


Abstract
Endoribonuclease RNase E has a central role in both processing and decay of RNA in Escherichia coli, and apparently in many other organisms, where RNase E homologs were identified or their existence has been predicted from genomic data. Although the biochemical properties of this enzyme have been already studied for many years, the substrate specificity of RNase E is still poorly characterized. Here, I have described a novel oligonucleotide-based assay to identify specific sequence determinants that either facilitate or impede the recognition and cleavage of RNA by the catalytic domain of the enzyme. The knowledge of these determinants is crucial for understanding the nature of RNA–protein interactions that control the specificity and efficiency of RNase E cleavage and opens new perspectives for further studies of this multi-domain protein. Moreover, the simplicity and efficiency of the proposed assay suggest that it can be a valuable tool not only for the characterization of RNase E homologs but also for the analysis of other site-specific nucleases.
INTRODUCTION
Escherichia coli RNase E is an essential endoribonuclease that is believed to initiate the decay of many, if not most, messenger RNAs (1) as well as being involved in the processing of ribosomal RNA (2), the maturation of tRNA precursors (3) and small non-coding RNAs (4,5).
The N-terminal half (residues 1–498) of E.coli RNase E contains the ribonucleolytic site (6) and its amino acid sequence is evolutionarily conserved among RNase E homologs (7). The C-terminal half of the protein contains extra RNA-binding domain(s) (6–9) as well as multiple sites that are involved in the assembly of the E.coli degradosome complex (7,10). In addition to RNase E, the other major components of the degradosome are the 3′ exonuclease polynucleotide phosphorylase (PNPase), the RhlB helicase and the glycolytic enzyme enolase (11–13). The discovery of the degradosome and its further characterization support the idea (11) that degradation and processing of RNA in E.coli is most likely performed by the coordinated activity of degradosomal components.
RNase E is known to cleave RNA substrates in single-stranded regions that are usually proceeded or followed by a stable stem–loop structure (11,14–18). Moreover, the stem–loop structures adjacent to the cleavage site do not affect the specificity of cleavages but can influence the rate of cleavage indirectly (16,19–21). Early efforts aimed to characterize the substrate specificity of this enzyme in a systematic fashion suggested the following consensus sequence: (G,A)AUU(U,A) (20). The conservation of this sequence, however, was not supported by the mutational analysis of RNAI (22,23). In contrast, it was proposed (22,23) that a high A,U content, rather than a specific order of nucleotides is required for efficient cleavage. The aforementioned discrepancy in the composition of the proposed recognition motifs is largely due to a limited number of the mutated sequences that were analyzed by different laboratories. For instance, in the initial work (20), only a small portion of the consensus sequence was determined by mutational analysis, whereas the rest of the consensus was deduced based on the sequence similarity of RNase E cleavage sites of various natural transcripts. It is worth mentioning, however, that some of these naturally occurring sites apparently have a sub-optimal sequence [see site b of 9S precursor of 5S ribosomal RNA (19)] and, therefore, are not suitable for the similarity search of a consensus motif. Furthermore, regarding the mutational analysis of RNAI (22,23), it is uncertain whether every observed difference in the efficiency and specificity of cleavages is related to the effect of the primary sequence per se rather than a consequence of structural changes induced by alternative base pairing within the mutated sequence.
An important finding that opened a new perspective for the analysis of substrate specificity, was the observation that the specificity of cleavages is not affected when the full-length substrate is represented only by a short oligonucleotide corresponding in sequence to the single-stranded region of its cognate RNA (21). Employing small oligonucleotides as substrates, we have recently shown that guanosines, which are 5′ and in close vicinity to the scissile bond can significantly affect the efficiency of cleavages (19). In this report, I developed and experimentally tested a new oligonucleotide-based approach and, for the first time, used this method to identify specific sequence determinants of RNase E cleavage sites.
MATERIALS AND METHODS
Purification of Rne498
Escherichia coli BL21(DE3) cells (Novagen) containing a pET16b-based plasmid encoding the first 498 amino acids of E.coli RNaseE (6) were used to produce the Rne498 polypeptide. This His-tagged polypeptide was purified by immobilized metal affinity chromatography (IMAC) (24) under denaturing conditions as described by the vendor of the chromatography column (Qiagen). The eluates were dialyzed against 50 mM Tris–HCl (pH 8.0), 0.1 mM EDTA, 0.5 M NaCl and 20% (v/v) glycerol, and dithiothreitol was added to a final concentration of 1 mM. The purity of the polypeptide in this preparation was >95% as judged by Coomassie blue staining of samples run in SDS–polyacrylamide gels.
Preparation of substrates
Oligoribonucleotides 5′-AUCCAAUAAA (9SBmut4C), 5′-AUCUAAUAAA (9SBmut4U), 5′-AUCGAAUAAA (9SBmut4G), 5′-AUCAGAUAAA (9SBmut5G), 5′-AUCAAGUAAA (9SBmut6G), 5′-AUCAAAGAAA (9SBmut7G), 5′-AAAAAAAAAAAAAAAAAAAAAAAAAAA (A27), 5′-AAAAAAAAAAAAAGAAUAAAAAAAAAA (BR27), 5′-AAA AAAAAAAAAAGCAAAAAAAAAAAA (GC27), 5′-AAAAAAAAAAAAAUAAAAAAAAAAAAA [(A)13U(A)13], 5′- AAAAAAAAAAAAAGAAAAAAAAAAAAA [(A)13G(A)13], 5′-AAAAAAAAAAAAACAAAAAAAAAAAAA [(A)13 C(A)13], 5′-UUUUUUUUUUUUUUUUUUUUUUUUUUU (U27); 5′-UUUUUUUUUUUUUAUUUUUUUUUUUUU [(U)13A(U)13] were synthesized using phosphoramidite chemistry (Vienna Biocenter Core Facility) and 5′ end-labeled as described (21).
Site-specific RNase E cleavage of oligonucleotides
RNase E cleavage reactions were performed as described previously (21) using Rne498 and 5′ labeled substrates. The 1 nt ladders were prepared by partial digestion of each 5′ labeled oligonucleotides with S1 nuclease in buffer provided by the vendor (MBI Fermentas).
RESULTS
Probing the specificity of RNase E cleavage by a novel oligonucleotide-based assay
The property of RNase E to cleave poly(A)- and poly(U)-homo-oligomers (25,26), prompted me to develop a simple approach that allows one to deduce specific sequence determinants of RNase E cleavage sites. According to this approach, a single nucleotide within a homo-oligomer is substituted at the very same position by either A, G, C or U. Then, the effect of each substitution is assessed by cleavage efficiency at multiple positions in the vicinity of the substituted nucleotide. By linking their relative location and efficiency of cleavage at the respective inter-nucleotide bonds, it is possible to assign each of four nucleotides to specific positions within a putative motif.
As seen in Figure 1, in comparison with the cleavage of the homo-oligomer A27 (Fig. 1A), the cleavage of (A)13G(A)13, (A)13C(A)13 and (A)13U(A)13 oligonucleotides (Fig. 1B, C and D, respectively) by an RNase E polypeptide (Rne498; residues 1–498) is not only enhanced but also inhibited at various positions (for details, see Table 1). Similarly, the effect of adenosine was determined using supplementary oligonucleotides U27 and (U)13A(U)13 (Fig. 2 and Table 2). To minimize the background, I analyzed the pattern of cleavage at early time-points (specifically shaded in gray boxes at the top of each selected lane), when the original substrate is still in a large excess over the cleavage products, and, therefore, their abundance reflects the efficiency of primary cuts at the corresponding locations of the full-length oligonucleotide.
Figure 1.


RNase E cleavage patterns of A27 (A), (A)13G(A)13 (B), (A)13C(A)13 (C) and (A)13U(A)13 (D). Each 5′ labeled substrate (∼1 pmol) was incubated without (control) or with affinity-purified RNase E (Rne498, residues 1–498) and aliquots withdrawn at times indicated above each lane were analyzed on 15% (w/v) polyacrylamide sequencing-type gels that were run for 3 (short run) or 4.5 (long run) h. The nucleotides at which the inhibition of RNase E cleavages takes place are shown by asterisks and also in red in the corresponding nucleotides within the portion of the cleaved sequence shown at the bottom of the corresponding panel. Also indicated are inter-nucleotide bonds that are moderately (open triangles) or highly (closed triangles) sensitive to RNase E cleavage. Specific time-points at the top of each panel that indicate lanes selected for analysis of the cleavage pattern are shaded in gray boxes. Lane S1, 1 nt ladders generated by partial digestion of each oligonucleotide with S1 nuclease.
Table 1. The experimentally determined variations in the nucleotide composition of RNase E cleavage sites.
| Nucleotide | N–5 | N–4 | N–3 | N–2 | N–1 | N+1 | N+2 | N+3 | N+4 | N+5 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| G | +/– | – | +/– | + | +/– | – | – | – | – | – | |
| C | – | +/– | +/– | – | – | – | + | – | +/– | +/– | |
| U | – | – | +/– | – | +/– | Cleavage | +/– | + | – | – | – |
| A | +/– | +/– | +/– | +/– | +/– | +/– | – | – | +/– | +/– | |
| G,A | C,A | Any | G | G,U,A | A,U | C,U | Any | C,A | C,A |
Depending on their relative location to the scissile bond, uridines (U), guanosines (G), adenosines (A) and cytosines (C) have either negative (–), positive (+) or neutral (+/–) effects on the efficiency of cleavage (Fig. 1). The locations which are downstream and upstream of the point of cleavage are referred to as N+1 to N+5 and N–5 to N–1, respectively. The optimal position of each nucleotide within a cleavage site is presented in the form of a partly degenerated sequence shown in the last row of the table. Several nucleotides of this sequence that confer enhancement of cleavage are shown in bold.
Figure 2.

RNase E cleavage patterns of U27 (A) and (U)13A(U)13 (B). Each 5′ labeled substrate (∼1 pmol) was incubated without (control) or with affinity-purified RNase E (Rne498, residues 1–498) and aliquots withdrawn at times indicated above each lane were analyzed on 15% (w/v) polyacrylamide sequencing-type gels that were run for 3 (short run) or 4.5 (long run) h. The nucleotides at which the inhibition of RNase E cleavages takes place are shown by asterisks and also in red in the corresponding nucleotides within the portion of the cleaved sequence shown at the bottom of the corresponding panel. Also indicated are inter-nucleotide bonds that are moderately (open triangles) or highly (closed triangles) sensitive to RNase E cleavage. Specific time-points at the top of each panel that indicate lanes selected for analysis of the cleavage pattern are shaded in gray boxes. Lane S1, 1 nt ladders generated by partial digestion of each oligonucleotide with S1 nuclease.
Table 2. Some of the well characterized RNase E cleavage sites that are identical both in vitro and in vivo.
| Origin | 10 nt sequence bracketing the point of cleavage (5′ to 3′) | Reference |
|---|---|---|
| pBR322 RNAI | ACAGU⇓AUUUG | (21) |
| pACYC RNAI | ACAAG⇓UUUUG | (21) |
| 9S rRNA precursor (site a) | ACAGA⇓AUUUG | (16) |
| 9S rRNA precursor (site b) | AUCAA⇓AUAAA | (16) |
| ompA mRNA (site C) | GAAGG⇓AUUUA | (11) |
| T4 gene 32 mRNA | UGCGA⇓AUUAU | (20) |
| rpsT mRNA | UAAGC⇓ACAAC | (18) |
| AACCG⇓A·UCGU | ||
| rpsO mRNA | ACUAG⇓AAAAA | (31) |
| AUGGU⇓UUCUC | ||
| GCGAG⇓UUUCA | ||
| pre-M1 RNA | ACCUG⇓AUUUA | (4) |
The length of regions flanking RNase E sites affects the efficiency of cleavages
The cleavage patterns of A27, (A)13U(A)13, (A)13C(A)13 and (A)13G(A)13 (Fig. 1) as well as U27 and (U)13A(U)13 oligomers (Fig. 2) revealed that RNase E progressively cleaves these substrates up to the point when the length of the upstream cleavage product reaches the size of a nonamer (Fig. 1) or an octamer (Fig. 2), respectively. The accumulation of these products and their resistance to cleavage [‘end-proximity effects’ (26)] suggest that the inter-nucleotide bonds of the oligomers are differentially accessible to the nucleolytic activity of RNase E. Cleavages occur at positions 9–23 (Fig. 1) or 8–22 (Fig. 2) but are hardly detectable at the 5′ end (bases 1–8, Fig. 1, or bases 1–7, Fig. 2) and at the very 3′ end (bases 24–27, Fig. 1, or bases 23–27, Fig. 2) of the substrates.
Further validation of the assay
The result of the assay (Fig. 1) strongly supports our previous idea (19) that a G nucleotide, which is positioned upstream and in close proximity to the scissile bond, can induce efficient cleavage. The previously documented enhancement of RNase E cleavage of the 9SB oligo (5′-AUCAAAUAAA-3′) caused by the substitution of the A nucleotide at position 4 by a G nucleotide (19) is, in fact, due to guanosine per se, and does not occur when the A at position 4 was replaced by a C or U nucleotide (Fig. 3B). Furthermore, this effect is context dependent. For example, the enhancement of RNase E cleavages is observed when a G nucleotide was placed in positions 4, 7 and partly 6, but not when it is in position 5 (Fig. 4B). Among the 4G, 5G, 6G and 7G derivatives of 9SB, the oligonucleotides 4G and 7G show the shortest length of the 5′ or 3′ sequences adjacent to the scissile bonds, and yet they are relatively well cleaved by RNase E. The intermediate position of 5G and 6G (in terms of the expected length of the 5′ and 3′ sequences bracketing their putative points of cleavage), therefore, suggest that they should be even less dependent on the ‘end-proximity effects’ (see above) than 4G and 7G. In other words, under precisely the same conditions and, provided that they have adequate similarity to the pattern shown in the last row of Table 1, 5G and 6G should be at least as good substrates as 4G and 7G. In contrast, 5G and 6G are relatively resistant (in comparison with 4G and 7G) to the nucleolytic activity of the enzyme, thereby suggesting that the presence of certain nucleotide(s) in their sequence negatively affects the efficiency of cleavage (for details, see Discussion).
Figure 3.

Mutational analysis of the importance of a G positioned 2 nt upstream from bonds cleaved by RNase E. To compare the efficiency of RNase E cleavage, 5′ labeled mutant variants (A) of the oligoribonucleotide 9SB, which is known to be cleaved by RNase E 10-fold slower than its mutant 4G (19), were incubated without or with affinity-purified Rne498, and aliquots withdrawn at times indicated above each lane were analyzed in a 20% (w/v) polyacrylamide sequencing-type gel. Lane S1, 1 nt ladders generated by partial digestion of the corresponding oligonucleotide.
Figure 4.
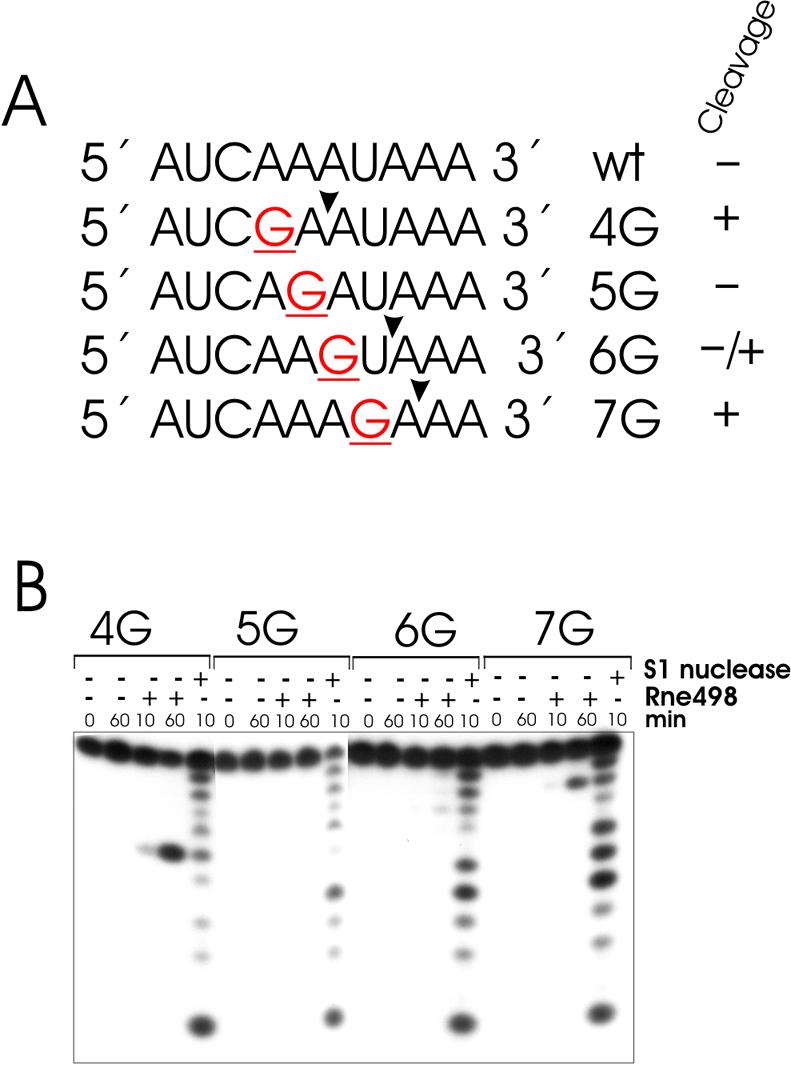
Effects of guanosine substitutions on RNase E cleavage of 9SB-derived oligonucleotides. Mutant variants of the wild-type decaoligonucleotide 9SB (A) (19) that contain single-base substitutions at positions 4, 5, 6 or 7, respectively, were synthesized and used in the RNase E cleavage assay (B). Each 5′ labeled oligoribonucleotide (∼1 pmol) was incubated with (+) or without (–) affinity-purified RNase E (Rne498, residues 1–498) and aliquots at time-points indicated at the top of each lane were withdrawn and analyzed in 20% (w/v) polyacrylamide sequencing-type gel. Lane S1, 1 nt ladders generated by partial digestion of the corresponding oligonucleotide.
This idea is in agreement with the finding that other nucleotides such as U and C occurring at certain locations of the cleavage site (for details, see Table 1) differentially affect the efficiency and specificity of RNase E cleavages, and is further supported by additional data illustrating the effects of double substitutions (Fig. 5). I found that replacement of the A at position 16 in (A)13G(A)13 with a U increases the specificity and efficiency of cleavages (Fig. 5A and C, respectively). On the other hand, substitution of the A at position 15 in (A)13G(A)13 with a C reduces the cleavages at positions 12, 14, 15 and 16 (Fig. 5B and C), which is fully consistent with the negative effect of cytosines when they are placed in certain locations relative to scissile bonds (Fig. 1C and Table 1). Likewise, the G at position 14 inhibits RNase E cleavage at position 13 (Fig. 5B), thereby counterbalancing the expected increase in the efficiency of cleavage, which should be observed because of the C at position 15 (compare Figs 5B and 1C).
Figure 5.


Effects of double substitutions on the specificity and efficiency of RNase E cleavages. RNase E cleavage patterns of BR27 (A) and GC27 (B). Each 5′ labeled substrate (∼1 pmol) was incubated without (control) or with affinity-purified RNase E (Rne498, residues 1–498) and aliquots withdrawn at times indicated above each lane were analyzed on 15% (w/v) polyacrylamide sequencing-type gels that were run for 3 (short run) or 4.5 (long run) h. The nucleotides at which the inhibition of RNase E cleavages takes place are shown by asterisks and also in red in the corresponding nucleotides within the portion of the cleaved sequence shown at the bottom of the corresponding panel. Also indicated are inter-nucleotide bonds that are moderately (open triangles) or highly (closed triangles) sensitive to RNase E cleavage. Specific time-points at the top of each panel that indicate lanes selected for analysis of the cleavage pattern are shaded in gray boxes. Lane S1, 1 nt ladders generated by partial digestion of each oligonucleotide with S1 nuclease. Longer exposure of the same ladder is indicated by an asterisk. (C) Relative efficiency of RNase E cleavage of A27, (A)13G(A)13, BR27 and GC27 derived by phosphorimaging of the gels shown in (A) and (B) of Figures 1 and 5, respectively. The graph is plotted as a function of time and demonstrates that higher similarity to the proposed motif results in faster cleavage of the corresponding substrate.
DISCUSSION
Previous work has shown that the use of short oligonucleotides corresponding in sequence to the single-stranded segments of their cognate RNAs and devoid of their natural flanking stem–loop structures can be used to determine the contribution of individual nucleotides within these segments to the efficiency and specificity of RNase E cleavages. However, the use of a conventional oligonucleotide-based approach to determine the optimal location of each particular nucleotide within RNase E cleavage sites would require the synthesis and the testing of a myriad of oligonucleotide substrates, thus making the task unbearable. To greatly simplify and accelerate the analysis, I proposed an alternative strategy that enables one to achieve the same goal by using only four specific oligonucleotide substrates. This strategy is based on the property of RNase E and other site-specific ribonucleases to randomly cleave homo-oligomers such as oligo(A) (25,27,28), oligo(U) (25,27,28), oligo(C) (27) or even oligo(I) (29). Substitution of a single base in one of these oligomers would therefore allow one to examine the effect of this nucleotide at multiple locations relative to the scissile bond.
To test this idea, I used derivatives of the homo-oligomers oligo(A) and oligo(U) to probe the motif which is specifically recognized by the catalytic domain of E.coli RNase E (amino acid residues 1–498) (6). By analyzing the cleavage patterns of these oligonucleotides (for details, see Table 1), it is possible to deduce the following, partly degenerated motif: (G,A) (C,A)N(G)(G,U,A) ⇓ (A,U)(C,U)N(C,A)(C,A). This motif, for example, suggests that a G, but not a A, a C or a U, at position –2 can significantly enhance the efficiency of cleavage reinforcing our previous observation (19) that a G nucleotide which is 5′ and in close vicinity to the scissile bond is an important determinant of cleavage. This conclusion is also supported by the experimental data shown in Figure 3. As anticipated, however, the effect of guanosine is context dependent (Fig. 4), and can therefore be insignificant when other nucleotides within the same sequence exert strong inhibitory effects. For instance, when the G of oligonucleotide 5G is in the most optimal location to the scissile bond (i.e. in position N–2, see Table 1), position N–5 is inevitably occupied by a U. However, any U in position N–5 should negatively affect the efficiency of cleavage (Table 1), thereby resulting in higher resistance to the nucleolytic activity of the enzyme as illustrated for oligonucleotide 5G in Figure 4. Likewise, the observed effects of double substitutions within A27 (Fig. 5A and B) are fully consistent with the individual contribution of nucleotides predicted from their location within the recognition motif (Table 1). Moreover, the higher the similarity to this motif the faster the cleavage of the corresponding oligonucleotide takes place (Fig. 5C).
There is a high homology between the well characterized RNase E cleavage sites selected on the basis of their identity in vitro and in vivo (Table 2) and the proposed recognition motif. Nevertheless, the enhancement of cleavage at some of these sites [see site b in 9S RNA (19)] may be mediated by an interaction of the adjacent regions of secondary structure with the centrally located arginine-rich domain of the enzyme, and, therefore, may not necessarily require a high similarity to this motif.
My experimental data and others (26) also show that the catalytic domain of RNase E senses the continuity of single-stranded regions bracketing the sites of cleavage. The requirement of at least 3–4 nt 3′ to the point of cleavage (Figs 1 and 2) is not surprising, as this region is part of the recognition motif. As to the lengthy 5′ segment, one can envisage that the extension of this region beyond the recognition motif may be required to enhance the affinity to the catalytic domain via an interaction with the so-called S1-like RNA-binding domain of the enzyme (30). However, given that 10 nt substrates (19,21,28) can still be cleaved by RNase E efficiently and, in fact, faster than homo-oligomers (28), the contribution of this interaction seems to be essential only for cleavages that occur within regions with rather low similarity to the consensus motif [e.g. poly(A) or poly(U) stretches], i.e. in the case of regions with relatively low affinity to the catalytic site of the enzyme.
In conclusion, giving the important role of RNase E cleavages in post-transcriptional control of gene expression, the knowledge of the specific sequence determinants not only allows the fine regulation of RNA stability by means of modification of RNase E cleavage sites but also adds to the understanding of how different elements of RNA structure and RNase E functional domains individually contribute to the efficiency and specificity of RNase E cleavages. Finally, the assay I propose is simple, fast and potentially applicable for the analysis of other site-specific RNases.
Acknowledgments
ACKNOWLEDGEMENTS
I thank Drs Udo Bläsi and Isabella Moll for critical reading of the manuscript. This work was supported by grant no. F1707 from the Austrian Science Foundation.
REFERENCES
- 1.Melefors O., Lundberg,U. and von Gabain,A. (1993) RNA processing and degradation by RNase K and RNase E. In Belasco,J. and Brawerman,G. (eds), Control of Messenger RNA Stability. Academic Press, San Diego, CA, pp. 53–70. [Google Scholar]
- 2.Misra T.K. and Apirion,D. (1979) RNase E, an RNA processing enzyme from Escherichia coli. J. Biol. Chem., 254, 11154–11159. [PubMed] [Google Scholar]
- 3.Li Z. and Deutscher,M.P. (2002) RNase E plays an essential role in the maturation of Escherichia coli tRNA precursors. RNA, 8, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sim S., Kim,K.S. and Lee,Y. (2002) 3′-End processing of precursor M1 RNA by the N-terminal half of RNase E. FEBS Lett., 529, 225–231. [DOI] [PubMed] [Google Scholar]
- 5.Lin-Chao S., Wei,C.L. and Lin,Y.T. (1999) RNase E is required for the maturation of ssrA RNA and normal ssrA RNA peptide-tagging activity. Proc. Natl Acad. Sci. USA, 96, 12406–12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McDowall K.J. and Cohen,S.N. (1996) The N-terminal domain of the rne gene product has RNase E activity and is non-overlapping with the arginine-rich RNA-binding site. J. Mol. Biol., 255, 349–355. [DOI] [PubMed] [Google Scholar]
- 7.Kaberdin V.R., Miczak,A., Jakobsen,J.S., Lin-Chao,S., McDowall,K.J. and von Gabain,A. (1998) The endoribonucleolytic N-terminal half of Escherichia coli RNase E is evolutionarily conserved in Synechocystis sp. and other bacteria but not the C-terminal half, which is sufficient for degradosome assembly. Proc. Natl Acad. Sci. USA, 95, 11637–11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leroy A., Vanzo,N.F., Sousa,S., Dreyfus,M. and Carpousis,A.J. (2002) Function in Escherichia coli of the non-catalytic part of RNase E: role in the degradation of ribosome-free mRNA. Mol. Microbiol., 45, 1231–1243. [DOI] [PubMed] [Google Scholar]
- 9.Taraseviciene L., Bjork,G.R. and Uhlin,B.E. (1995) Evidence for an RNA-binding region in the Escherichia coli processing endoribonuclease RNase E. J. Biol. Chem., 270, 26391–26398. [DOI] [PubMed] [Google Scholar]
- 10.Vanzo N.F., Li,Y.S., Py,B., Blum,E., Higgins,C.F., Raynal,L.C., Krisch,H.M. and Carpousis,A.J. (1998) Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. Genes Dev., 12, 2770–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carpousis A.J., Van Houwe,G., Ehretsmann,C. and Krisch,H.M. (1994) Copurification of E. coli RNase E and PNPase: evidence for a specific association between two enzymes important in RNA processing and degradation. Cell, 76, 889–900. [DOI] [PubMed] [Google Scholar]
- 12.Miczak A., Kaberdin,V.R., Wei,C.L. and Lin-Chao,S. (1996) Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc. Natl Acad. Sci. USA, 93, 3865–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Py B., Higgins,C.F., Krisch,H.M. and Carpousis,A.J. (1996) A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature, 381, 169–172. [DOI] [PubMed] [Google Scholar]
- 14.Lin-Chao S. and Cohen,S.N. (1991) The rate of processing and degradation of antisense RNAI regulates the replication of ColE1-type plasmids in vivo. Cell, 65, 1233–1242. [DOI] [PubMed] [Google Scholar]
- 15.Ehretsmann C.P., Carpousis,A.J. and Krisch,H.M. (1992) mRNA degradation in procaryotes. FASEB J., 6, 3186–3192. [DOI] [PubMed] [Google Scholar]
- 16.Cormack R.S. and Mackie,G.A. (1992) Structural requirements for the processing of Escherichia coli 5 S ribosomal RNA by RNase E in vitro. J. Mol. Biol., 228, 1078–1090. [DOI] [PubMed] [Google Scholar]
- 17.Melefors O. and von,G.A. (1988) Site-specific endonucleolytic cleavages and the regulation of stability of E. coli ompA mRNA. Cell, 52, 893–901. [DOI] [PubMed] [Google Scholar]
- 18.Mackie G.A. and Genereaux,J.L. (1993) The role of RNA structure in determining RNase E-dependent cleavage sites in the mRNA for ribosomal protein S20 in vitro. J. Mol. Biol., 234, 998–1012. [DOI] [PubMed] [Google Scholar]
- 19.Kaberdin V.R., Walsh,A.P., Jakobsen,T., McDowall,K.J. and von Gabain,A. (2000) Enhanced cleavage of RNA mediated by an interaction between substrates and the arginine-rich domain of E. coli ribonuclease E. J. Mol. Biol., 301, 257–264. [DOI] [PubMed] [Google Scholar]
- 20.Ehretsmann C.P., Carpousis,A.J. and Krisch,H.M. (1992). Specificity of Escherichia coli endoribonuclease RNase E: in vivo and in vitro analysis of mutants in a bacteriophage T4 mRNA processing site. Genes Dev., 6, 149–159. [DOI] [PubMed] [Google Scholar]
- 21.McDowall K.J., Kaberdin,V.R., Wu,S.W., Cohen,S.N. and Lin-Chao,S. (1995) Site-specific RNase E cleavage of oligonucleotides and inhibition by stem–loops. Nature, 374, 287–290. [DOI] [PubMed] [Google Scholar]
- 22.Lin-Chao S., Wong,T.T., McDowall,K.J. and Cohen,S.N. (1994) Effects of nucleotide sequence on the specificity of rne-dependent and RNase E-mediated cleavages of RNA I encoded by the pBR322 plasmid. J. Biol. Chem., 269, 10797–10803. [PubMed] [Google Scholar]
- 23.McDowall K.J., Lin-Chao,S. and Cohen,S.N. (1994) A+U content rather than a particular nucleotide order determines the specificity of RNase E cleavage. J. Biol. Chem., 269, 10790–10796. [PubMed] [Google Scholar]
- 24.Hochuli E., Dobeli,H. and Schacher,A. (1987) New metal chelate adsorbent selective for proteins and peptides containing neighbouring histidine residues. J. Chromatogr., 411, 177–184. [DOI] [PubMed] [Google Scholar]
- 25.Huang H.J., Liao,J. and Cohen,S.N. (1998). Poly(A)- and poly(U)-specific RNA 3′ tail shortening by E. coli ribonuclease E. Nature, 391, 99–102. [DOI] [PubMed] [Google Scholar]
- 26.Walsh A.P., Tock,M.R., Mallen,M.H., Kaberdin,V.R., von Gabain,A. and McDowall,K.J. (2001) Cleavage of poly(A) tails on the 3′-end of RNA by ribonuclease E of Escherichia coli. Nucleic Acids Res., 29, 1864–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savkur R.S. and Olson,M.O. (1998) Preferential cleavage in pre-ribosomal RNA by protein B23 endoribonuclease. Nucleic Acids Res., 26, 4508–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tock M.R., Walsh,A.P., Carroll,G. and McDowall,K.J. (2000) The CafA protein required for the 5′-maturation of 16 S rRNA is a 5′-end-dependent ribonuclease that has context-dependent broad sequence specificity. J. Biol. Chem., 275, 8726–8732. [DOI] [PubMed] [Google Scholar]
- 29.Przewlocki G., Lipecka,J., Edelman,A. and Przykorska,A. (1998) New sequence-specific human ribonuclease: purification and properties. Nucleic Acids Res., 26, 4047–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bycroft M., Hubbard,T.J.P., Proctor,M., Freund,S.M.V. and Murzin,A.G. (1997) The solution structure of the S1 RNA binding domain: a member of an ancient nucleic acid-binding fold. Cell, 88, 235–242. [DOI] [PubMed] [Google Scholar]
- 31.Braun F., Hajnsdorf,E. and Regnier,P. (1995) Polynucleotide phosphorylase is required for the rapid degradation of the RNaseE-processed rpsO messenger RNA of Eschericha coli devoid of its 3′ hairpin. Mol. Microbiol., 19, 997–1005. [DOI] [PubMed] [Google Scholar]