

Abstract
The translation of reverse transcriptase and other essential viral proteins from the HIV-1 Pol mRNA requires a programmed –1 ribosomal frameshift. This frameshift is induced by two highly conserved elements within the HIV-1 mRNA: a slippery sequence comprised of a UUUUUUA heptamer, and a downstream stem–loop structure. We have determined the structure of the HIV-1 frameshift inducing RNA stem–loop, using multidimensional heteronuclear nuclear magnetic resonance (NMR) methods. The 22 nucleotide RNA solution structure [root mean squared deviation (r.m.s.d.) = 1.2 Å] was determined from 475 nuclear Overhauser effect (NOE)-derived distance restrains, 20 residual dipolar couplings and direct detection of hydrogen bonds via scalar couplings. We find that the frameshift inducing stem–loop is an A-form helix capped by a structured ACAA tetraloop. The ACAA tetraloop is stabilized by an equilateral 5′ and 3′ stacking pattern, a sheared A–A pair and a cross-strand hydrogen bond. Unexpectedly, the ACAA tetraloop structure is nearly identical to a known tetraloop fold, previously identified in the RNase III recognition site from Saccharomyces cerevisiae.
INTRODUCTION
HIV-1 replication requires the expression of viral reverse transcriptase, protease and integrase which are produced from a Gag-Pol polyprotein that is the result of a –1 frameshift event (1). The frameshift event is essential for HIV-1 replication (2). Two RNA elements within the mRNA are required for frameshifting: (i) a heptanucleotide slippery sequence (UUUUUUA) within which the frameshift occurs, and (ii) a highly conserved downstream sequence that is predicted to form a stem–loop structure (3–5).
The frameshift inducing stem–loop element is predicted to be highly stable based on secondary structure prediction algorithms, and is strongly conserved among all known HIV-1 isolates (6). The stem–loop is predicted to contain at least 11 Watson–Crick base pairs, capped by an ACAA tetraloop sequence (1,4,5,7). The ACAA tetraloop is highly conserved, and found in 86.3% of plasma isolates (4). Thermodynamic stability appears to be important for the function of the frameshift inducing stem–loop, as the sequences found with highest frequency in plasma isolates are also predicted to be the most thermodynamically stable (4).
The frameshift inducing stem–loop is functionally critical for HIV-1 replication (5). The programmed –1 frameshift occurs at 5–10% efficiency (1), resulting in an approximately 10:1 or 20:1 ratio in the expression of Gag to Gag-Pol, depending on the assay used to measure the ratio (5,8,9). This stoichiometry of Gag/Gag-Pol proteins is conserved among retroviruses and is important for viral RNA dimerization and infectivity (10). Increasing or decreasing the frameshift efficiency significantly reduces the amount of infective virus particles produced (2). The murine leukemia virus frameshift inducing pseudoknot can be functionally replaced by the HIV-1 stem–loop, indicating that the HIV-1 frameshift inducing stem–loop can stimulate –1 frameshifting outside of its native context (11).
Structural information of the HIV-1 virus is necessary to fully understand its replication mechanism and potential weaknesses. The HIV-1 structural genome is incomplete (12), and no structural information for the HIV-1 frameshift inducing element exists. In order to understand the mechanism of stem–loop induced frameshifting in HIV-1 and other systems, it is necessary to gain an atomic-level understanding of the structures involved.
MATERIALS AND METHODS
RNA synthesis and purification
RNA was transcribed in vitro using purified His6-tagged T7 RNA polymerase and synthetic DNA oligonucleotides (Integrated DNA Technologies), as previously described (13–15). RNA was purified by denaturing 20% polyacrylamide gel electrophoresis, identified by UV absorbance and excised from the gel. RNA was recovered by diffusion into 0.3 M sodium acetate, ethanol precipitated, purified on a 6 ml DEAE anion exchange column, again ethanol precipitated and desalted on a 15 ml G-25 gel filtration column. The purified RNA was lyophilized, resuspended in water and brought to pH 6.8 by the addition of 1 M NaOH. 13C/15N-labeled RNA was prepared using 13C/15N-labeled rNTPs (Silantes GmbH, München, Germany). All nuclear magnetic resonance (NMR) samples were 1 mM RNA and 50 mM NaCl.
NMR spectroscopy
All NMR spectra were collected on Bruker DMX spectrometers at the National Magnetic Resonance Facility at Madison (NMRFAM). Spectrometers used for data collection were equipped with either a conventional proton, carbon, nitrogen (HCN) triple resonance, triple-axis pulsed field gradient probe, or cold (cryoprobe, Bruker) single Z-axis gradient HCN probes.
Exchangeable resonances were assigned by reference to 2D nuclear Overhauser effect spectroscopy (NOESY) (150 ms mixing time) in 90% H2O/10% D2O at 283 K. Non-exchangeable resonances were assigned by reference to 2D NOESY spectra (50, 75, 100, 150, 200, 300, 400 ms mixing times) and 2D total correlation spectroscopy (TOCSY), 2D 1H-13C heteronuclear single quantum correlation spectroscopy (HSQC), 3D 1H-13C-1H proton, carbon, carbon, proton (HCCH) TOCSY, 3D 1H-13C-1H HCCH correlation spectroscopy (COSY) and 3D 1H-13C-1H NOESY–heteronuclear multiple quantum correlation spectroscopy (HMQC) spectra of the RNA in 99.99% D2O at 303 K, as previously described (13,14). Water suppression for samples in 90% H2O/10% D2O was achieved with a 1–1 spin-echo or watergate pulse sequence. For experiments in 99.99% D2O, residual HDO resonance was suppressed with a low power presaturation pulse. Hydrogen bonds were detected for adenosine/uridine and guanosine/cytidine 13C,15N-labeled samples, respectively, in 90% H2O/10% D2O at 298 K, as described previously (16).
Partial alignment of RNA for residual dipolar coupling measurements was achieved by adding 17 mg/ml Pf1 filamentous bacteriophage (ASLA Ltd, Riga, Latvia) to the 13C/15N-labeled samples (17). 1H-13C residual dipolar couplings (RDCs) were measured in the carbon and proton dimensions of 2D 1H-13C constant time (CT)-HSQC spectra for adenosine/uridine and guanosine/cytidine 13C,15N-labeled samples in 90% H2O/10% D2O at 303 K, for both isotropic and partially oriented samples.
All data were processed using XWINNMR software from Bruker. Resonance assignments were completed using Sparky (http://www.cgl.ucsf.edu/home/sparky/).
Interproton distances
NOE distances were estimated from the integrated peak volumes obtained from 2D NOESY spectra. Intense NOEs observable at short mixing times (50 and 75 ms) were assigned as strong (1.8–3.6 Å), medium intensity NOEs observed at 75–200 ms mixing times were grouped as medium (1.8–5 Å), and low intensity NOEs observable only at long mixing times (200–400 ms) were grouped as weak (3–7 Å). The NOE peak volumes were also integrated and calibrated relative to the average pyrimidine H5-H6 NOE volume, corresponding to the fixed distance 2.4 Å. The integration and peak calibration supported the NOE assignments of strong, medium and weak distances.
Torsion angle constraints
The backbone torsion angles (α, β, γ, δ, ε and ζ) of the nucleotides in the stem (G1–C9, G14–C22) were constrained to A-form values (18), which were consistent with NOESY, proton, nitrogen, nitrogen (HNN) COSY and RDC data. The backbone torsion angles of the tetraloop were left unrestrained. 2D TOCSY with a 40 ms mixing time was used to analyze sugar pucker conformations. Nucleotides with strong H1′-H2′ and H1′-H3′ crosspeaks (C11, A12, A13) were restrained to the C2′-endo range; nucleotides within the stem did not have H1′-H2′ crosspeaks and were thus restrained to the C3′-endo range; and nucleotide A10, which displayed an intermediate H1′-H2′ coupling indicative of an averaging of sugar puckers, was left unrestrained. Analysis of the peak volumes of intranucleotide H1′-aromatic NOEs from 50 and 75 ms mixing time 2D NOESYs revealed that all nucleotides were in the anti range, and thus the torsion angle χ was restrained to 160 ± 15° for all nucleotides.
Residual dipolar coupling analysis
Residual Dipolar Couplings were measured using XWINNMR (Bruker) software by calculating the difference between 1H and 13C coupling for isotropic and partially aligned samples. PALES software (19) (http://spin.niddk.nih.gov/bax/software/PALES) was used to estimate the values for the axial (Da) and rhombic (R) components of the alignment tensor from converged, low energy structures calculated in the absence of RDCs. The PALES predicted values were found to be Da = –18 Hz and R = 0.28. A grid search (20) method was then employed to obtain the optimal Da and R values, using CNS 1.1(21) (http://cns.csb.yale.edu/v1.1). In the grid search, lowest overall energies were used as a target function and Da was varied from –14 to –23 Hz and R was varied from 0.20 to 0.40. The grid search identified optimal values of Da = –16 Hz and R = 0.28, very close to the original values predicted by the PALES analysis.
Structure calculations
CNS 1.1 (21) was used to calculate structures using NOE distance and dihedral restraints, and residual dipolar couplings. The structure calculations closely followed the default values for NMR structure determination of nucleic acids with CNS 1.1. First, an extended (completely unfolded) structure was generated, from which 100 structures were calculated from random initial velocities. The structures were subjected to 60 ps (15 fs timesteps) of restrained molecular dynamics in torsion angle space, followed by 90 ps of slow cooling. Finally, 30 ps (5 fs timesteps) of restrained molecular dynamics in Cartesian coordinate space were performed. For Watson–Crick base pairs that were detected by trans-hydrogen bond experiments, only very weak planarity restraints (10 kcal mol–1 Å–2) were enforced during calculations and hydrogen bonds were maintained by distance restraints. Structures were also calculated without RDCs for comparison. Acceptance criteria of converged structures were low overall energies and no significant NOE (>0.2 Å) or dihedral (>5 Å) violations. Structures were viewed and analyzed using MOLMOL (22). The final structures were used to back-calculate RDC values. The calculated RDCs were plotted against experimentally measured values and were found to be in excellent agreement.
RESULTS
The sequence of the HIV-1 frameshift inducing element is shown (Fig. 1), consisting of the slippery site heptamer and a downstream stem–loop of at least 11 predicted Watson–Crick base pairs and an ACAA tetraloop (6). The sequence investigated in this study corresponds to the most frequently occurring isolate among natural variants circulating in plasma (4). The construct used for NMR studies begins at the naturally occurring GG dinucleotide within the stem–loop, which facilitates large scale in vitro RNA preparation with T7 RNA polymerase. Thus, the NMR RNA construct is 22 nucleotides in length and contains the potential for nine Watson–Crick base pairs and an ACAA tetraloop (Fig. 1).
Figure 1.

Sequence and predicted secondary structure of the HIV-1 frameshift inducing site. The frameshift inducing site is made up of a slippery site heptamer (underlined), and a stem–loop structure. The NMR construct of the downstream stem–loop is boxed. For simplicity, the NMR construct is numbered 1–22 beginning with the 5′ guanine.
NMR experiments, including 2D 1H-1H NOESY, 2D 1H-15N HSQC and 2D HNN COSY were performed to analyze base pairing within the HIV-1 frameshift inducing stem–loop. The presence of all nine Watson–Crick base pairs within the stem were confirmed by the detection of trans-hydrogen bond scalar couplings for each of the base pairs in the stem (Fig. 2), indicating that the secondary structure proposed for the HIV-1 frameshift inducing stem–loop (Fig. 1) is forming in the NMR construct. The spectra exhibit dispersion of 1H, 13C, and 15N resonances throughout the RNA suggesting that the molecule is well-structured (Figures 2 and 3 and data not shown). Sequential 1H-1H NOEs, indicative of A-form helical geometry, are observed throughout the stem, with the exception of a break between nucleotides C11 and A12 within the tetraloop (Fig. 3). Additionally, NOEs indicative of base stacking are observed between the first and second bases, as well as the third and fourth bases of the tetraloop (data not shown). These data suggest that the backbone turns at the mid-point of the tetraloop between C11 and A12, and that an even base-stacking pattern occurs on either side of the turn. Additionally, an unusual NOE is observed between the H2 proton of the first A (A10) and the H8 proton of the last A (A13), indicating that the two closing nucleotides of the tetraloop are in close proximity and adopt a non-canonical geometry.
Figure 2.
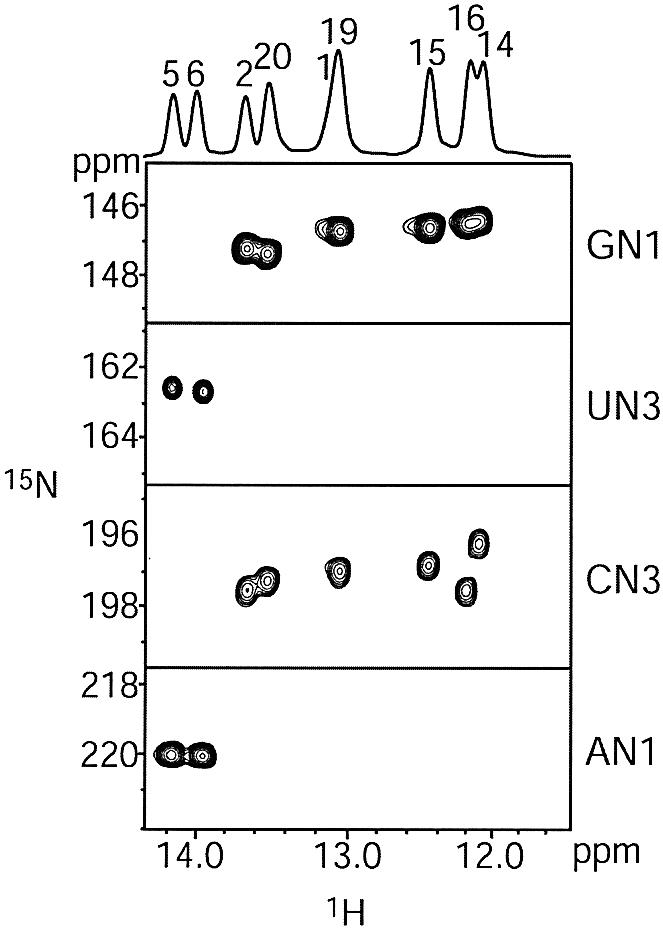
750 MHz 2D HNN COSY experiment to detect trans-hydrogen bond scalar couplings. The spectra were acquired at pH 6.8, 25°C, 1 mM RNA and 50 mM NaCl. Correlations are observed between the H1 of guanosine and the N3 of cytidine, and between the H3 of uridine and the N1 of adenosine for all Watson–Crick base pairs.
Figure 3.

600 MHz 1H-1H NOESY of the HIV-1 frameshift inducing stem–loop RNA. The spectrum was acquired at pH 6.8, 30°C, 1 mM RNA and 50 mM NaCl. The NOESY mixing time was 400 ms. The sequential H1′–H6/H8 assignments are traced with lines. A break in sequential NOEs is observed between C11 and A12 (highlighted in red).
The solution structure of the HIV-1 frameshift inducing stem–loop was calculated using 475 NOE restraints (an average of 21.6 per nucleotide), in addition to backbone torsion angle and hydrogen bond restraints for the A-form helical regions (Table 1). Twenty RDCs were used to improve the long range order of the structure. Back-calculated RDCs from the final structures agree well with the measured values (Fig. 4). The inclusion of RDCs in the structure calculation improved the long range order and A-form geometry of the structures, while reducing the root mean squared deviation (r.m.s.d.) to 1.24 Å (Table 1 and Fig. 5A), versus an r.m.s.d. of 1.93 Å for structures calculated without RDCs (Table 1).
Table 1. Structural statistics for the HIV-1 frameshift inducing stem–loop structurea.
| With dipolar coupling data | Without dipolar coupling data | |
|---|---|---|
| NOE-derived distance restraints | 475 | 475 |
| Intranucleotide | 221 | 221 |
| Internucleotide (|i – j| = 1) | 222 | 222 |
| Long-range (|i – n| ≥ 2) | 32 | 32 |
| Dihedral restraints | 181 | 181 |
| Hydrogen bond restraints | 26 | 26 |
| Dipolar coupling restraints | 20 | 0 |
| r.m.s.d. (for all heavy atoms to mean structure, residues 62–71,73,75–85) (Å) | 1.24 | 1.93 |
| NOE violations > 0.2 Å | 0 | 0 |
| Dihedral violations > 5° | 0 | 0 |
| Average NOE r.m.s.d. (Å) | 0.010 | 0.010 |
| Average dihedral r.m.s.d. (°) | 0.610 | 0.632 |
| Average RDC r.m.s.d. (Hz) | 2.79 | – |
aStructural statistics are given for the 20 lowest energy structures out of 100 calculated structures.
Figure 4.

Comparison of back-calculated (vertical) and experimental (horizontal) RDC values for the HIV-1 frameshift inducing stem–loop for the lowest energy structure. The straight line indicates experimental = calculated.
Figure 5.

NMR solution structure of the HIV-1 frameshift inducing stem–loop. (A) Superimposition over all heavy atoms of the 20 conformers with the lowest energies calculated with the incorporation of RDCs. The r.m.s.d. over all heavy atoms for these 20 structures is 1.24 Å. (B) Stereo view of the lowest energy structure of the HIV-1 frameshift inducing stem–loop. View is into the minor groove of the stem and the major groove of the tetraloop. (C) Stereo view of the tetraloop and adjacent Watson–Crick pair from the 20 lowest energy conformers superimposed over nucleotides 9–14 (r.m.s.d. 0.47 Å). View is into the minor groove of the tetraloop and the molecule is rotated 180° relative to (B).
The stem adopts an A-form helical geometry (Fig. 5B), as would be expected for an RNA helix of nine Watson–Crick pairs, and is consistent with the NOEs observed in the H1′-H6/H8 region of the 2D NOESY (Fig. 3). The ACAA tetraloop is highly structured (Fig. 5C). The ACAA tetraloop maintains an equilateral 5′ and 3′ stacking pattern, in which C11 is stacked upon A10, and A12 is stacked upon A13 (Fig. 6). This geometry is consistent with the break in sequential NOEs between C11 and A12 (Fig. 3) as well as the presence of base stacking NOEs between A10 and C11 and between A12 and A13. A10 and A13 are positioned in close proximity to each other and form a sheared A·A base pair, by hydrogen bonding between the N3 of A10 and the amino proton of A13 (Fig. 6). A hydrogen bond is also likely to form between the amino proton of A12 and the 2′ oxygen of A10, as these atoms are between 2 and 3 Å apart in the 10 lowest energy structures (Fig. 6). The sheared A·A base pair and the hydrogen bond between A10 and A12, along with the extensive base stacking likely act to stabilize the formation of this tetraloop structure.
Figure 6.

Adenine stacking and hydrogen bonding in the ACAA tetraloop. Structure of the ACAA tetraloop showing A10, A12 and A13 as viewed from the top of the stem–loop. For clarity, C11 is not shown. Hydrogen bonds between N3 of A10 and the amino proton (H61) of A13, and between O2′ of A10 and the amino proton (H61) of A12 are indicated as dashed lines.
DISCUSSION
We have solved the structure of the frameshift inducing stem–loop RNA of HIV-1, which forms a stable A-form helix capped by an ACAA tetraloop. The question remains, how does this structure function to induce frameshifting at the slippery site heptamer? One possibility is that the frameshift inducing stem–loop interacts with the ribosome during translation to facilitate the –1 frameshift. If this is the case, the structure of the ACAA tetraloop may provide a binding surface for this event; such an interaction may also explain the high degree of sequence conservation observed for this structure.
Interestingly, we find that the ACAA tetraloop assumes a conformation that is similar to a known RNA structure that serves as a recognition site for the RNase III protein Rnt1p in yeast (23,24). The RNA binding domains of RNase III interact with stem–loop structures possessing tetraloops with the consensus AGNN, and the AGNN fold is superimposable with the HIV-1 ACAA tetraloop structure (Fig. 7), with a r.m.s.d. of 3 Å. The major difference between the ACAA and AGAA tetraloop structures is that the cytosine base in the HIV-1 tetraloop has an anti glycosidic torsion angle, whereas the guanosine adopts a syn conformation (23,24). Nevertheless, the two structures are closed by the same sheared A–A pair and adopt the same overall equilateral stacking pattern. This tetraloop fold exposes the Watson–Crick and Hoogsteen faces of the bases, with the first two directed toward the major groove and the last two oriented in the minor groove. Therefore, this motif may provide a potential contact surface from either or both sides of the stem–loop. Previously, this family of tetraloops was thought to share the consensus (U/A)(G)NN (23–26); now, the consensus for this fold should be broadened to include cytosine at the second position (U/A)(G/C)NN.
Figure 7.

ACAA tetraloop of the lowest energy HIV-1 frameshift inducing stem–loop (colored as in Fig. 5) superimposed over the AGAA tetraloop from the RNase III recognition site (grey).
The ACAA tetraloop also occurs within a conserved stem–loop that is part of the rev responsive element (RRE) region of HIV-1, a RNA structure that plays an integral role in regulating the expression of viral structural proteins (6). ACAA is not a common tetraloop sequence (27), so it is curious that this tetraloop should be highly conserved at two functionally important regions of the HIV-1 RNA. The ACAA tetraloop may therefore be an attractive target for the development of antiviral agents specific for HIV-1, owing to its relative rarity and high level of conservation at two critical sites.
Saccharomyces cerevisiae RNase III catalyzes a double-stranded endonucleolytic break 13–16 base pairs away from AGNN tetraloops (28). The ACAA tetraloop is a poor substrate for native S.cerevisiae RNase III, presumably due to a lack of the G determinant at the second position (24). However, due to the structural similarities between the HIV-1 ACAA and AGNN tetraloops, it may be possible to engineer S.cerevisiae RNase III to selectively target HIV-1 mRNA, if it were possible to express or deliver this protein into HIV-1 infected cells.
Small molecules that alter HIV-1 frameshift inducing efficiency, presumably by interacting with the stem–loop, can significantly reduce viral replication (2). Therefore, computational searches for small molecules that may interact with the frameshift inducing stem–loop may have utility, as have been successfully applied with the HIV-1 TAR element (29,30).
Additional structure surrounding the HIV-1 frameshift inducing element may exist and contribute to the frameshift mechanism. Two alternative models have been proposed, which include additional elements of structure such as a triple helical structure (31) and an extended stem–loop structure (7). Both of these secondary structure models incorporate the highly conserved and stable stem–loop structure presented here. To test these models directly, we are currently in the process of determining the structures of larger HIV-1 RNA constructs containing the conserved frameshift inducing stem–loop element and surrounding sequences. The solution structure presented here marks a significant step in determining these larger structures and understanding how stem–loops induce frameshifting in HIV-1 and other systems.
Structure coordinates
Structure coordinates have been deposited to the Protein Data Bank (PDB) (http://www.rcsb.org), accession code 1PJY. Chemical shift data and NMR restraint files have been deposited to BioMagResBank (BMRB) (http://www.bmrb.wisc.edu), accession code 5834.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Anne Allmann, Nicole Aulik and Anne Riebau for technical assistance. NMR studies were carried out at the National Magnetic Resonance Facility at Madison with support from the NIH Biomedical Technology Program and additional equipment funding from the University of Wisconsin, NSF Academic Infrastructure Program, NIH Shared Instrumentation Program, NSF Biological Instrumentation Program, and the U.S. Department of Agriculture.
REFERENCES
- 1.Jacks T., Power,M.D., Masiarz,F.R., Luciw,P.A., Barr,P.J. and Varmus,H.E. (1988) Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature, 331, 280–283. [DOI] [PubMed] [Google Scholar]
- 2.Hung M., Patel,P., Davis,S. and Green,S.R. (1998) Importance of ribosomal frameshifting for human immunodeficiency virus type 1 particle assembly and replication. J. Virol., 72, 4819–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang S.Y., Sutthent,R., Auewarakul,P., Apichartpiyakul,C., Essex,M. and Lee,T.H. (1999) Differential stability of the mRNA secondary structures in the frameshift site of various HIV type 1 viruses. AIDS Res. Hum. Retroviruses, 15, 1591–1596. [DOI] [PubMed] [Google Scholar]
- 4.Telenti A., Martinez,R., Munoz,M., Bleiber,G., Greub,G., Sanglard,D. and Peters,S. (2002) Analysis of natural variants of the human immunodeficiency virus type 1 gag-pol frameshift stem-loop structure. J. Virol., 76, 7868–7873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parkin N.T., Chamorro,M. and Varmus,H.E. (1992) Human immunodeficiency virus type 1 gag-pol frameshifting is dependent on downstream mRNA secondary structure: demonstration by expression in vivo. J. Virol., 66, 5147–5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hofacker I.L., Fekete,M., Flamm,C., Huynen,M.A., Rauscher,S., Stolorz,P.E. and Stadler,P.F. (1998) Automatic detection of conserved RNA structure elements in complete RNA virus genomes. Nucleic Acids Res., 26, 3825–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dulude D., Baril,M. and Brakier-Gingras,L. (2002) Characterization of the frameshift stimulatory signal controlling a programmed –1 ribosomal frameshift in the human immunodeficiency virus type 1. Nucleic Acids Res., 30, 5094–5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park J. and Morrow,C.D. (1991) Overexpression of the gag-pol precursor from human immunodeficiency virus type 1 proviral genomes results in efficient proteolytic processing in the absence of virion production. J. Virol., 65, 5111–5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim Y.G., Maas,S. and Rich,A. (2001) Comparative mutational analysis of cis-acting RNA signals for translational frameshifting in HIV-1 and HTLV-2. Nucleic Acids Res., 29, 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shehu-Xhilaga M., Crowe,S.M. and Mak,J. (2001) Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J. Virol., 75, 1834–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunelle M.N., Brakier-Gingras,L. and Lemay,G. (2003) Replacement of murine leukemia virus readthrough mechanism by human immunodeficiency virus frameshift allows synthesis of viral proteins and virus replication. J. Virol., 77, 3345–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turner B.G. and Summers,M.F. (1999) Structural biology of HIV. J. Mol. Biol., 285, 1–32. [DOI] [PubMed] [Google Scholar]
- 13.Sashital D.G., Allmann,A.M., Van Doren,S.R. and Butcher,S.E. (2003) Structural basis for a lethal mutation in U6 RNA. Biochemistry, 42, 1470–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huppler A., Nikstad,L.J., Allmann,A.M., Brow,D.A. and Butcher,S.E. (2002) Metal binding and base ionization in the U6 RNA intramolecular stem-loop structure. Nat. Struct. Biol., 9, 431–435. [DOI] [PubMed] [Google Scholar]
- 15.Reiter N.J., Nikstad,L.J., Allmann,A.M., Johnson,R.J. and Butcher,S.E. (2003) Structure of the U6 RNA intramolecular stem-loop harboring an S(P)-phosphorothioate modification. RNA, 9, 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grzesiek S., Cordier,F. and Dingley,A.J. (2001) Scalar couplings across hydrogen bonds. Methods Enzymol., 338, 111–133. [DOI] [PubMed] [Google Scholar]
- 17.Hansen M.R., Hanson,P. and Pardi,A. (2000) Filamentous bacteriophage for aligning RNA, DNA and proteins for measurement of nuclear magnetic resonance dipolar coupling interactions. Methods Enzymol., 317, 220–240. [DOI] [PubMed] [Google Scholar]
- 18.Saenger W. (1984) Principles of Nucleic Acids. Springer-Verlag, New York, NY. [Google Scholar]
- 19.Zweckstetter M. and Bax,A. (2000) Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J. Am. Chem. Soc., 122, 3791–3792. [Google Scholar]
- 20.Clore G.M., Gronenborn,A.M. and Tjandra,N. (1998) Direct structure refinement against residual dipolar couplings in the presence of rhombicity of unknown magnitude. J. Magn. Reson., 131, 159–162. [DOI] [PubMed] [Google Scholar]
- 21.Brunger A.T., Adams,P.D., Clore,G.M., Delano,W.L., Gros,P., Grossekunstleve,R.W., Jiang,J.S., Kuszewski,J., Nilges,M., Pannu,N.S. et al. (1998) Crystallography and NMR system—a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr., 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 22.Koradi R., Billeter,M. and Wuthrich,K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph., 14, 51–55, 29–32. [DOI] [PubMed] [Google Scholar]
- 23.Lebars I., Lamontagne,B., Yoshizawa,S., Aboul-Elela,S. and Fourmy,D. (2001) Solution structure of conserved AGNN tetraloops: insights into Rnt1p RNA processing. EMBO J., 20, 7250–7258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu H., Yang,P.K., Butcher,S.E., Kang,S., Chanfreau,G. and Feigon,J. (2001) A novel family of RNA tetraloop structure forms the recognition site for Saccharomyces cerevisiae RNase III. EMBO J., 20, 7240–7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leeper T.C., Martin,M.B., Kim,H., Cox,S., Semenchenko,V., Schmidt,F.J. and Van Doren,S.R. (2002) Structure of the UGAGAU hexaloop that braces Bacillus RNase P for action. Nat. Struct. Biol., 9, 397–403. [DOI] [PubMed] [Google Scholar]
- 26.Butcher S.E., Dieckmann,T. and Feigon,J. (1997) Solution structure of the conserved 16 S-like ribosomal RNA UGAA tetraloop. J. Mol. Biol., 268, 348–358. [DOI] [PubMed] [Google Scholar]
- 27.Woese C.R., Winker,S. and Gutell,R.R. (1990) Architecture of ribosomal RNA: constraints on the sequence of ‘tetra-loops’. Proc. Natl Acad. Sci. USA, 87, 8467–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chanfreau G., Buckle,M. and Jacquier,A. (2000) Recognition of a conserved class of RNA tetraloops by Saccharomyces cerevisiae RNase III. Proc. Natl Acad. Sci. USA, 97, 3142–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lind K.E., Du,Z., Fujinaga,K., Peterlin,B.M. and James,T.L. (2002) Structure-based computational database screening, in vitro assay and NMR assessment of compounds that target TAR RNA. Chem. Biol., 9, 185–193. [DOI] [PubMed] [Google Scholar]
- 30.Du Z., Lind,K.E. and James,T.L. (2002) Structure of TAR RNA complexed with a Tat-TAR interaction nanomolar inhibitor that was identified by computational screening. Chem. Biol., 9, 707–712. [DOI] [PubMed] [Google Scholar]
- 31.Dinman J.D., Richter,S., Plant,E.P., Taylor,R.C., Hammell,A.B. and Rana,T.M. (2002) The frameshift signal of HIV-1 involves a potential intramolecular triplex RNA structure. Proc. Natl Acad. Sci. USA, 99, 5331–5336. [DOI] [PMC free article] [PubMed] [Google Scholar]