


Abstract
In mammalian cells, nucleotide excision repair (NER) is the major pathway for the removal of bulky DNA adducts. Many of the key NER proteins are members of the XP family (XPA, XPB, etc.), which was named on the basis of its association with the disorder xerodoma pigmentosum. Human replication protein A (RPA), the ubiquitous single-stranded DNA-binding protein, is another of the essential proteins for NER. RPA stimulates the interaction of XPA with damaged DNA by forming an RPA–XPA complex on damaged DNA sites. Binding of RPA to the undamaged DNA strand is most important during NER, because XPA, which directs the excision nucleases XPG and XPF, must bind to the damaged strand. In this study, nuclear magnetic resonance (NMR) spectroscopy was used to assess the binding of the tandem high affinity DNA-binding domains, RPA-AB, and of the isolated domain RPA-A, to normal DNA and damaged DNA containing the cyclobutane pyrimidine dimer (CPD) lesion. Both RPA-A and RPA-AB were found to bind non- specifically to both strands of normal and CPD- containing DNA duplexes. There were no differences observed when binding to normal DNA duplex was examined in the presence of the minimal DNA-binding domain of XPA (XPA-MBD). However, there is a drastic difference for CPD-damaged DNA duplex as both RPA-A and RPA-AB bind specifically to the undamaged strand. The strand-specific binding of RPA and XPA to the damaged duplex DNA shows that RPA and XPA play crucial roles in damage verification and guiding cleavage of damaged DNA during NER.
INTRODUCTION
In mammalian cells, nucleotide excision repair (NER) is the major repair pathway for the removal of bulky adducts induced by ultraviolet (UV) light or other environmental carcinogens (1–3). In the NER pathway, the detection of a single damaged site among an extensive background of undamaged DNA is accomplished via two distinct mechanisms, global genomic repair (GGR) and transcription-coupled repair (TCR). In the first step of the GGR pathway, a helix-distorting DNA lesion is bound by initial damage recognition proteins that recruit other repair proteins to the damaged site. A long DNA stretch is then unwound by the helicase activity of transcription factor IIH (TFIIH), and structure-specific endonucleases (XPG and XPF-ERCC1) are recruited to the repair complex. Double incision of the damaged strand releases 24–32 nucleotides (nt) that contain the DNA lesion. The resulting gap is filled in during the repair synthesis step by DNA polymerase δ or ε and sealed with a DNA ligase.
The eukaryotic single-stranded DNA (ssDNA)-binding protein, replication protein A (RPA), plays a crucial role in DNA replication, recombination and repair (4–6). RPA is a heterotrimer with 70, 32 and 14 kDa subunits. The high affinity DNA-binding activity is mediated by a pair of domains within the central part of the large subunit (RPA70): RPA-A (RPA70181–290) and RPA-B (RPA70300–422) (7,8). The X-ray crystallographic structure of the RPA-AB–dC8 complex revealed that each domain of RPA-AB (RPA-A and RPA-B) directly contacts 3 nt of ssDNA, with 2 nt filling the space between domains (9).
XPA is a 32 kDa protein that associates with the 70 and 32 kDa subunits of RPA (5,10) and on its own binds with a slight preference and modest affinity for damaged DNA (4,11). Although RPA itself is able to bind weakly to DNA containing various types of lesions (6), RPA stimulates stronger interaction of XPA with damaged DNA, which results in formation of an RPA–XPA complex at damaged DNA sites (12,13). The RPA–XPA complex also interacts with other DNA repair factors such as TFIIH, and the excision nucleases XPG and ERCC1-XPF (2). The solution structure of the minimal DNA-binding domain of XPA (XPA-MBD, XPA98–219) was recently determined (14,15) and shown to consist of a compact zinc-binding core and a loop-rich C-terminal subdomain connected by a linker.
Recently, it was suggested that in GGR, XPC-hHR23B is the primary damage recognition protein that initiates the NER pathway through binding to the damaged site (16,17). XPC-hHR23B binds preferentially to 3 and 5 nt bubbles with or without damaged bases (18). However, dual incision takes place only when a damaged base is present in the bubble structure (18). This implies that the progress of NER is determined not by the binding of XPC-hHR23B, but by the presence of DNA damage. Sugasawa et al. suggested a two-stage damage recognition model for NER (16). The lesion is first recognized by XPC-HR23B in GGR. XPC may induce some conformational changes in the DNA helix near the lesions. Then XPA, possibly together with RPA, is recruited to verify the substrate specificity of the lesion.
Cleavage of an undamaged DNA strand would cause severe problems with respect to maintenance of the integrity of the genome. However, only the DNA strand that contains the lesion is removed in mammalian NER, even though the dual incision takes place far from the damaged site. By comparing the affinity of RPA for artificial DNA hairpin structures with 3′- or 5′-protruding single-stranded arms, it was suggested that RPA binds to the undamaged strand during NER (19). However, there is no direct evidence for the preferential binding of RPA to the undamaged strand.
Binding of RPA to the undamaged strand of a damaged DNA duplex is most important during NER, because the dual incision must take place in the strand containing the lesion. In order to decipher the mechanism of damage recognition as well as the specific mechanism of cleavage of the damaged strand during dual incision, it is crucial that the specificity of RPA binding to the undamaged strand be determined. To this end, we used nuclear magnetic resonance (NMR) spectroscopy to compare the binding modes of RPA and XPA to normal DNA and DNA duplexes containing the cyclobutane pyrimidine dimer (CPD) lesion. Because XPC-hHR23B shows similar binding modes with normal and CPD-damaged DNA, but there are different patterns of dual incision, it is best to compare the modes of binding for RPA and XPA after XPC-hHR23B is bound to the DNA. In this study, NMR chemical shifts of RPA-A and RPA-B were monitored to determine the differences between the interactions with normal and CPD-containing DNA duplexes. The influence of the XPA-MBD in the ternary complex was then investigated. These results provide information on the mode of interaction of RPA and XPA, which has direct implications for the mechanism of damage verification and the subsequent strand-specific dual incision of NER.
MATERIALS AND METHODS
DNA preparation
The DNA oligonucleotides 5′-CGCATTACGC-3′ (TT-10) and 5′-GCGTAATGCG-3′ (AA-10) were synthesized on a DNA synthesizer (Applied Biosystem, model 391). The synthesized DNA oligonucleotides were purified by reverse-phase HPLC and desalted on a Sephadex G-25 column. The CPD-containing DNA decamer (CPD-10) was prepared by direct 254-nm UV irradiation of a TT-10 oligomer in an aqueous solution and purified as described (20). The double-stranded DNAs (dsDNAs), TT/AA-10 and CPD/AA-10, were prepared by dissolving each strand at a 1:1 stoichiometry in an NMR buffer solution.
Preparation of RPA-A, RPA-AB and XPA-MDB
The genes encoding human RPA-A (RPA70181–304) and RPA-AB (RPA70181–422) were cloned into the T7 polymerase expression vector pET14b (Novagen) using the recombinant RPA expression vector kindly provided by Dr M. Wold. An XPA-MBD (XPA98–219) expression vector was kindly provided by Dr K. Tanaka. All proteins were expressed in Escherichia coli strain BL21(DE3)pLysS as fusions with an N-terminal His6 tag. All proteins were purified with nickel-chelating affinity chromatography and, after removal of the histidine tag, by digestion with thrombin followed by a Superdex-75 (Pharmacia) gel filtration FPLC. Uniformly 15N- and 15N/13C-labeled RPA-A and RPA-AB were obtained by growing cells in M9-minimal medium containing 15NH4Cl and unlabeled/13C-labeled glucose as the sole nitrogen and carbon sources. The purity and homogeneity of all samples were assessed using SDS–PAGE. The NMR assignments of the free proteins were made using separate samples that were produced and purified as described elsewhere (21).
NMR experiments
NMR experiments were performed on a Varian 600 MHz (KAIST, Daejon) spectrometer and Bruker spectrometers operating at 600 and 800 MHz (Vanderbilt University, Nashville, TN). Complete backbone and side chain assignments for RPA-A, RPA-B and RPA-AB are described elsewhere (21,22). Backbone amide proton and amide nitrogen chemical shifts of RPA-A and RPA-AB in complexes with DNA and the XPA-MBD were assigned by comparison with 1H–15N HSQC spectra of the free proteins.
Two-dimensional 1H–15N HSQC spectra were acquired on uniformly 15N-labeled samples of RPA-A and RPA-AB in a 90% H2O/10% D2O solution containing 20 mM Tris–HCl, 100 mM NaCl, 2 mM dithiothreitol (DTT), pH 7.0 at 27°C. Titrations of three different ssDNA decamers, TT-10, CPD-10 and AA-10, were carried out with both RPA-A and RPA-AB. In all experiments, the concentrations of RPA-A and RPA-AB were 0.3 and 0.2 mM, respectively. Aliquots of 2 mM ssDNA, dissolved in the same buffer used for the RPA-A and RPA-AB samples, were added directly to the NMR cell, and the samples were allowed to equilibrate for several minutes. Changes in average amide chemical shifts (Δδavg) were calculated using equation 1,
![]()
where ΔδN and ΔδH are the differences in amide nitrogen and amide proton chemical shifts, respectively, between the free and bound states of the protein.
We carried out titrations of TT/AA-10 and CPD/AA-10 with RPA-A and RPA-AB in the presence and absence of the equimolar XPA-MBD. The strategy of this titration was the same as the one used for the ssDNA titrations. The amide chemical shifts of the mixed state of the RPA-DBDs (i.e. free and bound to each strand of dsDNA) are represented by equation 2, based on the consideration that the chemical exchange between the free and bound states of the protein is very fast:
δNobs = (1 – RA – RB) × δNfree + RA × δNA + RB × δNB
δHobs = (1 – RA – RB) × δHfree + RA × δHA + RB × δHB 2
(where δNobs/δHobs are the observed amide 15N/1H chemical shifts of the protein, δNfree/δHfree are the amide 15N/1H chemical shifts of the free state of the protein, δNA/δHA and δNB/δHB are the amide 15N/1H chemical shifts of the proteins saturated by A and B strands, respectively, and RA and RB are the molecular populations of protein bound to A and B strands). The population of each state is calculated by fitting the data of the 12 amino acids in RPA-A and RPA-AB that have well-resolved amide signals that change significantly during the course of the titration.
RESULTS
Binding of RPA-A and RPA-AB to ssDNA
Heteronuclear NMR spectroscopy has been used to monitor the DNA-binding activity of the RPA-AB domains. Using the backbone amide proton and amide nitrogen assignments (Fig. 1), it is possible to determine what residues are perturbed by binding of ssDNA and to map these on the structure (21). A subset of the 1H–15N HSQC cross-peaks observed for free RPA-A (black) shifted upon the addition of an equimolar concentration of TT-10 (blue) and CPD-10 (red) (Fig. 2). The residues perturbed significantly by TT-10 and CPD-10 are located on the β2, β3, β4′ and β5′ strands and loop L45 of RPA-A (Figs 3 and 4A). In the crystal structure, the side chain of F269 in the flexible loop (L45) is involved in a direct stacking interaction with the base, and this loop undergoes a significant conformational change upon binding to ssDNA (9). Thus, the neighboring Q268 residue is the most perturbed (Fig. 3). Interestingly, CPD-10, which is the same DNA sequence as TT-10 but contains a CPD formed at the central T–T sequence, causes a change in the chemical shift similar to that observed with RPA-A binding to TT-10 (Fig. 4A). However, the chemical shift perturbations induced by the binding of TT-10 are slightly different from those induced by binding of AA-10 (data not shown), reflecting the subtle adjustments that must be made in the binding site. These results show that the NMR chemical shift is a sensitive indicator of differences in RPA-A structure.
Figure 1.

The 1H–15N HSQC spectrum of RPA-AB, 25°C. Most of the amide resonances have been assigned. Two resonances of amino side chains are represented by lines. The amide resonances from the histidine tag are represented by the hash symbol.
Figure 2.

Comparison of the 1H–15N HSQC spectra of RPA-A in the absence (black) and presence of TT-10 (blue) or CPD-10 (red), 27°C.
Figure 3.

The average chemical shift changes (Δδavg) in the 1H and 15N resonances of RPA-AB upon addition of TT-10 and CPD-10. The residues whose cross-peaks disappear upon the addition of ssDNA are represented as dots.
Figure 4.

(A) Comparison of the 1H–15N HSQC peaks of S223, E232, Q268 and N274 of RPA-A in the absence (black) and presence of the TT-10 (blue) or CPD-10 (red), 27°C. (B) Comparison of the 1H–15N HSQC peaks of I332, V356, V383 and L394 of RPA-AB in the absence (black) and presence of the TT-10 (blue) or CPD-10 (red).
In the 15N–1H HSQC spectrum of RPA-AB, the chemical shifts of all of the cross-peaks of residues of the A domain are very close to those of the cross-peaks in the corresponding spectrum of isolated RPA-A, which indicates that RPA-A and RPA-B are connected with a flexible linker and do not interact with each other in the absence of ssDNA (23,24). We also observed that the chemical shift changes that occur upon addition of TT-10 or CPD-10 to RPA-AB are the same as those for RPA-A. This indicates that the RPA-A, whether alone or in the context of RPA-AB, binds to the same DNA sequence in TT-10 and CPD-10. In the crystal structure of the dC8–RPA-AB complex, two aromatic side chains of RPA-A (F238 and F269) directly stack with two of the cytosine bases (C1 and C3) in dC8 (12). The cyclobutane ring of the CPD lesion would interfere with this stacking interaction, and we find that RPA-A does indeed bind to the 10mer so that it does not sterically clash with the bulky lesion.
The pattern of changes in the amide resonances of the B domain in RPA-AB was similar upon binding to TT-10 and CPD-10, except for a few residues. Like RPA-A, the residues perturbed upon addition of TT-10 and CPD-10 were located in the corresponding β2, β3, β4′ and β5′ strands and the L45 loop of the RPA-B domain (Fig. 3). Residues whose chemical shift perturbations are different are located in β2, β5 and β5′ of RPA-B (Figs 4B and 5). These differences are rather small and appear to reflect subtle adjustments in the structure to accommodate conformational biases in the DNA induced by the lesion.
Figure 5.
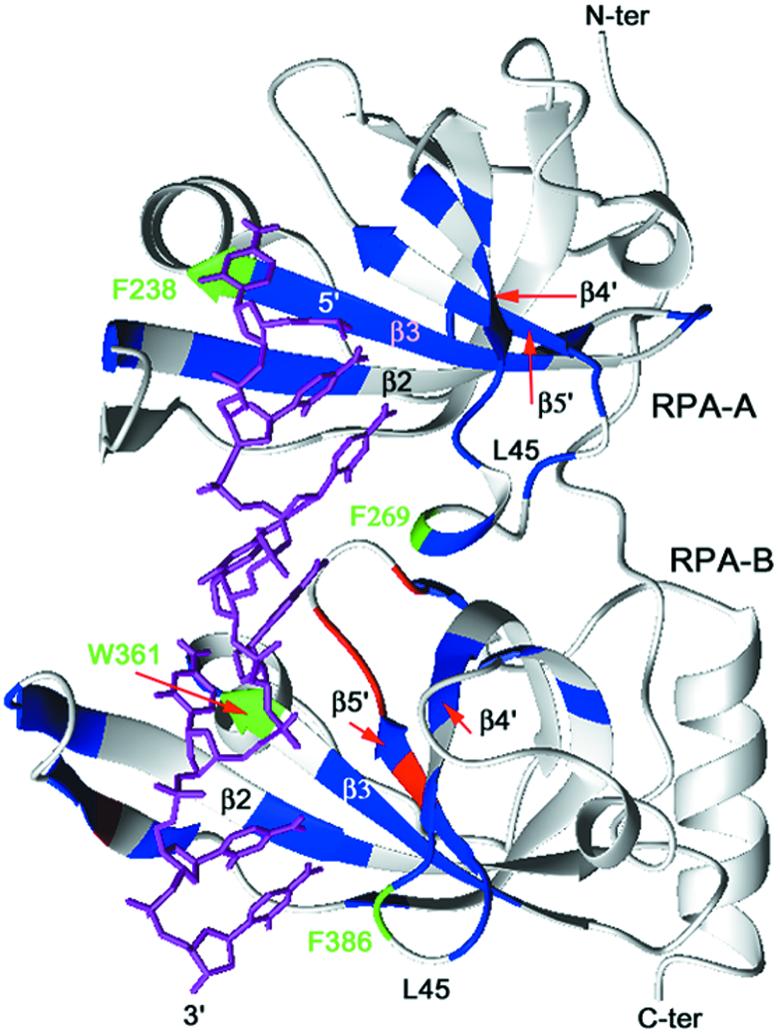
Mapping of the 15N-labeled RPA-AB residues with the chemical shift perturbation effect observed in the 15N–1H HSQC spectra. Residues with the largest chemical shift changes (Δδavg > 0.07 p.p.m.) following titration with ssDNA are indicated in blue. Residues with the largest differences in the chemical shift changes (Δδavg > 0.04 p.p.m.) observed with TT-10 versus CPD-10 are indicated in red. The four residues that have aromatic side chains involved in stacking interactions with bases in the ssDNA are represented in green.
Binding of RPA-A and RPA-AB to dsDNA
The binding affinity of RPA for a 49 nt dsDNA fragment is 1000-fold lower than the affinity of binding to the corresponding ssDNA (25). The X-ray crystal structure of the RPA-AB–dC8 complex revealed that RPA-AB directly interacts not only with the bases of ssDNA but also its phosphate backbone (9). Taken together, the in vitro binding and X-ray data suggest that when RPA binds to dsDNA, RPA first partially unwinds dsDNA and then binds to the resulting single-stranded region of the dsDNA. This model explains the in vitro binding assay results reported recently (25), in which the binding affinity of RPA to dsDNA correlated with the melting temperature of the bound dsDNA.
We tested this hypothesis by performing 1D NMR experiments on duplex DNA added to RPA. No imino proton resonances from the duplex form were observed when the TT/AA-10 duplex was added to the RPA-A solution, until the molar amounts of the RPA-A and TT/AA-10 were equivalent (data not shown). Conversely, 1D NMR experiments on duplex DNA at a different ratio of RPA-A (from 0 to 3) were performed. As the concentrated RPA-A was added to the TT/AA-10 duplex, imino proton resonances were broader and lower (see Supplementary Material available at NAR Online). These indicate that RPA-A unwinds the TT/AA-10 duplex before binding to the resulting single strands. In this binding mode, the length of TT/AA-10 is so short that the duplex melts completely to form two pieces of ssDNA rather than being partially unwound by RPA-A. To understand this observation in more detail, we fitted the data to equation 2 in order to quantitatively analyze the populations of RPA-A binding to the TT-10 and AA-10 strands. This analysis is based on the premise that the NMR resonance of one molecule in equilibrium between two states has the averaged value of the chemical shifts in each state when the exchange between the two states is sufficiently fast. RPA-A and RPA-AB bind to ssDNA in this regime, which enables the use of this method of analysis. Figure 6A shows the percentages of RPA-A binding to the TT-10 and AA-10 strands as a function of the molar ratio of the added TT/AA-10 duplex relative to RPA-A. At DNA/RPA molar ratios lower than 0.5, most of the TT-10 and AA-10 strands are bound by RPA-A (Fig. 6A). At the DNA:RPA-A molar ratio of 2:1, the percentages of RPA-A bound to TT-10 and AA-10 are 73 and 20%, respectively, reflecting the known preference for pyrimidine-rich versus purine-rich ssDNA (25). The same results were observed with the CPD/AA-10 duplex (Fig. 6B). The characteristics of binding of RPA-AB to the TT/AA-10 and CPD/AA-10 duplexes are very similar to those of RPA-A (data not shown). These results show that the formation of a CPD at the central T–T site of TT-10 does not affect the binding of DNA by RPA-AB.
Figure 6.

(A) Binding of RPA-A to the TT/AA-10 duplex as a function of DNA concentration in the absence (upper) and presence (lower) of equimolar XPA-MBD. Populations of RPA-A binding to the TT-10 and AA-10 strands are represented by white and black bars, respectively. (B) Binding of RPA-A to the CPD/AA-10 duplex as a function of DNA concentration in the absence (upper) and presence (lower) of equimolar XPA-MBD. Populations of RPA-A binding to the CPD-10 and AA-10 strands are represented by white and black bars, respectively. (C) Binding of RPA-AB to the CPD/AA-10 duplex as a function of DNA concentration in the absence (upper) and presence (lower) of equimolar XPA-MBD. Populations of RPA-AB binding to the CPD-10 and AA-10 strands are represented by white and black bars, respectively.
Effect of XPA-MBD in the binding of RPA-A and RPA-AB to dsDNA
We also performed NMR chemical shift perturbation experiments on RPA-A binding of TT/AA-10 and CPD/AA-10 in the presence of XPA-MBD. For undamaged DNA, the perturbations of amide resonances of RPA-A induced by the addition of TT/AA-10 were the same, whether or not RPA-A was pre-loaded with XPA-MBD (Fig. 6A), showing that no direct interaction exists between RPA-A and XPA-MBD, and that XPA-MBD does not affect the binding of RPA-A to non-damaged DNA. In contrast, the presence of the XPA-MBD results in reduced changes in the amide resonances of RPA-A upon addition of the CPD/AA-10 duplex. Thus, when the molar ratio of CPD/AA-10 to RPA-A is 0.3, the population of RPA-A binding to CPD-10 is significantly decreased from 40 to 21% by the presence of XPA-MBD (Fig. 6B). In contrast, under the same conditions, the population of RPA-A binding to AA-10 is barely affected (Fig. 6B). These findings imply that XPA-MBD interferes only with the binding of RPA-A to the CPD-10 strand.
The effect becomes even more complex when the dsDNA/RPA-A molar ratio is more than 0.5, as the pattern of RPA-A binding to each strand differs from that observed with lower molar ratios. When the molar ratio is 0.6, pre-loading XPA-MBD significantly decreases the population of RPA-A bound to CPD-10 from 64 to 39%, whereas the population of RPA-A bound to AA-10 is increased from 40 to 51% (Fig. 6B). These observations can be explained as follows. When the dsDNA/RPA-A molar ratio is more than 0.5, some of the AA-10 remains in the free ssDNA form in the absence of XPA-MBD, because RPA-A prefers to bind to the pyrimidine rich CPD-10 strand rather than AA-10, which is purine rich. XPA-MBD interferes with the binding of RPA-A to CPD-10, so that the population of RPA-A binding to CPD decreases. RPA-A therefore binds to the AA-10 strand, i.e. the population of RPA-A bound to AA-10 is higher. This incremental change in the bound population of AA-10 cannot be observed at molar ratios lower than 0.5 because, under these conditions, AA-10 only rarely exists in the single-stranded form.
To verify these results, we performed a titration of XPA-MBD to RPA-A in the presence of the CPD/AA-10 duplex (Fig. 7). At a molar ratio of XPA-MBD to RPA-A of 3, the population of RPA-A bound to CPD-10 is 10%, whereas the population of RPA-A binding to AA-10 was not changed (Fig. 7).
Figure 7.

Binding of RPA-A to the CPD/AA-10 duplex as a function of the concentration of XPA-MBD. Populations of RPA-A binding to the CPD-10 and AA-10 strands are represented by white and black bars, respectively. The molar ratio of the DNA duplex and RPA-A is 0.3.
XPA-MBD exhibited the same effect upon the binding of RPA-AB to the CPD/AA-10 duplex (Fig. 6C). As noted above, RPA-AB binds to just one strand, and RPA-AB itself cannot distinguish between the damaged and normal strands. Our results show that in the presence of RPA-AB, XPA-MBD prefers to bind to the damaged strand, so that RPA-AB localizes on the complementary undamaged strand.
DISCUSSION
NER in mammalian cells involves recognition of DNA damage and dual incision on the strand containing the lesion, then DNA synthesis and ligation to replace the excised oligonucleotide (1–3). The ability to differentiate between the damaged and undamaged strand is a crucial requirement of the NER machinery. It is generally accepted that RPA has a crucial role in stimulating the activity and coordinating the action of the NER nucleases XPG and the XPF-ERCC1 complex (1–3,17). The binding of RPA is proposed to be the determining factor in the decision of which strand to cleave; when RPA binds to an undamaged strand, the opposing damaged strand is cleaved by nucleases (19).
RPA-AB does not show a preference for binding to damaged versus undamaged ssDNA (25). In fact, we found that RPA-AB interacts directly with a 10 nt fragment of ssDNA that contains a CPD lesion at the central site. It is important to note that in our study, the CPD lesion was positioned across from the linker region between RPA-A and RPA-B and not within the binding site of either domain. Consequently, the large CPD adduct does not interfere with stacking interactions or hydrogen bonding in the binding sites in each domain. We also observed the previously reported preference for pyrimidine-rich versus purine rich ssDNA (26), whether or not the CPD adduct was present. Although additional studies will be required to confirm our findings, our results indicate that the binding properties of RPA-AB alone cannot induce the preference for undamaged versus damaged strand observed during NER.
Surprisingly, it was found that in the presence of XPA-MBD, the binding affinity of RPA-AB for the damaged strand is decreased significantly. This result implies that XPA provides the driving force for the specificity of RPA binding to the undamaged strand during NER. Our finding is consistent with photo-induced, protein–DNA cross-linking experiments, which showed that XPA contacts both the damaged and undamaged strands of the DNA duplex, and RPA preferentially binds to the undamaged strand of the damaged duplex DNA (27).
The model DNA duplexes used in this study are of the order of the shortest substrates that RPA alone can unwind completely in order to then bind to the resulting ssDNA fragment. Therefore, we can think of this model as a 10 nt bubble (formed by XPC-hHR23B and/or TFIIH in NER), even though the TT/AA-10 DNA duplex has no mismatch. RPA-AB (and RPA-A alone) binds to one strand of the DNA duplex or bubble structure with a relatively small preference for polypyrimidine over polypurine sequences, but independent of the presence of CPD damage. This implies that RPA alone cannot verify the presence of a DNA lesion.
In the presence of XPA-MBD, RPA-A and RPA-AB bind preferentially to TT-10 over AA-10, as expected based on the preference for pyrimidines over purines. In contrast, both the isolated A domain and the intact RPA-AB bind CPD-10 and AA-10 with equivalent affinity. From this result, we conclude that in NER, RPA-AB first binds non-specifically to single-stranded regions of a bubble structure formed by XPC-hHR23B. When RPA binds to a strand opposite a lesion, XPA, which is recruited along with RPA, is positioned to bind to the damaged site (Fig. 8). Our results support the concept that strand-specific binding of the RPA-AB and XPA-MBD complex is a major determinant in the decision to continue or abort NER and in the selection of the cleavage site.
Figure 8.

Model for the molecular mechanism of damage recognition and verification in GGR. XPC-hHR23B first recognizes the site where the helix distortion is induced. RPA, XPA and TFIIH then may be recruited to the suspected site of damage to verify the presence of a lesion. If there is a lesion (left), RPA binds to the undamaged DNA strand, and XPA binds to the damaged strand specifically and interacts with TFIIH. The pre-incision complex containing the fully opened DNA would then be assembled. If there is no lesion (right), the strand-specific binding of RPA and XPA proteins to DNA does not occur, and the NER process is aborted.
The functional roles of the protein–protein interactions of the XPA–RPA complex with other repair factors are 2-fold: (i) verification of the DNA lesion and (ii) deposition of the nucleases on the appropriate cleavage sites (17). Thus, we suggest a model for the DNA damage verification step in NER after the formation of a bubble structure by XPC-hHR23B binding to a damaged DNA duplex (Fig. 8). First, RPA binds to the undamaged strand of the bubble structure, and XPA binds to the damaged strand. Then, XPA interacts with TFIIH, which is recruited to the damaged site by XPC-hHR23B (28). If all these interactions occur properly, then an open DNA complex is formed and the NER nucleases are recruited; XPG is recruited by RPA, and ERCC1-XPF is recruited by XPA and RPA. If one of these interactions does not occur, NER is aborted. Our results support a model where DNA damage recognition in the NER progresses contains two steps: (i) DNA binding of XPC-hHR23B and (ii) damage verification by RPA-XPA via strand-specific DNA binding and protein–protein interactions.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank M. Wold for providing the RPA expression vectors and K. Tanaka for providing the XPA-MBD expression vector. This work was supported by the National Creative Research Initiative Program to B.-S.C. from the Ministry of Science and Technology, Korea and an operating grant (RO1 GM65484) to W.J.C. from the National Institutes of Health, USA. C.-J.P. was supported partially by the BK21 project.
REFERENCES
- 1.Wood R.D. (1999) DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie, 81, 39–44. [DOI] [PubMed] [Google Scholar]
- 2.deLaat W.L., Jaspers,N.G.J. and Hoeijmakers,J.H. (1999) Molecular mechanism of nucleotide excision repair. Genes Dev., 13, 768–785. [DOI] [PubMed] [Google Scholar]
- 3.Batty D.P. and Wood,R.D. (2000) Damage recognition in nucleotide excision repair of DNA. Gene, 241, 193–204. [DOI] [PubMed] [Google Scholar]
- 4.Jones C.J. and Wood,R.D. (1993) Preferential binding of the xeroderma pigmentosum group A complementing protein to damaged DNA. Biochemistry, 32, 12096–12104. [DOI] [PubMed] [Google Scholar]
- 5.He Z., Henricksen,L.A., Wold,M.S. and Ingles,C.J. (1995) RPA involvement in the damage-recognition and incision steps of nucleotide excision repair. Nature, 374, 566–569. [DOI] [PubMed] [Google Scholar]
- 6.Burns J.L., Huzder,S.N., Sung,P., Prakash,S. and Prakash,L. (1996) An affinity of human replication protein A for ultraviolet-damaged DNA. J. Biol. Chem., 271, 11607–11610. [DOI] [PubMed] [Google Scholar]
- 7.Gomes X.V. and Wold,M.S. (1996) Functional domains of the 70-kilodalton subunit of human replication protein A. Biochemistry, 35, 10558–10568. [DOI] [PubMed] [Google Scholar]
- 8.Pfuetzner R.A., Bochkarev,A., Frappier,L. and Edwards,A.M. (1997) Replication protein A. Characterization and crystallization of the DNA binding domain. J. Biol. Chem., 272, 430–434. [DOI] [PubMed] [Google Scholar]
- 9.Bochkarev A., Pfuetzner,R.A., Edwards,A.M. and Frappier,L. (1997) Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature, 385, 176–181. [DOI] [PubMed] [Google Scholar]
- 10.Matusa T., Saijo,M., Kuraoka,I., Kobayashi,T., Nakatsu,Y., Nagai,A., Enjosi,T., Masutani,C., Sugasawa,K. and Hanaoka,F. (1995) DNA repair protein XPA binds replication protein A (RPA). J. Biol. Chem., 270, 4152–4157. [DOI] [PubMed] [Google Scholar]
- 11.Buschta-Hedayat N., Buterin,T., Hess,M.T., Missura,M. and Naegeli,H. (1999) Recognition of nonhybridizing base pairs during nucleotide excision repair of DNA. Proc. Natl Acad. Sci. USA, 96, 6090–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li L., Lu,X., Peterson,C.A. and Legerski,R.J. (1995) An interaction between the DNA repair factor XPA and replication protein A appears essential for nucleotide excision repair. Mol. Cell. Biol., 15, 5396–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stigger E., Drissi,R. and Lee,S.-H. (1998) Functional analysis of human replication protein A in nucleotide excision repair. J. Biol. Chem., 273, 9337–9343. [DOI] [PubMed] [Google Scholar]
- 14.Ikegami T., Kuraoka,I., Saijo,M., Kodo,N., Kyogoku,Y., Morikawa,K., Tanaka,K. and Shirakawa,M. (1998) Solution structure of the DNA- and RPA-binding domain of the human repair factor XPA. Nature Struct. Biol., 5, 701–706. [DOI] [PubMed] [Google Scholar]
- 15.Buchko G.W., Ni,S., Thrall,B.D. and Kennedy,M.A. (1998) Structural features of the minimal DNA binding domain (M98–F219) of human nucleotide excision repair protein XPA. Nucleic Acids Res., 26, 2779–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugasawa K., Ng,J.M.Y., Masutani,C., Iwai,S., van der Spek,P.J., Eker,A.P.M., Hanaoka,F., Bootsma,D. and Hoeijmakers,J.H. (1998) Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol. Cell, 2, 223–232. [DOI] [PubMed] [Google Scholar]
- 17.Lindahl T. and Wood,R.D. (1999) Quality control by DNA repair. Science, 286, 1897–1905. [DOI] [PubMed] [Google Scholar]
- 18.Sugasawa K., Okamoto,T., Shimizu,Y., Masutani,C., Iwai,S. and Hanaoka,F. (2001) A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev., 15, 507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.deLaat W.L., Appeldoorn,E., Sugasawa,K., Weterings,E., Jaspers,N.G.J. and Hoeijmakers,J.H.J. (1998) DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev., 12, 2598–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee J.-H., Choi,Y.-J. and Choi,B.-S. (2000) Solution structure of the DNA decamer duplex containing a 3′-T·T base pair of the cis-syn cyclobutane pyrimidine dimer: implication for the mutagenic property of the cis-syn dimer. Nucleic Acids Res., 28, 1794–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhattacharya S., Botuyan,M.-V., Hsu,F., Shan,X., Arunkumar,A.I., Arrowsmith,C.H., Edwards,A.M. and Chazin,W.J. (2002) Characterization of binding-induced changes in dynamics suggests a model for sequence-nonspecific binding of ssDNA by replication protein A. Protein Sci., 11, 2316–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhattacharya S., Arunkumar,A.I., Sullivan,S.L., Botuyan,M.-V., Arrowsmith,C.H. and Chazin,W.J. (2003) Complete assignment of the 1H, 13C and 15N resonances of the tandem high affinity binding domains of replication protein A. J. Biomol. NMR, in press. [DOI] [PubMed] [Google Scholar]
- 23.Bochkareva E., Belegu,V., Korolev,S. and Bochkarev A. (2001) Structure of the major single-stranded DNA-binding domain of replication protein A suggests a dynamic mechanism for DNA binding. EMBO J., 20, 612–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arunkumar A.I., Stauffer,M.E., Bochkareva,E., Bochkarev,A. and Chazin,W.J. (2003) Independent and coordinated functions of the replication protein A tandem high affinity ssDNA binding domains. J. Biol. Chem., in press. [DOI] [PubMed] [Google Scholar]
- 25.Lao Y., Gomes,X.V., Ren,Y., Taylor,J.-S. and Wold,M.S. (2000) Replication protein A interactions with DNA. III. Molecular basis of recognition of damaged DNA. Biochemistry, 39, 850–859. [DOI] [PubMed] [Google Scholar]
- 26.Kim C., Snyder,R.O. and Wold,M.S. (1992) Binding properties of replication protein A from human and yeast cells. Mol. Cell. Biol., 12, 3050–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hermanson-Miller I.L. and Turchi,J.J. (2002) Strand-specific binding of RPA and XPA to damaged duplex DNA. Biochemistry, 41, 2402–2408. [DOI] [PubMed] [Google Scholar]
- 28.Yokoi M., Masutani,C., Maekawa,T., Sugasawa,K., Ohkuma,Y. and Hanaoka,F. (2000) The xeroderma pigmentosum group C protein complex XPC-HR23B plays an important role in the recruitment of transcription factor IIH to damaged DNA. J. Biol. Chem., 275, 9870–9875. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.