


Abstract
The bacteriophage lambda recombination system has proven to be a valuable tool for engineering bacterial artificial chromosomes (BAC). Due to its high efficiency, subtle alterations in the BACs can be generated using oligonucleotides as targeting vectors. Since no selection marker is used, recombinant clones are identified utilizing a selective PCR screening method. However, occasionally the selective PCR screening is not feasible. We describe here a two-step ‘hit and fix’ method that can be reliably used for generating any subtle alteration in BACs using short denatured PCR fragments as targeting vectors. In the first step of this method, 6–20 nucleotides are changed around the base where the mutation has to be generated. In the second step, these altered nucleotides are reverted to the original sequence and simultaneously a subtle alteration is introduced. Since in each step several nucleotides are changed, PCR primers specific for such alterations can be designed. This two-step method provides a simple and efficient tool for generating subtle alterations in BACs that can be very valuable for functional analysis of genes.
INTRODUCTION
Several methods to engineer genomic DNA cloned into bacterial artificial chromosome (BAC) vectors have been described (1–4). One of these methods utilizes the bacteriophage lambda Red recombination system to modify large genomic DNA fragments (see refs 5 and 6 for review). The bacteriophage λ genes involved in recombineering are exo, bet and gam. The exo gene product has 5′-3′ exonuclease activity and the bet gene product is a single-strand DNA binding protein that promotes annealing. The gam gene product inhibits the recBCD nuclease, thus preventing the degradation of linear DNA fragments. This recombination system has been used to disrupt genes present in the BAC insert by inserting selectable markers, e.g. neomycin resistance gene. We have used the Red recombination system to generate single-base alterations, small deletions and insertions, using oligonucleotides as targeting vectors (7). Due to the high recombination efficiency, modified BACs could be identified without the use of any selectable marker. In the absence of such markers, a PCR-based screening method called mismatch amplification mutation assay-PCR (MAMA-PCR) was used to identify single-base alterations (7,8). In the MAMA-PCR method, one of the two PCR primers, the ‘mismatch detection’ primer, has two mismatched bases at the 3′ end with respect to the wild-type sequence (ultimate and penultimate 3′ base), but a single mismatch with the mutated allele (the penultimate 3′ base). The two mismatched bases at the 3′ end of the primer, when annealed to the wild-type template, fail to amplify a PCR product. However, in the case of the mutant DNA, the primer anneals to the template and allows selective amplification and detection of the targeted clone.
Although the MAMA-PCR method has been successfully used to generate multiple mutations, it has considerable limitations. For example, occasionally the mismatch detection primer is able to amplify a PCR product from the wild-type DNA, in spite of the two-base mismatch at the 3′ end, resulting in false positive clones (8). Also, mutations involving deletion, insertion or alteration of a base that is identical to one of the flanking bases can result in non-specific amplification of the mismatched PCR product. Similarly, when a unit of a small repetitive sequence (e.g. a di- or a tri-nucleotide repeat) has to be deleted or inserted, it is not possible to generate primers specific for the mutated sequence. In some cases where the targeting efficiency is high (1 per 100 electroporated cells), we have identified the recombinant clones by directly sequencing the BAC DNA from electroporated cells (S.Swaminathan and S.K.Sharan, unpublished data). However this approach is not very useful when 500–1000 colonies have to be screened to identify a single targeted clone. Methods like the SacB or the tetracycline resistance gene counter selection can be used as an alternative approach (2,9–12), but these screening methods result in unwanted rearrangement (13).
To tackle these problems, we have developed a simple method that can be consistently and efficiently used to generate any subtle alterations in the BACs without the use of a selectable marker. It is a two-step, ‘hit and fix’ method that utilizes short denatured PCR fragments as targeting vectors to modify BACs. In the first step of this method, a stretch of about 6–20 nucleotides is changed, including the nucleotide(s) that must be mutated (Fig. 1A). In the second step, the modified bases generated in the first step are restored to the original sequence, except for the insertion of the desired mutation. Since several nucleotides are changed in each step, the recombinant BACs can be identified by the standard PCR method using a primer specific for the altered bases. In addition, a restriction enzyme site is included in the nucleotides that are inserted in the first step; the presence of this site provides an additional tool to rapidly confirm the targeting event. In the second step, loss of this restriction enzyme site serves to confirm correct targeting. Due to the specificity of the screening method, the ‘hit and fix’ approach provides an efficient technique to generate subtle alterations in the BACs.
Figure 1.

‘Hit and fix’ method to generate subtle mutations in BACs using short denatured PCR fragments as targeting vectors. (A) Schematic representation of the two steps involved in the ‘hit and fix’ method to generate a single base deletion in codon 339 (nucleotide A, shown in box) of BRCA1 cDNA. In step 1, 180 bp denatured PCR fragments are used to change 20 nucleotides in the BAC DNA including codon 339. Primers specific for these 20 nucleotides are used to identify the targeted clone by PCR. The targeting is confirmed by testing for the presence of an XhoI restriction site (underlined) that is included in the 20 nt sequence. In step 2, these 20 nucleotides are restored to the original sequence, except for the single-base deletion. Such clones can be selected by PCR using primers specific to the 19 nucleotides that are reintroduced. (B) Generating 180mer single-stranded targeting vectors. Targeting vectors containing 160 bases of homology and 20 unique bases were generated by using two 100mer oligonucleotides in a PCR reaction. The two 100mer oligonucleotides had 20 complementary bases (including an XhoI recognition site, underlined) at the 3′ end. A 180 bp PCR product was generated. The PCR product was denatured to obtain single-stranded targeting vector.
MATERIALS AND METHODS
Bacterial strain
DY380 cells containing the λ prophage that provides the recombination function were used for recombineering (11). 100 ng of purified BAC DNA (HB1-812) with a 200 kb insert that includes the human breast cancer susceptibility gene BRCA1 (14) were electroporated into DY380 cells and used for recombineering. In addition, DH10B cells containing the HB1-812 BAC were directly used for recombineering after introducing a mini-λ DNA circle containing the Red recombination function into these cells (15). The mini-λ DNA integrates into the bacterial chromosome as a defective prophage. Cells containing the mini-λ were selected on LB agar plates containing tetracycline, since the mini-λ DNA carries the tetracycline resistance gene. The Red recombination genes exo, bet and gam in the λ prophage present in DY380 cells as well as in the mini-λ DNA are under the control of a temperature-sensitive repressor and are expressed at 42°C but remain repressed at 32°C.
Targeting vector
The targeting vectors used in step 1 and step 2 were 180mer PCR fragments that were denatured to obtain single-stranded DNA prior. The PCR fragments were generated by using two 100mer oligonucleotides containing 20 complementary bases at the 3′ end (shown in bold below) in a PCR reaction (Fig. 1B). No additional template DNA was included. The two 100mer oligonucleotides used to generate the step 1 targeting vector included an XhoI restriction site (underlined): E11F252 (forward): 5′-AAGCAAACAGCCTGGCTTAGCAAGGAGCCAACATAACAGATGGGCTGGAAGTAAGGAAACATGTAATGATAGGCGGACTCAAGCTTACTGTCAGCTCGAG-3′; E11R431 (reverse): 5′-GTATCTCTAGGATTCT CTGAGCATGGCAGTTTCTGCTTATTCCATTCTTTTCTCTCACACAGGGGATCAGCATTCAGATCCTCGAGCTGACAGTAAGCTT-3′.
The two 100mer oligonucleotides used to generate the step-2 targeting vector were: DeltA1129F2 (forward): 5′-AAGCAAACAGCCTGGCTTAGCAAGGAGCCAACATAACAGATGGGCTGGAAGTAAGGAAACATGTAATGATAGGCGGACTCCCAGCACAGAAAAAAGGTAG-3′; DeltA1129R2 (reverse): 5′-GTATCTCTAGGATTCTCTGAGCATGGCAGTTTCTGCTTATTCCATTCTTTTCTCTCACACAGGGGATCAGCATTCAGATCTACCTTTTTTCTGTGCTGGG-3′.
To generate the targeting vector, 300 ng of the forward and reverse 100mers were used in a 50 µl PCR reaction using the Expand™ High Fidelity PCR system (Boehringer-Mannheim). The PCR condition included initial denaturation for 4 min at 94°C followed by 35 cycles of 94°C for 30 s, 55°C for 45 s and 72°C for 1 min; and a final extension at 72°C for 5 min. The resultant 180 bp PCR product was purified by using the QIAquick PCR Purification kit (Qiagen) and reconstituted in sterile water. The PCR product was denatured at 94°C and chilled on ice before electroporation.
Induction of Red recombination genes and preparation of electro-competent cells
Bacterial cells containing the λ prophage were induced and made electro-competent as described previously (4). Briefly, 10 ml of cells, grown at 32°C to an OD600 of 0.6, were induced at 42°C for 15 min to express the exo, bet and gam genes in a 50 ml glass flask. After being chilled on ice for 10 min, the cells were washed with ice-cold water three times and resuspended in 50 µl of ice-cold, sterile water. A 50-µl aliquot of the electrocompetent cells was used for each electroporation.
Electroporation of targeting vector and identification of targeted clones
300 ng of the denatured PCR products was then electroporated into induced bacterial cells containing the BAC HB1-812. The electroporated cells were grown at 32°C in 1 ml of SOC medium for 1.5 h. To remove excess targeting vector from the media, the cells were washed three times with LB and then diluted to obtain about 20–30 cells per ml of LB containing chloramphenicol (20 µg/ml). The cells were diluted based on the assumption that 10 ml of culture at OD600 of 0.6 contains 108 cells. The cells were grown in a 96-deep well plate in 500 µl of culture medium, with 10–15 cells per well for 24 h at 32°C. To determine the exact number of cells per well, the dilutions were also plated on LB agar plates containing chloramphenicol. The next day, 8 µl of cell culture from each well was analyzed by standard PCR in a 50 µl reaction volume to identify the pool containing the targeted clone. The primers used in the PCR reaction were: HX (forward primer) 5′-AAG CTTACTGTCAGCTCGAG-3′; 31219R (reverse primer) 5′-CTCTCTACTGATTTGGAGTGAAC-3′. The primers amplified a 363 bp product from positive pools. Once the positive pools were identified, cells were diluted and plated on agar plates to obtain isolated colonies. Forty-eight colonies from each pool were screened by PCR using HX and 31219R primers. Targeting was confirmed by amplifying a 624 bp fragment including the targeted site using two primers, 30596F (forward primer) 5′-TGAACACCACTGAGAAG CGT-3′ and the 31219R reverse primer. The PCR product was purified by ethanol precipitation. About 300–500 ng of the PCR product was digested with XhoI restriction enzyme and analyzed on a 2.0% agarose gel.
To detect recombinant clones in step 2, the primers used in the PCR reaction were: DeltA 1129F (forward primer) 5′-CTCCCAGCACAGAAAAAAGG-3′ and the 31219R (reverse primer). The primers amplified a 363 bp product. The targeting was confirmed by amplifying a 624 bp fragment using the primers 30596F and 31219R and digesting with XhoI restriction enzyme as described above. In addition, about 60 ng of the 624 bp PCR product was sequenced using a primer, 30661F: 5′-ACTTGCATGTGGAGCCATGT-3′ to confirm that no additional mutations were generated during the recombination process.
RESULTS AND DISCUSSION
Several mutations have been identified in the BRCA1 gene that result in susceptibility to breast and ovarian cancer in humans (Breast Cancer Information Core, http://research.nhgri.nih.gov/bic/). To understand how these mutations result in tumorigenesis, we are generating some of the mutations in a human BAC clone, HB1-812, containing the full-length BRCA1 gene. These BACs will be used for functional analysis in transgenic mice lacking endogenous BRCA1 protein. We have utilized the ‘hit and fix’ method to generate mutations in the BAC. We describe here the application of this method to delete a single base, an ‘A’ nucleotide in codon 339 (nucleotide 1129 of the cDNA, U14680). This nucleotide is part of a stretch of seven ‘A’ nucleotides present in the cDNA and the mutation causes a frame shift, resulting in a truncated protein (16). Recombinant bacterial clones containing such an alteration are difficult to identify by the method described previously (7). However, the two-step method is designed to overcome such problems.
Denatured PCR fragments (180 bp), with 160 bases of homology (80 bases on each side) flanking 20 non-homologous bases to substitute the 20 nucleotides around codon 339, were used as a targeting vector. Two 100mer oligonucleotides with 20 complementary bases at the 3′ end were used in the PCR reaction to generate a 180 bp product (Fig. 1B). The 20 unique bases included an XhoI restriction site. The PCR product was denatured and electroporated into bacterial cells containing the BAC HB1-812 and the λ recombination proteins Exo, Beta and Gam. The cells were cultured in a 96-deep well plate and analyzed by PCR as described in Materials and Methods. The forward primer used in the PCR reaction was specific for the 20 bases that were unique to the targeted clone and the reverse primer was specific to the BRCA1 gene. The positive pools were identified by the presence of a 363 bp product (Fig. 2A). Twenty positive pools were identified out of a total 93 pools that were analyzed. Cells from three positive pools were diluted and plated to obtain isolated colonies. About 48 individual colonies from each pool were screened by PCR. Once individual recombinant clones were identified, the targeting was confirmed by detecting the presence of the XhoI restriction site in a 624 bp PCR fragment that was amplified by PCR (Fig. 2B). This provided additional evidence that clones obtained by PCR screening were correctly targeted recombinant clones and contained the desired ‘intermediate BAC’.
Figure 2.

Generation of a single-base deletion in codon 339 of the BRCA1 cDNA. (A) Bacterial cells that were electroporated with the targeting vector were analyzed in pools of 14 cells/well of culture plate, grown at 32°C in a 96-deep well plate. Using a PCR primer specific for the 20 nucleotides that were altered, positive pools containing the targeted clone were identified (shown by an arrow) by the amplification of a 363 bp product as observed on an agarose gel. (B) The targeting in step 1 was confirmed by the presence of an XhoI restriction site in a 624 bp product that was amplified from the PCR-positive clones. PCR products from correctly targeted clones, containing an XhoI site, showed the presence of two bands (lanes 1–3), whereas the PCR product from the wild-type BAC remained undigested (lane 4). (C) The targeting in step 2 was confirmed by the loss of the XhoI restriction site in the 624 bp PCR product amplified from positive clones. PCR products from correctly targeted clones remained undigested (lanes 1–3), whereas the PCR product from the ‘intermediate BAC’ clone showed the 348 bp and 276 bp bands (lane 4). (D) Deletion of a single nucleotide in the BRCA1 gene in the codon 339 was confirmed by sequencing. The upper panel shows the wild-type (WT) BAC sequence (an arrow marks the nucleotide A that is deleted in the mutant BAC) and the lower panel shows the sequence of the mutant (Delt A) BAC.
One of the ‘intermediate BAC’ clones was used in the second targeting step aimed to restore the 20 bases that were altered and at the same time introduce the desired mutation. The targeting vector was generated as described for the first step. However, in this case the 20 unique sequences were replaced with the wild-type sequence, with the exception of the single-base deletion in codon 339. The bacterial cells containing the prophage were induced, electroporated with a single-strand targeting vector; subsequently, 10–15 cells/well were cultured overnight in a 96-deep well plate, as described in the previous step. The pools of cells were screened to identify the recombinant clone by PCR, using a forward primer specific to the 19 nucleotides that were substituted in step 2 and the same reverse primer used in step 1. Positive pools were identified by the presence of a 363 bp PCR product. Again, cells from positive pools were diluted and individual clones were identified by PCR. To confirm that these clones were the desired recombinants, a 624 bp region including the codon 339 was amplified and digested with the XhoI restriction enzyme. As expected, PCR products from the correctly targeted clones remained undigested, while the PCR product from the intermediate clone was digested by XhoI (Fig. 2C). Finally, clones identified by PCR and XhoI digestion were confirmed by sequencing to be correctly targeted (Fig. 2D). To examine the structural integrity of the BACs, we digested the BAC DNA obtained from various clones containing the desired mutation with restriction enzymes and compared it to the parental BAC DNA digestion pattern (Fig. 3). No rearrangement was detected in any of the BAC clones.
Figure 3.
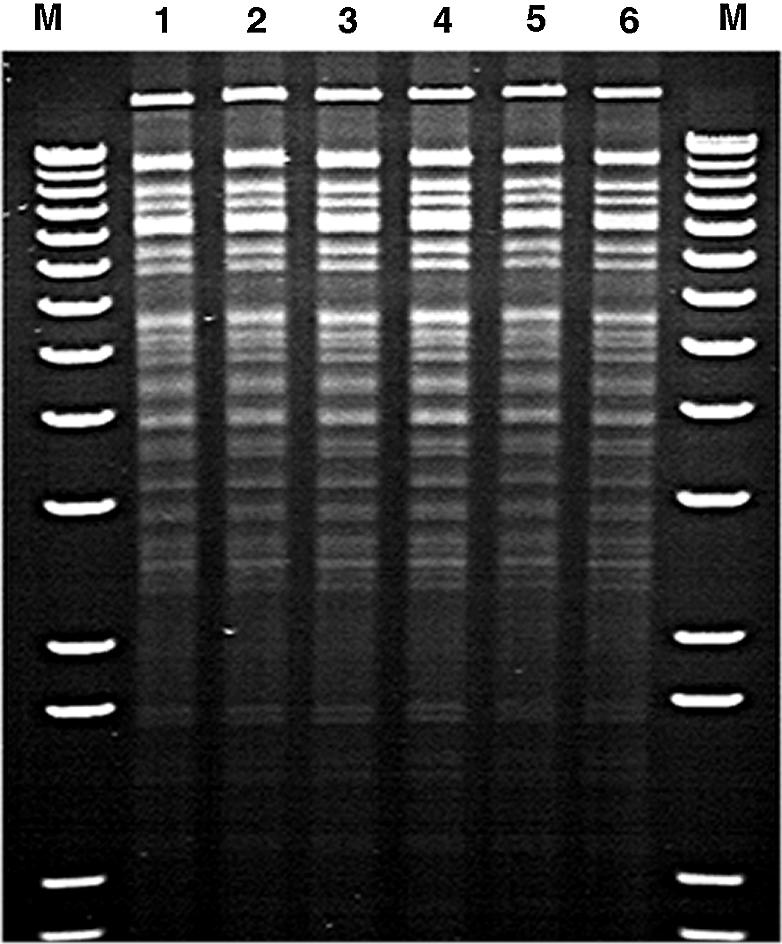
Stability of the BAC DNA after recombineering. An agarose gel picture of various BAC DNA samples digested with EcoRV and SpeI restriction enzymes showed the absence of any unwanted rearrangement after the genetic manipulation. Lane 1 contains the parental BAC HB1-812 DNA, lanes 2 and 3 contain ‘intermediate BAC’ DNA from two different clones and lanes 4–6 contain DNA from three different mutant (Delt A) BAC clones. A 1 kb plus DNA ladder (Invitrogen) was used as molecular weight marker (M). The smallest marker band on the gel represents an 850 bp fragment.
We then used the ‘hit and fix’ method to generate two additional mutations in the BRCA1 gene. These were missense mutations in codons 1495 and 1823 of the BRCA1 cDNA. The mutation in codon 1495 (G to T) results in an arginine (AGG) to methionine (ATG) change, and the mutation in codon 1823 (G to A) causes an alanine (GCA) to threonine (ACA) amino acid alteration (Breast Cancer Information Core). These mutations have been predicted to disrupt the normal function of the BRCA1 protein and to result in tumorigenesis. Earlier, we attempted to generate these mutations in BAC by using the MAMA-PCR screening method (7). We failed to identify any recombinant clone due to the non-specific amplification of the PCR product (data not shown). However, as summarized in Table 1, using the two-step method, both mutations were successfully generated and confirmed by sequence analysis.
Table 1. Summary of targeting efficiency of mutations generated in the BRCA1 gene.
| Mutation | Codon | Nucleotide | Amino acid change | Targeting frequencya | Clones confirmed by sequencing | |
|---|---|---|---|---|---|---|
| Step 1 | Step 2 | |||||
| delt A | 339 | 1129 | Stop 340 | 1/65 | 1/68 | 3/3 |
| G to T | 1495 | 4603 | Arg to Met | 1/32 | 1/32 | 3/3 |
| G to A | 1823 | 5586 | Ala to Thr | 1/840 | 1/1003 | 2/2 |
aNumber of PCR positive clones/number of electroporated cells.
The results clearly demonstrate that the two-step method can be used to generate a subtle alteration in a large genomic DNA. Although this method involves two separate targeting steps and therefore takes a longer time than the single step method in which mutant clones are identified by MAMA-PCR, this approach is much more efficient. Due to the high specificity of the primers to the recombinant clones, no false positive results are obtained. In addition to using a PCR-based screening method, colony hybridization using 32P-labeled oligonucleotides, specific to the 20 nucleotides that are replaced in each step, can also be used for screening purposes. Targeted clones have been successfully identified with high specificity using this method (S.Kuznetsov and S.K.Sharan, unpublished results).
Since the BAC clones maintain their structural integrity and do not show unwanted rearrangements after the genetic manipulation, the method described here provides a valuable tool for the functional analysis of genes. For example, to understand the function of cis-regulatory elements or the biological significance of various structural and functional domains of proteins or to understand the relevance of missense mutations identified in human disease-causing genes, subtle alterations can be rapidly generated in BACs carrying these genes. Their functional significance can then be examined by introducing the BACs into mammalian cells or by generating transgenic mice for in vivo analysis.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Donald Court, Srividya Swaminathan and Sergey Kuznetsov for helpful discussions and critical review of the manuscript and Linda Cleveland for help with DNA sequencing. We also thank the publication department of NCI-Frederick for help with the illustrations. Research sponsored by the National Cancer Institute, DHHS.
REFERENCES
- 1.Yang X.W., Model,P. and Heintz,N. (1997) Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nat. Biotechnol., 15, 859–865. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y., Buchholz,F., Muyrers,J.P. and Stewart,A.F. (1998) A new logic for DNA engineering using recombination in Escherichia coli. Nature Genet., 20, 23–128. [DOI] [PubMed] [Google Scholar]
- 3.Muyrers J.P., Zhang,Y., Testa,G. and Stewart,A.F. (1999) Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res., 27, 1555–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu D., Ellis,H.M., Lee,E.C., Jenkins,N.A., Copeland,N.G. and Court,D.L. (2000) An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl Acad. Sci. USA, 97, 5978–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Copeland N.G., Jenkins,N.A. and Court,D.L. (2001) Recombineering: a powerful new tool for mouse functional genomics. Nature Rev. Genet., 2, 769–779. [DOI] [PubMed] [Google Scholar]
- 6.Court D.L., Sawitzke,J.A. and Thomason,L.C. (2002) Genetic engineering using homologous recombination. Annu. Rev. Genet., 36, 361–388. [DOI] [PubMed] [Google Scholar]
- 7.Swaminathan S., Ellis,H.M., Waters,L.S., Yu,D., Lee,E.C., Court,D.L. and Sharan,S.K. (2001) Rapid engineering of bacterial artificial chromosomes using oligonucleotides. Genesis, 29, 14–21. [DOI] [PubMed] [Google Scholar]
- 8.Cha R.S., Zarbl,H., Keohavong,P. and Thilly,W.G. (1992) Mismatch amplification mutation assay (MAMA): application to the c-H-ras gene. PCR Methods Appl., 2, 14–20. [DOI] [PubMed] [Google Scholar]
- 9.Blomfield I.C., Vaughn,V., Rest,R.F. and Eisenstein,B.I. (1991). Allelic exchange in Escherichia coli using the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Mol. Microbiol., 5, 1447–1457. [DOI] [PubMed] [Google Scholar]
- 10.Muyrers J.P., Zhang,Y., Benes,V., Testa,G., Ansorge,W. and Stewart,A.F. (2000) Point mutation of bacterial artificial chromosomes by ET recombination. EMBO Rep., 1, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee E.C., Yu,D., Martinez de Velasco,J., Tessarollo,L., Swing,D.A., Court,D.L., Jenkins,N.A. and Copeland,N.G. (2001) A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics, 73, 56–65. [DOI] [PubMed] [Google Scholar]
- 12.Nefedov M., Williamson,R. and Ioannou,P.A. (2000) Insertion of disease-causing mutations in BACs by homologous recombination in Escherichia coli. Nucleic Acids Res., 28, e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imam A.M., Patrinos,G.P., de Krom,M., Bottardi,S., Janssens,R.J., Katsantoni,E., Wai,A.W., Sherratt,D.J. and Grosveld,F.G. (2000) Modification of human beta-globin locus PAC clones by homologous recombination in Escherichia coli. Nucleic Acids Res., 28, e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandler J., Hohenstein,P., Swing,D.A., Tessarollo,L. and Sharan,S.K. (2001) Human BRCA1 gene rescues the embryonic lethality of Brca1 mutant mice. Genesis, 29, 72–77. [DOI] [PubMed] [Google Scholar]
- 15.Court D.L., Swaminathan,S., Yu,D., Wilson,H., Baker,T., Bubunenko,M., Sawitzke,J. and Sharan,S.K. (2003) Mini-lambda: a tractable system for chromosome and BAC engineering. Gene, in press. [DOI] [PubMed] [Google Scholar]
- 16.Hogervorst F.B., Cornelis,R.S., Bout,M., van Vliet,M., Oosterwijk,J.C., Olmer,R., Bakker,B., Klijn,J.G., Vasen,H.F., Meijers-Heijboer,H. et al. (1995) Rapid detection of BRCA1 mutations by the protein truncation test. Nature Genet., 10, 208–212. [DOI] [PubMed] [Google Scholar]