucleotides (9.6 pmol) as described in Materials and Methods. After 30 min of incubation at 4°C the entire binding sample was applied to polyacrylamide gel electrophoresis. The arrows indicate the shift bands.
FP-4B contains a single SP1/SP3 binding site and EMSA experiments with HEK293 and J774 nuclear extracts (Fig. 6C) indicated strong single shift signals (lanes b and d), which were competed off by unlabeled SP1/SP3 consensus oligonucleotides (lanes c and e). Moreover, we found (data not shown) that the signals disappeared after addition of unlabeled FP-1 competitor (contains a SP1/SP3 site). In contrast, a FP-4A oligonucleotide (contains a NF-Y and SMAD binding site) or a mutated FP-4B probe (see Table S2 in the Supplementary Material) did not induce any alterations in the shift pattern (data not shown). These data suggest that FP-4B may be due to the binding of a SP1/SP3 family member.
To obtain more detailed information on the identity of the trans-regulatory proteins, supershift experiments were carried out in which specific antibodies against SP1, SP3 and NF-Y were added to the incubation mixture. From Figure 7A it can be seen that the SP1/SP3 shift signal obtained for FP-1 was not detectable when SP1 antibody was added. In contrast, addition of SP3 and NF-Y antibodies did not alter the shift pattern. Similar results were obtained for FP-3 and FP-4B. Our failure to alter the shift pattern by addition of SP3-specific antibodies suggests that SP1 but not SP3 belongs to the dominant promoter binding protein. The NF-Y shift signal obtained with an oligonucleotide representing FP-2 was quenched by the addition of an NF-Y antibody. In contrast, the SP1 antibody did not impact the shift pattern. Similar results were obtained for the NF-Y signal in FP-4A. It should be noted that the SMAD shift signal in FP-4A was not impacted either by the SP1 or the NF-Y antibody.
Figure 7.
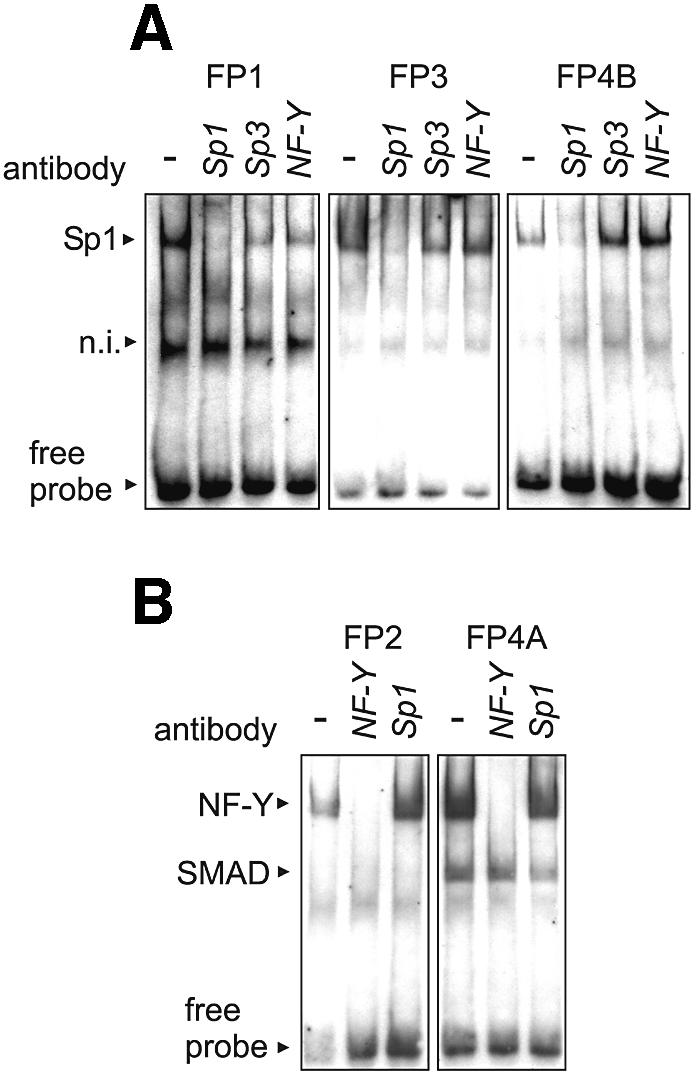
Supershift assays using antibodies against SP1, SP3 and NF-Y. EMSA experiments were carried out as described in the captions to Figures 3–6. For the supershift experiments shown in this figure, specific antibodies raised against SP1, SP3 and NF-Y were added to the incubation mixtures. (A) Footprints FP-1, FP-3 and FP-4B containing SP1/SP3 binding sites were analyzed. (B) Footprints FP-2 and FP-4A containing NF-Y binding sites were analyzed. n.i., not identified.
Functional promoter studies with mutated reporter gene constructs
To quantify the relative contribution of the various cis-regulatory elements we constructed reporter genes, in which the consensus binding sequences were mutated (see Table S2 in the Supplementary Material). For the reporter gene constructs only mutations that did not bind the corresponding transcription factors were used. To investigate separately the regulatory elements for the mitochondrial and cytosolic phGPx isoform (Fig. 8) we mutated the most active reporter gene constructs from Figure 1 (construct B1 for the mitochondrial isoform and construct C1 for the cytosolic isoform). Mutation of the SP1/SP3 binding site in FP-1 caused a strong inhibition in the promoter activity of both constructs (B1a versus B1b and C1a versus C1b). These data suggest that this binding site may be of major importance for expression regulation of both the mitochondrial and cytosolic phGPx isoforms. It has been reported before that mutation of the NF-Y binding site of FP-2 of the human ph/snGPx gene impaired the promoter activity of corresponding reporter genes by ∼40% (23). We also observed a decreased promoter activity when the NF-Y binding site of the murine gene was mutated (B1a versus B1c and C1a versus C1c). However, this difference was not statistically significant (P = 0.07, n = 3). Mutation of the SP1/SP3 binding site in FP-3 (B1a versus B1d and C1a versus C1d) did not induce a major alteration in the promoter activity for either construct. Inactivation of the consensus binding sequences in FP-4A (B1a versus B1e) and FP-4B (B1a versus B1f) slightly reduced the promoter activity of the B1 constructs (residual promoter activity of 89 and 78%), whereas inhibition observed with C1 mutants (C1a versus C1e and C1a versus C1f) was more pronounced (47 and 48%).
Figure 8.

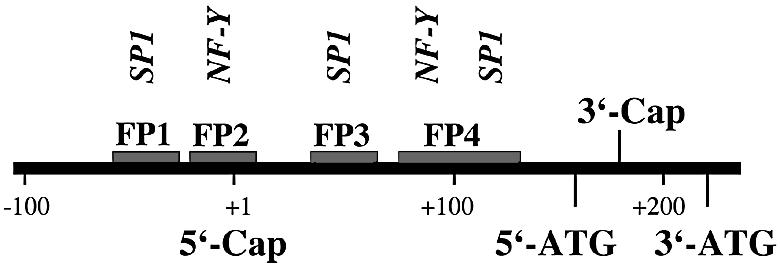
Functional promoter studies using mutated reporter gene constructs. Site-directed mutagenesis of the phGPx promoter was performed as described in Materials and Methods. A mixture of each of 1 µg of β-galactosidase promoter construct and 1 µg of pGL3 luciferase plasmid were transfected into HEK293 cells with FuGene 6 (see Materials and Methods). The β-galactosidase activity was normalized for the luciferase activity. Values are means ± SEM for three determinations given as a percentage compared with the wild-type construct. (Top) Localization of footprints (FP-1 to FP-4) and functional transcription factor binding sites in the 5′-flanking region of the sn/phGPx gene and the length of the reporter gene constructs B1 and C1. The major transcription initiation site for the mitochondrial phGPx isoform was set +1. Rectangular arrows indicate heterogeneity of transcription initiation sites. 5′-ATG (translational initiation site for the mitochondrial phGPx) and 3′-ATG (translational initiation site for the cytosolic phGPx) are also indicated. (Bottom) Relative promoter activity of wild-type (WT) and mutant reporter gene constructs. *P < 0.05; **P < 0.01 compared with the wild-type construct.
To test the regulatory interplay of different cis-acting elements we carried out simultaneous mutations of different consensus sequences and with this strategy we observed remarkable differences between the two sets of constructs. When the two functional binding sequences present in FP-1 and FP-2 were mutated simultaneously, no promoter activity was detectable at all with the B1 construct (B1g). In contrast, a residual promoter activity of >80% was measured when the same mutations were introduced into the C1 reporter gene (C1g). Thus, preventing NF-Y binding to FP-2 further impaired the promoter activity of the FP-1-deficient B1 construct (B1b versus B1g) but reversed inhibition of the promoter activity of the FP-1-deficient C1 reporter genes (C1b versus C1g). Since the mutated B1 constructs lack the mitochondrial insertion sequence (between 5′- and 3′-ATG) and the mutated C1 reporter genes contain it (the only structural difference between the B1 and C1 constructs) one may conclude that the inverse regulatory effect must be related to this stretch of nucleotides. It might be possible that NF-Y may interact directly or indirectly with the mitochondrial insertion sequence and depending on this interaction its regulatory impact may be modified. Additional experiments are required to further elucidate this interesting regulatory effect.
When the transcription factor binding sites of FP-2 + FP-3 (B1h) and FP-2 + FP-4A (B1i) were simultaneously mutated in B1 the inhibition of the promoter activity was somewhat more pronounced when compared with the separate mutations, suggesting additive effects of the two regulatory elements. In the C1 construct simultaneous mutation of FP-2 and FP-3 (C1h) was without additive effects on promoter activity when compared with the separate mutations (C1c and C1d). Similarly, no additive effect was observed for this construct when FP-2 and FP-4A were mutated jointly (C1i) or separately (C1 c and C1e). In contrast, when the consensus binding sequences in FP-3 + FP-4A (C1k) and FP-4A + FP-4B (C1m) were simultaneously mutated we observed an additive reduction of the promoter activity. For the corresponding B1 constructs (B1k, B1m) such an additive effect was not observed.
Taken together these data allow the following conclusions: (i) the SP1/SP3 binding site in FP-1 is of major regulatory importance for expression of both the mitochondrial and cytosolic phGPx isoform; (ii) simultaneous mutation of this sequence in connection with the NF-Y site of FP-2 completely wiped out the promoter activity of the B1 construct (mitochondrial isoform) and strongly reduced the inhibitory effect of separate mutations of the SP1/SP3 site in reporter gene C1 (cytosolic isoform); (iii) simultaneous mutations of the distal cis-regulatory elements (FP-1, FP-2, FP-3) impaired the promoter activity of the mitochondrial phGPx constructs more severely than that of the cytosolic isoform. In contrast, mutation of the proximal cis-regulatory elements (FP-4A and FP-4B) preferentially inhibited the activity of the cytosolic constructs (C1).
In vivo binding of SP1 and NF-Y to the FP-1–FP-2 region
The EMSA experiments including the supershift assays indicated that SP1 and NF-Y are present in the nuclei of testis and HEK293 cells and that they bind in vitro to the corresponding cis-regulatory sequences in the phGPx promoter. To find out whether they also bind under in vivo conditions, chromatin immunoprecipitation was carried out. For this purpose DNA-binding proteins were covalently linked to genomic DNA by treatment with formaldehyde. The DNA/protein complexes were then sheared by sonication, and specific DNA/protein complexes were immunoprecipitated with antibodies against the two transcription factors. Covalent linkage was reversed and the precipitated double-stranded DNA was amplified by PCR using binding-site-specific primers. From Figure 9B it can be seen that PCR signals were obtained when the DNA/protein adducts were immunoprecipitated. In contrast, the corresponding controls, in which immunoprecipitation was performed without specific antibodies, did not show any PCR signal.
Figure 9.

Chromatin immunoprecipitation of relevant transcription factors. Chromatin immunoprecipitation was carried out as described in Materials and Methods. (A) Localization of the relevant transcription factor binding sites in the promoter of the ph/snGPx gene. The arrows indicate the positions of the primers used for amplification of the immunoprecipitated DNA. (B) Sheared DNA/protein complexes were precipitated with antibodies against NF-Y and SP1 and precipitated genomic DNA was amplified. Input, sheared DNA prior to immunoprecipitation. A control immunoprecipitation using an irrelevant antibody (anti-15-LOX antibody) did not show any PCR signal (see Supplementary Material).
DISCUSSION
Regulation of expression of the ph/snGPx gene is a complex process, which involves transcriptional and post-transcriptional events (4,14). Unfortunately, to date their mechanisms remain largely elusive. In mature rat testis the enzyme is expressed at high levels (17,21) but in prepuberal testicular tissue only small amounts can be detected (19). These biological dynamics suggested a causal relation between phGPx expression and sperm development (25). In fact, there is a striking burst of phGPx expression during the transition of round to elongated spermatids (19), but it remains unclear which regulatory mechanisms might contribute. One reason for this lack of information may be the fact that the cis- and trans-regulatory elements involved in phGPx expression have not been investigated in detail. The only information currently available was the description of a CCAAT box in the 5′-flanking region of the human ph/snGPx gene, which binds NF-Y in vitro (23). Here we characterized several cis-regulatory elements in the putative promoter of the murine ph/snGPx gene and characterized the corresponding trans-regulatory proteins. Interestingly, we found that the SP1/SP3 binding site detected in FP-1 appears to be of higher regulatory importance than the above-mentioned CCAAT box, which is also conserved in mice.
According to the conventional view of the structure of mammalian genes cis-regulatory sequences are localized in the non-transcribed 5′-flanking region. However, there is an increasing number of reports describing identification of functional transcription factor binding sites in intronic (26,27) or even exonic (28,29) sequences and recently, the existence of regulatory sequences has been reported in intron 1 of the ph/snGPx gene (30). The transcription factor binding sites identified here for the murine phGPx are localized in both, the 5′-UTR of the mRNA (exonic sequences) and/or in the non-transcribed 5′-flanking region of the gene. Their exact location relative to the transcriptional initiation site (Cap) is rather variable because several Cap sites have been described. As indicated above, there are two major phGPx mRNA populations (a mitochondrial and a cytosolic mRNA family) and both of them are heterogeneous with respect to their 5′ termini (11). The cytosolic transcript family lacks the mitochondrial insertion sequence and utilizes the 3′-ATG for translational initiation. In contrast, mitochondrial mRNA population contains both, the 5′- and the 3′-ATG as well as the complete mitochondrial targeting sequence. Translation of the latter mRNA species is initiated at the 5′-ATG. Our reporter gene assays (Fig. 1) revealed significant promoter activity in the DNA region encoding for the mitochondrial insertion peptide. However, in this study we did not address the question of whether or not functional transcription factor binding sites can be found. This might be the subject of a follow-up study.
Transcriptional regulation of the ph/snGPx gene is highly complex since at least three different proteins can be synthesized. Apart for the cytosolic and mitochondrial phGPx isoforms the gene also encodes for the snGPx (31) and there appear to be tissue-specific regulatory mechanisms of gene expression. The 34 kDa snGPx is predominantly expressed in the nuclei of late spermatids of various mammals. Protein chemical characterization indicated that the cytosolic phGPx is identical with the C-terminal two thirds of the snGPx. However, the N-terminal peptide of the snGPx carries an arginine-rich nuclear targeting sequence, which is responsible for nuclear import of the mature protein (31). Interestingly, this N-terminal peptide is encoded for by an alternative exon located in intron 1 of the joint ph/snGPx gene (31). Recently, it was reported that the 5′-flanking region of alternative exon 1, which constitutes the potential snGPx promoter, lacks major promoter activity but strongly suppresses the activity of the ph/snGPx promoter suggesting the existence of negative cis-regulatory elements in the first intron of the gene (30). EMSAs and chromatin immunoprecipitation revealed the in vitro (e.g. CRE-binding proteins) and in vivo (e.g. Egr1 and SREBP1) binding of several transcription factors suggesting their involvement in transcriptional silencing (30). The question as to whether the transcripts encoding for the snGPx are generated by transcriptional initiation at an alternative promoter or by alternative splicing of a joint precursor is currently discussed in the literature (30,32). However, the experimental data available at the moment are not sufficient to answer this mechanistic question and more work is needed to finally resolve it.
The relative lack of information on the structural basis of transcriptional regulation of the joint ph/snGPx gene on the one hand and the functional importance (14) of the enzyme in anti-oxidative defense and spermatogenesis on the other, prompted us to study the regulatory mechanisms in more detail. Our attempts at designing an experimental strategy for this purpose were aggravated by the complex expression pattern (at least three different proteins originate from the ph/snGPx gene), which is due to the existence of multiple transcription initiation sites and/or to alternative splicing mechanisms. Since for each transcriptional initiation site regulatory sequences were supposed to exist in the 5′-flanking region, a complex pattern of cis-regulatory elements was expected in the putative promoter. When we investigated the 5′-flanking region of the sn/phGPx gene starting at the 3′-ATG (translational initiation of the cytosolic enzyme) we observed five functional protein-binding sites (FP-1 to FP-4B) and site-directed mutagenesis (Fig. 7) suggested a clustering of these cis-regulatory elements. Separate mutation of FP-1 reduced the promoter activity of mitochondrial and cytosolic constructs. Thus, this SP1/SP3 binding site appears to be essential for expression regulation of both isoforms. Simultaneous inactivation of the transcription factor binding sites localized in FP-1 and FP-2 induced a complete loss in promoter activity of the B1 construct (block of transcription of the mitochondrial isoform). In contrast, the C1 construct that contains the regulatory elements for transcription of the cytosolic messenger exhibited a residual promoter activity of ∼80%. These data suggest that simultaneous functional silencing of the distal protein-binding sites (5′-cluster containing FP-1 and FP-2) abolished the promoter activity for the mitochondrial isoform but hardly impacts that for the cytosolic enzyme. Inversely, simultaneous mutation of the proximal protein-binding sites (3′-cluster containing FP-4A and FP-4B) strongly reduces the promoter activity of construct C1 (cytosolic isoform) but had only minor effects on construct B1 (mitochondrial isoform).
It should be stressed at this point that this scenario might be an oversimplification of the in vivo situation since it does not consider the interactions of the identified trans-regulatory elements with other regulatory proteins. Moreover, nothing is known about spatial arrangements of the initiation complexes and the existence of far distal enhancer and silencer sequences cannot be excluded. Finally, covalent modification (phosphorylation, acetylation, methylation, etc.) of cis- and trans-regulatory elements may impact their regulatory importance and this aspect has not been addressed in our study.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Scheme 2. Functional cis-acting elements involved in transcriptional regulation of mitochondrial and cytosolic phGPx isoforms. The 5′-ATG represents the translational initiation site for the mitochondrial isoform whereas translation of the cytosolic enzyme starts at the 3′-ATG. The transcriptional initiation sites for the long (mitochondrial isoform) and the short messenger (cytosolic messenger) are indicated by the 5′- and the 3′-Cap, respectively.
REFERENCES
- 1.Flohé L. (1989) The selenoprotein glutathione peroxidase. In Dolphin,D., Poulson,R. and Avramovic,O. (eds), Glutathione: Chemical, Biochemical and Medical Aspects—Part A. John Wiley and Sons, New York, pp. 643–731. [Google Scholar]
- 2.Arthur J.R. (2000) The glutathione peroxidases. Cell. Mol. Life Sci., 57, 1825–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Behne D. and KyriakopoulosA. (2001) Mammalian selenium-containing proteins. Annu. Rev. Nutr., 21, 453–473. [DOI] [PubMed] [Google Scholar]
- 4.Kuhn H. and Borchert,A. (2002) Regulation of enzymatic lipid peroxidation. The interplay of peroxidizing and peroxide reducing enzymes. Free Radic. Biol. Med., 33, 154–172. [DOI] [PubMed] [Google Scholar]
- 5.Ursini F., Maiorino,M., Valente,M., Ferri,L. and Gregolin,C. (1982) Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim. Biophys. Acta, 710, 197–211. [DOI] [PubMed] [Google Scholar]
- 6.Roveri A., Maiorino,M., Nisii,C. and Ursini,F. (1994) Purification and characterization of phospholipid hydroperoxide glutathione peroxidase from rat testis mitochondrial membranes. Biochim. Biophys. Acta, 1208, 211–221. [DOI] [PubMed] [Google Scholar]
- 7.Maiorino M., Thomas,J.P., Girotti,A.W. and Ursini,F. (1991) Reactivity of phospholipid hydroperoxide glutathione peroxidase with membrane and lipoprotein lipid hydroperoxides. Free Radic. Res. Commun., 12–13 (Pt 1), 131–135. [DOI] [PubMed] [Google Scholar]
- 8.Schnurr K., Belkner,J., Ursini,F., Schewe,T. and Kühn,H. (1996) The selenoenzyme phospholipid hydroperoxide glutathione peroxidase controls the activity of the 15-lipoxygenase with complex substrates and preserves the specificity of the oxygenation products. J. Biol. Chem., 271, 4653–4658. [DOI] [PubMed] [Google Scholar]
- 9.Sattler W., Maiorino,M. and Stocker,R. (1994) Reduction of HDL- and LDL-associated cholesterylester and phospholipid hydroperoxides by phospholipid hydroperoxide glutathione peroxidase and Ebselen (PZ 51). Arch. Biochem. Biophys., 309, 214–221. [DOI] [PubMed] [Google Scholar]
- 10.Thomas J.P., Maiorino,M., Ursini,F. and Girotti,A.W. (1990) Protective action of phospholipid hydroperoxide glutathione peroxidase against membrane-damaging lipid peroxidation. In situ reduction of phospholipid and cholesterol hydroperoxides. J. Biol. Chem., 265, 454–461. [PubMed] [Google Scholar]
- 11.Pushpa-Rekha T.R., Burdsall,A.L., Oleksa,L.M., Chisolm,G.M. and Driscoll,D.M. (1995) Rat phospholipid-hydroperoxide glutathione peroxidase. cDNA cloning and identification of multiple transcription and translation start sites. J. Biol. Chem., 270, 26993–26999. [DOI] [PubMed] [Google Scholar]
- 12.Arai M., Imai H., Koumura T., Yoshida M., Emoto K., Umeda M., Chiba N. and Nakagawa Y. (1999) Mitochondrial phospholipid hydroperoxide glutathione peroxidase plays a major role in preventing oxidative injury to cells. J. Biol. Chem., 274, 4924–4933. [DOI] [PubMed] [Google Scholar]
- 13.Arai M., Imai,H., Sumi,D., Imanaka,T., Takano,T., Chiba,N. and Nakagawa,Y. (1996) Import into mitochondira of phospholipid hydroperoxide glutathione peroxidase requires a leader sequence. Biochem. Biophys. Res. Commun., 227, 433–439. [DOI] [PubMed] [Google Scholar]
- 14.Brigelius-Flohé R. (1999) Tissue specific function of individual glutathione peroxidases. Free Radic. Biol. Med., 27, 951–965. [DOI] [PubMed] [Google Scholar]
- 15.Knopp E.A., Arndt,T.L., Eng,K.L., Caldwell,M., LeBoeuf,R.C., Deeb,S.S. and O’Brien,K.D. (1999) Murine phospholipid hydroperoxide glutathione peroxidase: cDNA sequence, tissue expression and mapping. Mamm. Genome, 10, 601–605. [DOI] [PubMed] [Google Scholar]
- 16.Dreher I., Schmutzler,C., Jakob,F. and Kohrle,J. (1997) Expression of selenoproteins in various rat and human tissues and cell lines. J. Trace Elem. Med. Biol., 11, 83–91. [DOI] [PubMed] [Google Scholar]
- 17.Ursini F., Heim,S., Kiess,M., Maiorino,M., Roveri,A., Wissing,J. and Flohe,L. (1999) Dual function of the selenoprotein PHGPx during sperm maturation. Science, 285, 1393–1396. [DOI] [PubMed] [Google Scholar]
- 18.Roveri A., Casasco,A., Maiorino,M., Dalan,P., Calligaro,A. and Ursini,F. (1992) Phospholipid hydroperoxide glutathione peroxidase of rat testis. Gonadotropin dependence and immunocytochemical identification. J. Biol. Chem., 267, 6142–6146. [PubMed] [Google Scholar]
- 19.Maiorino M., Wissing,J.B., Brigelius-Flohé,R, Calabrese,F., Roveri,A., Steinert,P., Ursini,F. and Flohé,L. (1998) Testosterone mediates expression of the selenoprotein PHGPx by induction of spermatogenesis and not by direct transcriptional gene activation. FASEB J., 12, 1359–1370. [DOI] [PubMed] [Google Scholar]
- 20.Brigelius-Flohe R., Aumann,K.D., Blocker,H., Gross,G., Kiess,M., Kloppel,K.D., Maiorino,M., Roveri,A., Schuckelt,R., Ursini,F., Wingender,E. and Flohé,L. (1994) Phospholipid-hydroperoxide glutathione peroxidase. Genomic DNA, cDNA and deduced amino acid sequence. J. Biol. Chem., 269, 7342–7348. [PubMed] [Google Scholar]
- 21.Kelner M.J. and Montoya,M.A. (1998) Structural organization of the human selenium-dependent phospholipid hydroperoxide glutathione peroxidase gene (GPX4): chromosomal localization to 19p13.3. Biochem. Biophys. Res. Commun., 249, 53–55. [DOI] [PubMed] [Google Scholar]
- 22.Borchert A., Schnurr,K., Thiele,B. and Kühn,H. (1999) Cloning of the mouse phospholipid glutathione peroxidase gene. FEBS Lett., 446, 223–227. [DOI] [PubMed] [Google Scholar]
- 23.Huang H.S., Jiunn,C. and Chang,W.C. (1999) The CCAAT-box binding factor NF-Y is required for the expression of the phospholipid hydroperoxide glutathione peroxidase in human epidermoid carcinoma A431 cells. FEBS Lett., 455, 111–116. [DOI] [PubMed] [Google Scholar]
- 24.Quandt K., Frech,K., Karas,H., Wingender,E. and Werner,T. (1995) MatInd and MatInspector—new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res., 23, 4878–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strauss E. (1999) Selenium’s role in infertility explained. Science, 285, 1339. [DOI] [PubMed] [Google Scholar]
- 26.Frederickson R.M., Micheau,M.R., Iwamoto,A. and Miyamoto,N.G. (1989) 5′ Flanking and first intron sequences of the human beta-actin gene required for efficient promotor activity. Nucleic Acids Res., 17, 253–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wandersee N.J., Ferris,R.C. and Ginder,G.D. (1996) Intronic and flanking sequences are required to silence enhancement of an embryonic β-type globin gene. Mol. Cell. Biol., 16, 236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao J., Bilsland,A., Hoare,S.F. and Keith,N. (2003) Involvement of NF-Y and Sp1 binding sequences in basal transcription of the human telomerase RNA gene. FEBS Lett., 536, 111–119. [DOI] [PubMed] [Google Scholar]
- 29.Farina A., Manni,I., Fontemaggi,G., Tiainen,M., Cenciarelli,C., Bellorini,M., Mantovani,R., Sacchi,A. and Piaggio,G. (1999) Down-regulation of cyclin B1 gene transcription in terminally differentiated skeletal muscle cells is associated with loss of functional CCAAT-binding NF-Y complex. Oncogene, 18, 2818–2827. [DOI] [PubMed] [Google Scholar]
- 30.Borchert A., Savaskan,N. and Kuhn,H. (2003) Regulation of expression of the phospholipid hydroperoxide/sperm nucleus glutathione peroxidase gene. J. Biol. Chem., 278, 2671–2580. [DOI] [PubMed] [Google Scholar]
- 31.Pfeifer H., Conrad,M., Roethlein,D., Kyriakopouluos,A., Brielmeier,M., Bornkamm,G.W. and Behne,D. (2001) Identification of a specific sperm nuclei selenoenzyme necessary for protamine thiol cross-linking during sperm maturation. FASEB J., 15, 1236–1238. [PubMed] [Google Scholar]
- 32.Moreno S.-G., Laux,G., Brielmeier,M., Bornkamm,G.W. and Conrad,M. (2003) Testis-specific expression of the nuclear form of phospholipids hydroperoxide glutathione peroxidase (PHGPx). Biol. Chem., 384, 635–643. [DOI] [PubMed] [Google Scholar]
- 33.Nam S., Nakamuta,N., Kurohmaru,M. and Hayashi,Y. (1997) Cloning and sequencing of the mouse cDNA encoding a phospholipid hydroperoxide glutathione peroxidase. Gene, 198, 245–249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.