

Summary
Growth factor-dependent accumulation of the cyclin D1 proto-oncogene is balanced by its rapid phosphorylation-dependent proteolysis. Degradation is triggered by threonine 286 phosphorylation, which promotes its ubiquitination by an unknown E3 ligase. We demonstrate that Thr286 phosphorylated cyclin D1 is recognized by a SCF ubiquitin ligase where FBX4 and αB crystallin govern substrate specificity. Overexpression of FBX4 and αB crystallin triggered cyclin D1 ubiquitination and increased cyclin D1 turnover. Impairment of SCFFBX4-αBcrystallin function attenuated cyclin D1 ubiquitination, promoting cyclin D1 overexpression and accelerated cell cycle progression. Purified SCFFBX4-αBcrystallin catalyzed polyubiquitination of cyclin D1 in vitro. Consistent with a putative role for a cyclin D1 E3 ligase in tumorigenesis, FBX4 and αB crystallin expression was reduced in tumor-derived cell lines and a subset of primary human cancers that overexpress cyclin D1. We conclude that SCFFBX4-αB crystallin is an important E3 ubiquitin ligase that promotes ubiquitin-dependent degradation of Thr286 phosphorylated cyclin D1.
Keywords: cyclin D1, CDK4, FBX4, αB crystallin, E3 ubiquitin ligase
INTRODUCTION
Cyclin D1, the allosteric regulator of CDK4/6, is an integral mediator of growth factor-dependent G1 phase progression. Cyclin D1 expression, activation and nuclear accumulation occur during mid-G1 phase(Marshall, 1999); nuclear cyclin D1/CDK complexes trigger G1 progression via inactivation of the retinoblastoma protein and related family members (Sherr, 1996). During S-phase, cyclin D1 activation is opposed by Pro287-directed phosphorylation of Thr286 by glycogen synthase kinase 3β (GSK3β); phosphorylation triggers cyclin D1 nuclear export and ubiquitination-mediated proteolysis (Alt et al., 2000).
While phosphorylation of Thr286 directs CRM1 binding to cyclin D1, it is also an essential signal for ubiquitin-dependent destruction (Diehl et al., 1997). Ubiquitination is catalyzed through a pathway involving the E1 (ubiquitin activating enzyme), E2 (ubiquitin conjugating enzyme), and an E3 (ubiquitin ligase). The latter enzyme is the most diverse and it determines substrate-specificity and the rate of ubiquitin conjugation. The E3 itself can be either a single protein or a multi-protein complex. An E3 that recognizes Thr286 phosphorylated cyclin D1 has not been identified.
Regulated proteolysis is commonly utilized to maintain threshold levels of critical cell cycle regulators. Two distinct classes of E3 ligases participate in the regulation of cell cycle progression. The Skp1-Cul1-F-box (SCF) family of E3 ubiquitin ligases promote ubiquitination of phosphorylated substrates and typically target mediators of the G1-S phase transition (Skowyra et al., 1997). While the F-box component determines substrate specificity, additional co-factors may also facilitate substrate recognition (Ganoth et al., 2001; Hao et al., 2005; Spruck et al., 2001). The G2-M transition requires the E3 ligase activity of the Anaphase Promoting Complex (APC/C) (Harper et al., 2002); CDC20 and CDH1 subunits of APC/C direct substrate recognition (Burton et al., 2005; Kraft et al., 2005; Pfleger et al., 2001).
Cyclin D1 is frequently overexpressed in human cancer; overexpression is often associated with increased gene expression due to gene amplification or oncogene induced signaling (Sherr, 1996). Accumulation of cyclin D1 in cancer can also result from disruption of cyclin proteolysis. Cyclin D1 proteolysis can be inhibited through several distinct mechanisms. First, recent work revealed that human cancer cells harbor mutations in cyclin D1 that disrupt Thr286 phosphorylation (Benzeno, 2006) and prevent ubiquitin-mediated proteolysis. Second, upstream oncogenic events, such as those targeting Wnt or Ras inhibit GSK3β activity thereby decreasing cyclin D1 turnover (Diehl et al., 1998; Rimerman et al., 2000). Finally, it remains plausible that loss of a D1-specific E3 ligase might contribute to D1 overexpression in human cancer. The latter mechanism emphasizes the importance of identifying the cyclin D1-specific E3 ligase. Given the necessity of Thr286 phosphorylation for cyclin D1 proteolysis (Diehl et al., 1997), an unidentified SCF complex is a likely candidate cyclin D1 ubiquitin ligase.
We have identified a SCF E3 ligase, wherein the F-box protein, FBX4, targets Thr286 phosphorylated cyclin D1. FBX4 belongs to the Fbx class of F-box proteins, which lack known structural motifs in their C-terminus. Cyclin D1 recognition by FBX4 requires αB crystallin which functions in a manner analogous to CKS1 in Skp2-dependent recognition of phosphorylated p27Kip1. The data presented reveal that both FBX4 and αB crystallin are required for rapid ubiquitination and degradation of cyclin D1 in vivo and that purified SCFFBX4-αB crystallin complexes can direct cyclin D1 ubiquitination in vitro. Strikingly, analysis of breast cancer-derived cells revealed loss αB crystallin in a subset of cell lines. Loss of αB crystallin resulted in cyclin D1 stabilization revealing the requirement of the SCFFBX4-αB crystallin ligase for cyclin D1 proteolysis.
RESULTS
Identification of αB crystallin as a major component of cyclin D1 complexes
To identify a putative cyclin D1 E3 ubiquitin ligase, we purified cyclin D1-complexes by affinity chromatography. Due to the short half-life of phosphorylated cyclin D1, cells were treated with proteasome inhibitors to enrich for phospho-T286 cyclin D1 and stabilize interactions with a putative E3 ligase. A major protein of ~22–23 kDa (identified by mass spectrometric analysis as αB crystallin) co-purified with Flag-cyclin D1, but not from parental NIH3T3 cells (Fig. 1A) or with a non-degradable cyclin D1 mutant (data not shown). We also noted a protein of ~46Kd that co-purified with cyclin D1; the identity of this protein could not be confirmed due to contaminating actin peptides. Association between endogenous cyclin D1 and αB crystallin under conditions of proteasome inhibition was confirmed by precipitation with a cyclin D1 specific antibody followed by immunoblot with αB crystallin antibodies (Fig. 1B). Binding of cyclin D1-αB crystallin depended upon the integrity Thr286 since the cyclin D1T286A mutant did not co-precipitate with αB crystallin (Fig. 1C).
Fig. 1. Identification of αB crystallin as a cyclin D1-associated protein.
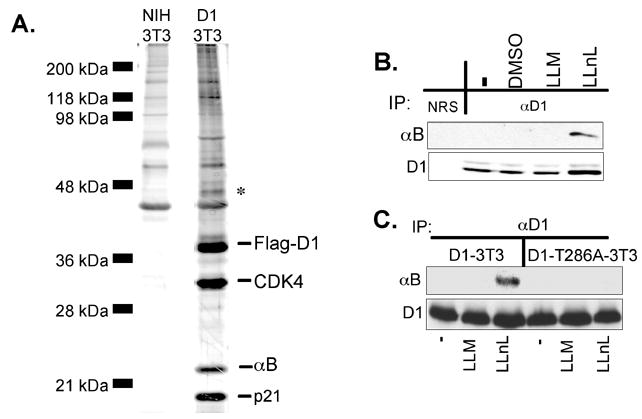
(A) Silver stain of affinity-purified cyclin D1 complexes prepared from parental NIH3T3 cells or Flag-D1 3T3 cell lines treated with the proteasome inhibitor, LLnL (leucyl-leucyl-norleucinal). Cyclin D1-interacting proteins were identified by mass spectrometry. (B) Western blot of endogenous cyclin D1 precipitates prepared from proliferating NIH3T3 cells treated with DMSO, LLM (N-acetyl-leucinyl-leucinyl-methioninal) or LLnL. Normal rabbit serum (NRS) precipitates served as a negative control. (C) The cyclin D1-αB crystallin interaction is dependent on cyclin D1 phosphorylation at Thr286. NIH3T3 cells expressing either wild-type cyclin D1 or the D1T286A mutant were treated with vehicle, LLM or LLnL for 6hrs and cell lysates prepared from these cells were precipitated with a cyclin D1 specific antibody (13-17G) and probed for cyclin D1 and αB crystallin.
Phospho-T286 dependent interaction between cyclin D1 and the SCFFBX4-αB crystallin ligase
αB crystallin associates with a 46 KD protein, FBX4, in vivo and increases ubiquitin-conjugation of unknown proteins (den Engelsman et al., 2003). We therefore considered whether αB crystallin might facilitate targeting of FBX4 to phosphorylated cyclin D1. Indeed, endogenous cyclin D1 and αB crystallin do co-precipitate with FBX4 (Fig. 2A and Fig. S1A). The binding of cyclin D1 to FBX4 and αB crystallin was observed only under conditions of proteasome inhibition likely reflecting the stabilization labile complexes.
Fig. 2. Phosphorylation-dependent interaction between cyclin D1 and the SCFFBX4-αB crystallin complex.
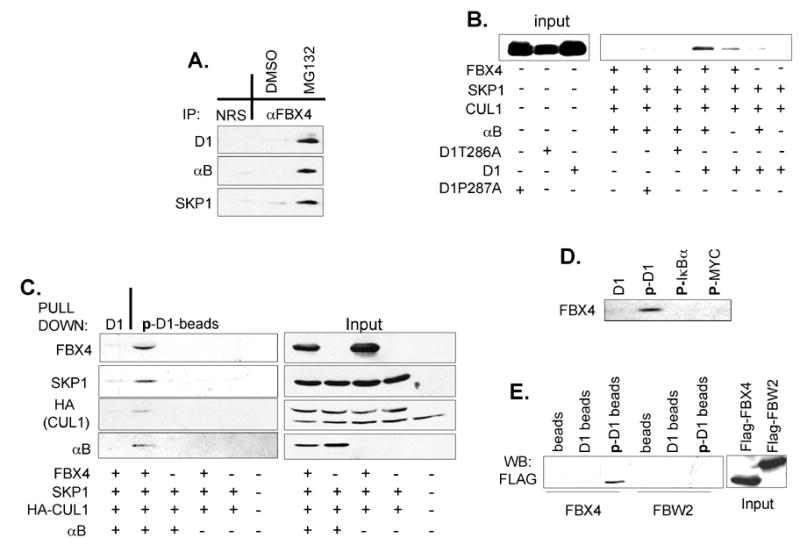
(A) Endogenous FBX4, cyclin D1 and αB crystallin associate in vivo. Cell lysates prepared from proliferating NIH3T3 cells treated with DMSO or MG132 were precipitated with antibodies directed against FBX4 or normal rabbit serum (NRS) and subjected to immunoblot with the indicated antibodies. (B) Phospho-T286-dependent binding of cyclin D1 to SCFFBX4-αB crystallin in vitro. Cyclin D1 or phosphorylation-deficient derivatives, D1T286A and D1P287A, were mixed with SCFFBX4-αB crystallin complexes and purified by immuno-affinity chromatography from 293T cells; where indicated, FBX4 and αB crystallin were omitted. (C) Cyclin D1-FBX4 binding is dependent on αB crystallin and Thr286 phosphorylation. 293T lysates containing the indicated proteins were incubated with beads coupled to the phosphorylated cyclin D1 peptide or to unphosphorylated cyclin D1 peptide. Bound proteins were detected by immunoblot. (D) Phospho-MYC and -IκKα do not interact with SCFFBX4-αB crystallin. Beads coupled to the indicated peptides were incubated with 293T lysates expressing SCFFBX4-αB crystallin complexes as in (C) and bound FBX4 was detected by immunoblot. (E) Beads coupled to the indicated peptides were incubated with 293T lysates harboring either SCFFBX4-αB crystallin or SCFFBW2 complexes and bound FBX4 or FBW2 were detected by anti-FLAG western blot. Uncoupled beads served as negative (background) control.
To determine whether Thr286 phosphorylation is required for recognition by the SCFFBX4-αBcrystallin complex, wild type cyclin D1 or phosphorylation deficient cyclin D1 mutants produced in SF9 insect cells (cyclin D1 is readily phosphorylated at Thr286 in Sf9 cells (Diehl et al., 1997)) was mixed with immobilized SCFFBX4-αB crystallin complexes produced in 293T cells. Wild-type cyclin D1 bound to the SCFFBX4-αB crystallin complex in vitro; omission of αB crystallin inhibited cyclin D1 binding to FBX4 (Fig. 2B). Binding of SCFFBX4-αB crystallin to two phosphorylation-deficient D1T286A and D1P287A (Benzeno, 2006) mutants was not observed (Fig. 2B). These results demonstrate that FBX4 binds to cyclin D1 in a phospho-Thr286 dependent manner and that αB crystallin increases the affinity of cyclin D1 for the SCFFBX4 complex.
The role of Thr286 phosphorylation in mediating recognition by FBX4-αB crystallin, SKP1 and CUL1 complexes was determined using a phospho-T286 peptide corresponding to the C-terminus of cyclin D1. FBX4-αB crystallin-SKP1-CUL1 complexes specifically associated with phospho-T286 peptides, but not to unphosphorylated peptides. Omission of either FBX4 or αB crystallin eliminated binding of SCFFBX4-αBcrystallin to the phospho-D1 peptides (Fig. 2C). FBX4 did not bind to other phosphodegrons (i.e. phospho-IκBα or phospho-MYC peptides) excluding the possibility that the SCFFBX4-αB crystallin complex simply has an affinity for phosphorylated Ser/Thr residues (Fig. 2D). Additionally, phosphorylated cyclin D1 peptides did not recruit other F-box proteins such as FBW2 (Fig. 2E) or SKP2 (data not shown). These results suggest that both αB crystallin and FBX4 are required for the recognition of phospho-T286 cyclin D1 by the SCFFBX4-αB crystallin complex.
The SCFFBX4-αB crystallin complex regulates cyclin D1 protein stability in vivo
To determine whether the SCFFBX4-αBcrystallin complex regulates cyclin D1 proteolysis, a dominant-negative FBX4 allele was created (ΔF)FBX4. F-box deletion abrogates SKP1 binding (Zheng et al., 2002), but not substrate or αB crystallin recognition (Fig. S1B; data not shown). Dominant-negative F-box mutants have been used to assess substrate relationships (Carrano et al., 1999; Latres et al., 1999; Montagnoli et al., 1999). Expression of (ΔF)FBX4 increased the half-life of endogenous cyclin D1 from <30 minutes to greater than 60 minutes (Fig. 3A). Expression of (ΔF)FBX4 also induced accumulation of cyclin D1 in an αB crystallin-dependent manner, but did not increase cyclin D1-T286A levels (Fig. 3B). The effect of αB crystallin likely reflects the capacity to promote (ΔF)FBX4 binding to phosphorylated cyclin D1 in cells.
Fig. 3. The SCFFBX4-αB crystallin ubiquitin ligase regulates cyclin D1 stability and protein levels in vivo.
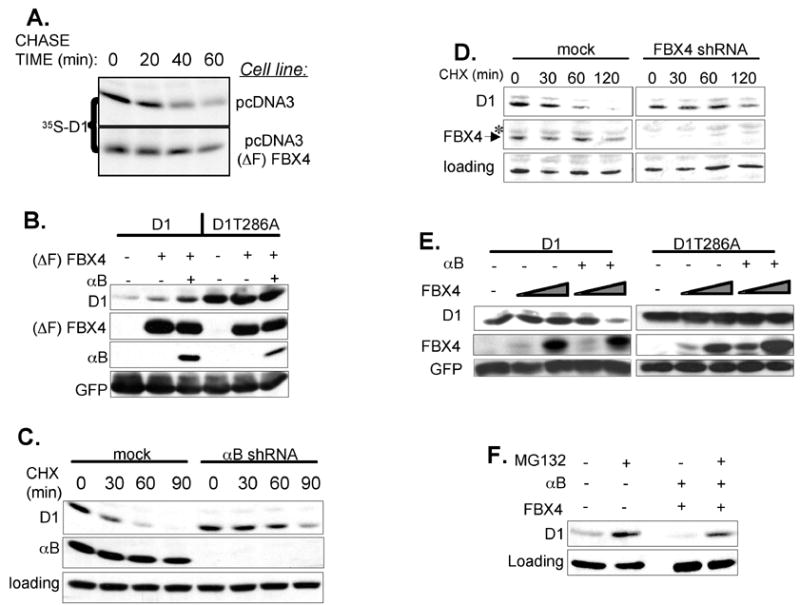
(A) NIH3T3 cells expressing empty vector or (ΔF)FBX4 were pulse-labeled with 35S methionine/cysteine and “chased” for the indicated time periods. Cyclin D1 was precipitated and visualized by autoradiography. (B) Western analysis of whole cell extracts prepared from 293T cells transfected with cyclin D1, CDK4, (ΔF) FBX4 and αB crystallin. (C) (D) NIH3T3 cells expressing either control, αB crystallin (C) or FBX4 shRNAs (D) were treated with 100 μg/mL of cycloheximide for the indicated time periods, and cyclin D1 half-life was determined by western blotting. (E) Expression of FBX4 and αB crystallin promotes cyclin D1 proteolysis. 293T cells were transfected with cyclin D1, CDK4, αB crystallin, FBX4 and GFP. Cell lysates were prepared and immunoblotted for cyclin D1, FBX4 and GFP. (F) Proteasome inhibition inhibits FBX4-dependent cyclin D1 proteolysis.
As independent assessment of FBX4 and αB crystallin function, we utilized short-hairpin vectors to target either component. Knockdown of FBX4 or αB crystallin extended the half-life of cyclin D1 to greater than 60 min whereas the half-life remained <30 minutes in cells expressing control shRNA (Fig. 3C, 3D). Knockdown of FBX4 also stabilized cyclin D2 (data not shown) suggesting that SCFFBX4-αBcrystallin may regulate the proteolysis of all D-type cyclins. Knockdown of FBX4 in human U2OS cells also triggered accumulation of cyclin D1 (Fig. S1C) compared to cells expressing shRNAs directed against another F-box protein, β-TrCP (Jin et al., 2003) demonstrating that FBX4 regulates cyclin D1 proteolysis in mouse and human cells. Conversely, knockdown of FBX4 did not effect cyclin E or A accumulation (Fig. 5C; S1C), which are ubiquitinated by the SCFFBW7 and APC/C complexes respectively (Harper et al., 2002; Koepp et al., 2001).
Fig. 5. Cytoplasmic localization of FBX4 and αB crystallin.
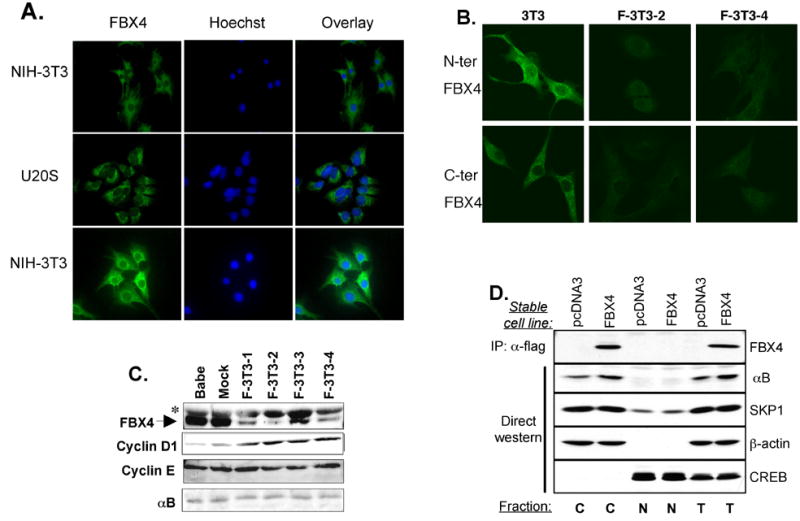
(A) Immunofluorescence of asynchronously growing NIH3T3 and U2OS cells with N-terminus (top) or C-terminus (middle and bottom panels) FBX4-specific antibodies. (B) Immunofluorescence of asynchronously growing mouse fibroblasts stably expressing FBX4-specific hairpins (F-3T3-2, F-3T3-4). (C) Western analysis confirming knockdown of FBX4 in NIH3T3 cells. Asterisk indicates a nonspecific band detected by the secondary antibody. (D) Stable NIH3T3 cells expressing empty vector or Flag-tagged FBX4 were fractionated into cytoplasmic (C), nuclear (N) and total (T) extracts. Fractions were used for immunoprecipitation with an anti-flag antibody or for direct western blots as indicated.
The ability of FBX4 and αB crystallin to drive cyclin D1 proteolysis was also investigated. 293T cells were transfected with cyclin D1, FBX4 and αB crystallin, and cyclin D1 abundance was assessed by immunoblot. Ectopic expression of FBX4 and αB crystallin decreased levels of cyclin D1 in a dose-dependent manner (Fig. 3E). Reduction of cyclin D1 was dependent on Thr286 phosphorylation (Fig. 3E) and proteasome activity (Fig. 3F). These data suggest the SCFFBX4-αBcrystallin ligase regulates cyclin D1 stability and levels in vivo.
The SCFFBX4-αB crystallin ubiquitin ligase catalyzes cyclin D1 ubiquitination
Reduced cyclin D1 turnover in cells expressing either (ΔF) FBX4 or knockdown of FBX4 suggested that FBX4 directs cyclin D1 polyubiquitination. Consistent with this notion, (ΔF)FBX4 expression decreased cyclin D1 polyubiquination in vivo and this effect was potentiated by co-expression of αB crystallin (Fig. 4A, 4B). Similar results were obtained when these reactions were analyzed by immunoblot with either a HA antibody (to detect HA-ubiquitin) or a cyclin D1 antibody (Fig. 4A, 4B) excluding the possibility that these results reflected ubiquitination of co-precipitating proteins. Reduced cyclin D1 ubiquitination was also observed in cells expressing shRNAs directed against FBX4 (Fig. 4C). Thus, impairment of SCFFBX4-αB crystallin function via either (ΔF)FBX4 or FBX4 knockdown stabilizes cyclin D1 as a result of decreased cyclin D1 polyubiquitination.
Fig. 4. The SCFFBX4-αB crystallin complex catalyzes cyclin D1 ubiquitination in vivo and in vitro.
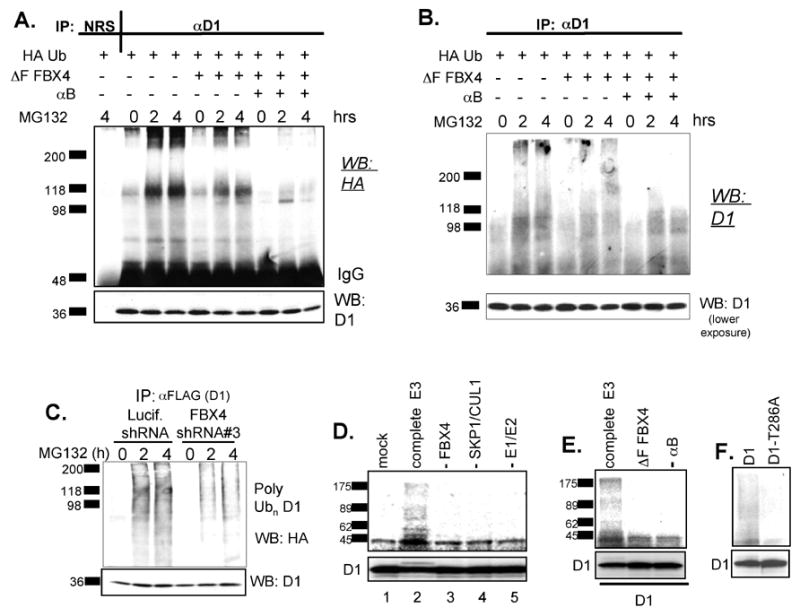
(A) NIH3T3 cells expressing Flag-cyclin D1 were transfected with HA-ubiquitin, (ΔF) FBX4 and αB crystallin and treated with 20 μM MG132 for 0, 2 and 4 hours. Cell lysates were subjected to precipitation with either normal rabbit serum (NRS) or with a cyclin D1 specific antibody and ubiquitinated proteins were visualized by anti-HA immunoblot. (B) Same as in (A) except that cyclin D1 immunoprecipitates were probed with a cyclin D1 antibody. (C) Human U2OS cells were co-transfected with HA-tagged ubiquitin, cyclin D1 and shRNAs specific for firefly luciferase or FBX4. Cyclin D1 ubiquitination was detected by immunoprecipitation with an anti-flag antibody followed by anti-HA western blotting. (D) The SCFFBX4-αB crystallin complex catalyzes ubiquitination of cyclin D1 in vitro. Ubiquitination of in vitro transcribed and translated cyclin D1 was assessed by mixing cyclin D1 with GSK3β ATP, ubiquitin, E1, E2, and a complete SCFFBX4 αBcrystallin complex (lane 2). The reactions were also carried out omitting the following: the entire E3 ligase (lane1), FBX4 (lane 3), Cul1, Skp1, Roc 1(lane 4) or E1, E2 (lane 5). (E) The F-box of FBX4 and αB crystallin are required for cyclin D1 polyubiquitination in vitro. Ubiquitination reactions were initiated by addition of an ATP, ubiquitin, E1 and E2 enzymes. Where indicated wild-type FBX4 was replaced with (ΔF)FBX4 (lane 2), αB crystallin was omitted from the reaction (lane 3). (F) Phosphorylation deficient cyclin D1-T286A is refractory to SCFFBX4-αB crystallin-dependent ubiquitination. Cyclin D1 ubiquitination D-F was assessed by autoradiography (upper panels) and input is shown (lower panels).
In vitro transcribed and translated 35S labeled cyclin D1 was mixed with purified SCFFBX4-αB crystallin complexes to determine whether cyclin D1 was a direct substrate. After addition of ATP, ubiquitin, E1, E2, and purified SCFFBX4-αB crystallin, we detected higher molecular cyclin D1 species consistent with cyclin D1 polyubiquitination (Fig. 4D, lane 2). In the absence of FBX4 (lane 3), Cul1/Skp1/Roc1 (lane 4), or E1/E2 (lane 5), cyclin D1 ubiquitination was eliminated. In addition, a mono-ubiquitinated cyclin D1 form was also seen in our in vitro ubiquitination reactions (Fig. 4D, lane 2, lower panel) but not in control reactions lacking FBX4 (Fig 4D, lanes 1 and 3, lower panel), supporting the role of SCFFBX4-αB crystallin as an E3 ligase for cyclin D1. Replacement of wild-type FBX4 with (ΔF) FBX4 impaired polyubiquitination of cyclin D1 (Fig. 4E, lane 2) demonstrating that ubiquitination depended on Cul1-Skp1-Roc1 and excluding the possibility that ubiquitination was mediated by another contaminating F-box protein. Similarly omission of αB crystallin inhibited cyclin D1 ubiquitination (Fig. 4E, lane 3) demonstrating that αB crystallin is an essential co-factor. Ubiquitinating activity was also dependent on Thr286 phosphorylation of cyclin D1 (Fig. 4F). Similar results were obtained using SCFFBX4-αB crystallin produced in Sf9 cells (data not shown). These results provide direct biochemical evidence that cyclin D1 is a bona fide substrate for SCFFBX4-αB crystallin.
Cytoplasmic localization of FBX4 and αB crystallin
Cyclin D1 ubiquitination and proteolysis are postulated cytoplasmic events (Diehl et al., 1998), suggesting the involvement of a cytoplasmic E3 ligase. Immunofluorescence staining, utilizing two distinct FBX4 antibodies, revealed cytoplasmic FBX4 in both NIH3T3 and U2OS cells (Fig. 5A). Knockdown of FBX4 dramatically reduced staining with either antibody confirming antibody specificity (Fig. 5B, 5C). The cytoplasmic localization was confirmed by biochemical fractionation (Fig. 5D). While our results reveal that FBX4 and αB crystallin are largely cytoplasmic, they do not exclude the possibility that a small fraction of FBX4 and αB crystallin transiently shuttles between nuclear and cytoplasmic compartments (den Engelsman et al., 2004).
Cell cycle dependent binding of FBX4 to cyclin D1
Following re-entry from a G0, cyclin D1 induction is limited by growth factor-dependent gene expression (Matsushime et al., 1991). At the G1/S-phase transition, cyclin D1 is phosphorylated at Thr286 by GSK3β, exported from the nucleus and degraded in the cytoplasm (Diehl et al., 1998). We hypothesized that following G0 release FBX4 would not limit cyclin D1 accumulation until the G1/S phase transition when GSK3β phosphorylates cyclin D1 inducing its cytoplasmic re-distribution. To test this hypothesis, we assessed the cell cycle dependence of cyclin D1-FBX4 binding following G0 release. NIH3T3 fibroblasts were cultured for 24 hrs in medium containing 0.1% FBS and released in medium containing 10% FBS. Lysates were prepared from NIH3T3 cells synchronized at G0 (0hrs), G1 (8hrs), and S (18hrs) phases of the cell cycle. Four hours prior to harvesting, cells were treated with MG132 to enrich for cyclin D1-FBX4 complexes. Immunoblot of FBX4 precipitates revealed enrichment of cyclin D1-FBX4 complexes during S-phase following cell cycle reentry (Fig. 6A). To assess the dependence of cyclin D1-FBX4 interaction on GSK3β, we transfected NIH3T3 cells with dominant negative GSK3β or empty vector and assessed binding. Expression of this GSK3β mutant dramatically reduced cyclin D1-FBX4 binding (Fig. 6B) as well as cyclin D1 phosphorylation (data not shown).
Fig. 6. The SCFFBX4-αB crystallin ubiquitin ligase regulates cell cycle progression.
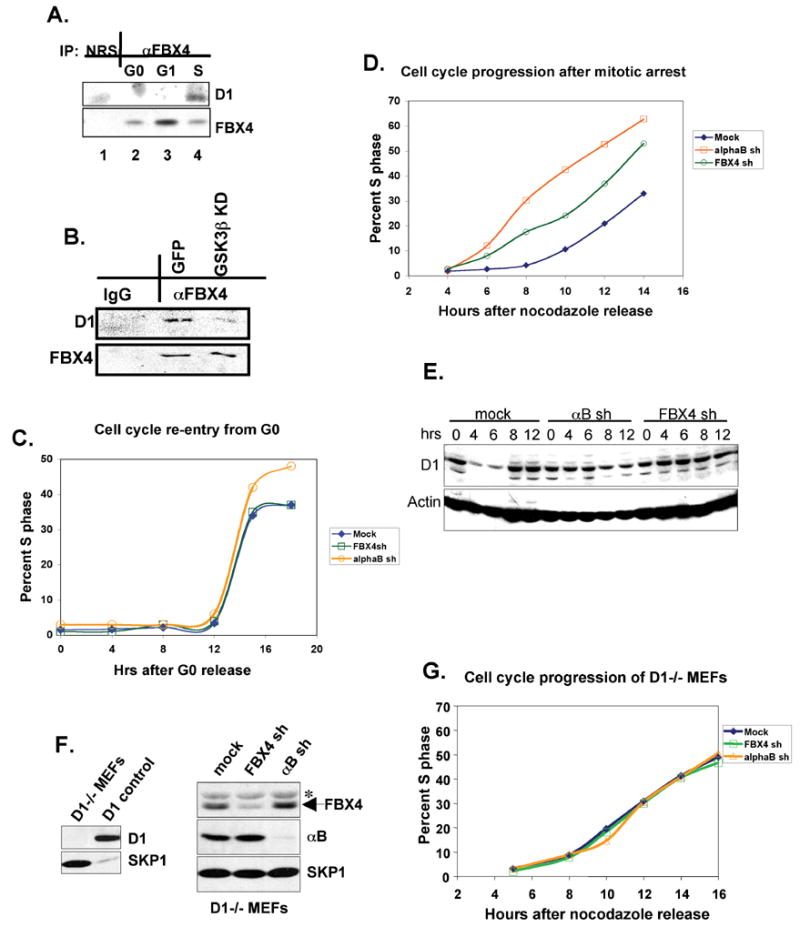
(A) Cell cycle dependent binding between FBX4 and cyclin D1. NIH3T3 cell were released from a G0 (serum starvation) block and binding between cyclin D1 and FBX4 was assessed by IP-western. (B) Lysates prepared from NIH-3T3 cells transfected with GFP or GSK3β kinase-dead (KD) constructs and treated with MG132 were precipitated with FBX4 antiserum or rabbit anti-mouse IgG and subjected to immunoblot with indicated antibodies. (C) Kinetics of cell cycle re-entry from G0 in NIH3T3 cells shRNAs against FBX4 or αB crystallin (D) NIH3T3 cells expressing shRNAs against FBX4/αB crystallin were released from a nocodazole (G2/M) block in media containing BrDU and percentage of cells in S phase assessed by FACS. (E) Immunoblot (cyclin D1) of NIH3T3 cells expressing shRNAs against FBX4/αB crystallin released from a nocodazole (G2/M) block for the indicated time. (F) Knockdown of FBX4/αB crystallin in D1−/− fibroblasts (right panel); control blot of D1 and Skp1 (left panel). (G) D1−/− cells expressing shRNAs against FBX4/αB crystallin were released from a nocodazole and S-phase entry was assessed as in D.
Knockdown of the SCFFBX4-αB crystallin ligase accelerates G1 phase progression
Because overexpression of cyclin D1 correlates with increased rates of cell cycle progression (Ohtsubo and Roberts, 1993; Quelle et al., 1993; Resnitzky et al., 1994), we expected that compromised cyclin D1 proteolysis would accelerate G1 transition. However, because G0-G1-S-phase movement is largely dependent upon cyclin D1 gene expression, we reasoned that reentry rates would be refractory to FBX4/αB crystallin levels. To test this, cell lines wherein either αB crystallin or FBX4 are knocked down were synchronized by serum starvation. Cells were released in complete media containing BrdU, and S-phase entry was assessed by FACS. Knockdown of αB crystallin or FBX4 did not accelerate the G0 to S-phase transition (Fig. 6C). Similarly, expression of (ΔF)FBX4 did not accelerate S phase entry following a serum starvation (G0) block (data not shown).
We next assessed whether loss of FBX4 or αB crystallin would accelerate progression from G2 to S-phase. Cells were arrested at the G2/M transition by nocodazole treatment and released once again in medium containing BrDU. Whereas control NIH3T3 cell lines entered S-phase between 9–10 hours post-nocodazole (G2/M) release, knockdown of αB crystallin or FBX4 reproducibly accelerated G1-phase traverse resulting in S-phase entry 4–6 hours post-nocodazole release (Fig. 6D and Supplemental Table 1). Cyclin D1 levels remained elevated in knockdown cells relative to controls following nocodazole release (Fig. 6E). Consistent with D1 as the primary target, knockdown of FBX4 or αB crystallin did not accelerate cell cycle progression in D1−/− fibroblasts (Fig. 6F, G) or D1-T286A expressing 3T3 cells (data not shown).
FBX4 and αB crystallin levels are altered in human cancers
Overexpression of cyclin D1 in various human malignancies suggests that impairment of SCFFBX4-αBcrystallin function might contribute to cyclin D1 overexpression in cancer. Indeed, the αB crystallin locus maps to chromosome 11q22.3-q23.1, a region deleted in human cancer (Bax et al., 2001; Dohner et al., 1999). To quantify FBX4 and αB crystallin levels in human malignancies, we assessed expression in a matched tumor vs. normal mRNA array (materials and methods). αB crystallin mRNA levels were low in 40% of tumors examined as compared to corresponding normal controls (Fig. 7A); in contrast, FBX4 was down regulated in 24% of tumors (Fig. 7A). mRNA levels of FBX4 and αB crystallin mRNA were down regulated in a broad spectrum of tumors including prostate, thyroid and breast adenocarcinomas and lymphoma. The reduced frequency of FBX4 loss may reflect a necessity for FBX4-mediated degradation of additional substrates (Lee et al., 2006).
Fig. 7. FBX4 and αB crystallin levels are altered in human cancers.
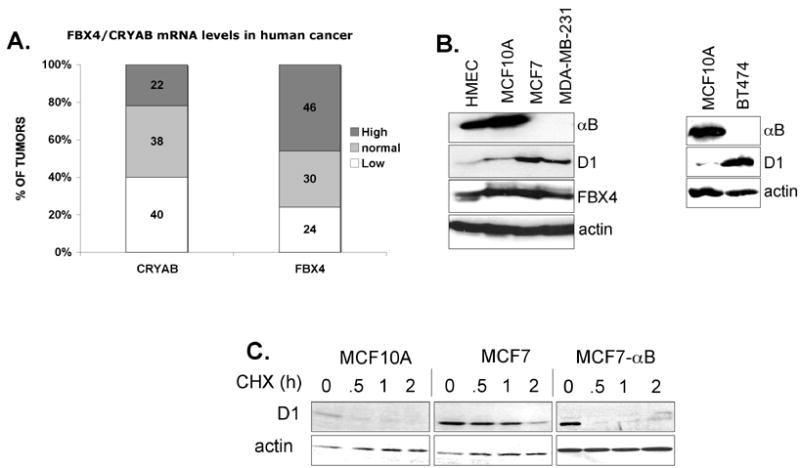
(A) Quantification of FBX4 and αB crystallin mRNA levels in a tumor vs. matching normal tissue in an mRNA array. (B) Expression of αB crystallin, cyclin D1 and FBX4 in normal human mammary epithelial (HMEC), non-tumorigenic breast MCF10A cells and tumorigenic breast cancer cell lines (MCF7, MDA-MB-231, BT474). (C) MCF10A, MCF7 or MCF7 cells infected with a retrovirus encoding αB crystallin were treated with cycloheximide for the indicated intervals and levels of cyclin D1, αB crystallin and actin were assessed by immunoblot.
We also assessed protein levels of FBX4, αB crystallin and cyclin D1 by immunohistochemical analysis of primary esophageal tumors (n=36), a cancer in which cyclin D1 is frequently deregulated (Sherr, 1996). In normal esophageal tissue, accumulation of cyclin D1 was mutually exclusive with FBX4 and αB crystallin (Fig. S2A, S2B); cyclin D1 was highest in proliferative basal cells. In tumors, αB crystallin and FBX4 levels were negative or very weak in 64% and 52% of adenocarcinomas and in 54% and 64% squamous cell carcinomas respectively (Fig. S2A, S2B). A subset of these tumors (24%, 6/25 adeno, 54%, 6/11 squamous cell carcinomas) exhibited negative or low αB crystallin or FBX4 protein levels with concomitant cyclin D1 overexpression (Fig. S2A, arrows and S2B).
In addition, screening of 10 tumor-derived cell lines revealed loss of αB crystallin in breast cancer derived, MCF7, MDA-MB-231, and BT474 cells (Fig. 7B). αB crystallin loss in MCF7 cells results from loss of chromosome 11 (www.path.cam.ac.uk/~pawefish/BreastCellLineDescriptions/mcf7.htm). The absence of αB crystallin correlated with decreased cyclin D1 proteolysis and increased D1 levels relative to control breast epithelial cells (Fig. 7B, 7C and S3). Reintroduction of αB crystallin into MCF7 cells (Fig. S1D) restored the rapid kinetics of cyclin D1 proteolysis (Fig. 7C) suggesting that stabilization of cyclin D1 in these cells resulted from loss of αB crystallin. Thus, impairment of SCFFBX4-αB crystallin function is a potential mechanism underlying cyclin D1 overexpression in human cancer.
DISCUSSION
Cyclin D1 is a substrate for the SCFFBX4-αB crystallin complex
Herein, we describe experiments revealing the following: 1) αB crystallin and FBX4 are components of phospho-T286 cyclin D1 containing protein complexes; 2) αB crystallin is necessary for recognition of phosphorylated cyclin D1 by the SCFFBX4 ubiquitin ligase; 3) the SCFFBX4-αB crystalline complex ubiquitinates phosphorylated cyclin D1 and targets it for proteasomal degradation; 4) the αB crystallin accessory component is lost in multiple breast cancer cell lines resulting in cyclin D1 stabilization and overexpression. This data supports the notion that reduced cyclin D1 proteolysis likely contributes to cyclin D1 overexpression in certain malignancies.
Several lines of evidence suggest that cyclin D1 is a true substrate for the SCFFBX4-αB crystalline ubiquitin ligase. First, cyclin D1, FBX4 and αB crystallin are present in endogenous complexes in vivo. Second, the interaction between cyclin D1 and the SCFFBX4-αB crystallin complex is dependent on cyclin D1 phosphorylation (a characteristic of SCF-substrate interactions). Third, overexpression of FBX4 and αB crystallin triggers the destruction of wild-type cyclin D1, but not of the phosphorylation-deficient cyclin D1 mutant, D1-T286A. Fourth, cyclin D1 is stabilized in vivo by the expression of either a dominant-negative FBX4 or by shRNAs against FBX4 and αB crystallin mRNAs. Finally, purified SCFFBX4-αB crystallin complexes catalyze ubiquitination of cyclin D1 in vitro providing direct biochemical evidence that cyclin D1 is a bona fide substrate for the SCFFBX4-αB crystallin complex.
The SCFSKP2 ligase has also been implicated in the ubiquitination of cyclin D1 (Ganiatsas et al., 2001; Yu et al., 1998). Knockdown of SKP2 promoted accumulation of cyclin D1(Yu et al., 1998). However, evidence for direct ubiquitination by the SCFSKP2 is absent. Indeed a physical association between SKP2 and either cyclin D1 or phospho-T286 cyclin D1, has not been reported. The stabilization of cyclin D1 observed in cells expressing antisense oligos against SKP2 likely reflects stabilization of other substrates such as p21Cip1 or p27Kip1 (Bornstein et al., 2003; Carrano et al., 1999), both of which can decrease cyclin D1 proteolysis (Alt et al., 2002; Cheng et al., 1999; Coleman et al., 2003).
Possible roles of αB crystallin within the SCFFBX4-αB crystallin complex
Our data suggests that αB crystallin acts as a specificity determinant that facilitates FBX4 recognition of Thr286 phosphorylated cyclin D1. It is possible that it is the chaperone-like activity of αB crystallin SCFFBX4-αBcrystallin provides this function. However, there is no data to suggest that the chaperoning function of αB crystallin depends upon substrate phosphorylation. Thus, we favor an explanation wherein targeting of phosphorylated cyclin D1 to SCFFBX4-αB crystallin represents a distinct function for αB crystallin. Three distinct mechanisms for αB crystallin function in this process can be envisioned. In the first, αB crystallin might directly bind to phospho-Thr286 of cyclin D1 and bridge D1 to SCFFBX4. Such a mechanism is analogous to the function provided by CKS1 in the SCFSKP2-CKS1 complex where CKS1 binds to Thr-187 phosphorylated p27Kip1 (Bashir et al., 2004; Bornstein et al., 2003; Ganoth et al., 2001; Hao et al., 2005). In an alternative model, the phospho-acceptor site within SCFFBX4-αB crystallin is cooperatively recognized by both FBX4 and αB crystallin. Finally, it remains formally possible that αB crystallin may not participate in phospho-acceptor recognition site. Rather, αB crystallin might function as an allosteric regulator of FBX4 by inducing a conformational change of FBX4 thereby increasing the affinity of FBX4 for phosphorylated cyclin D1.
Spatial control of cyclin D1 ubiquitination
Previous work suggested that phosphorylation-dependent ubiquitination of cyclin D1 occurs in the cytoplasm (Diehl et al., 1998). Indeed our analysis revealed that both FBX4 and αB crystallin are exclusively cytoplasmic. Based on the steady state cytoplasmic localization of FBX4 and the fact that, following a G0 release, cyclin D1 binds to FBX4 during S-phase (a cell cycle phase wherein cyclin D1 is cytoplasmic (Diehl et al., 1998)), we suggest FBX4-dependent ubiquitination of cyclin D1 is a cytoplasmic event. Although our data suggest FBX4 is the major E3 ligase, it remains possible that alternative cytoplasmic adaptor mechanisms exist that target cyclin D1 for destruction. Consistent with this, cyclin D1T286A exhibits reduced turnover relative to wild-type cyclin D1 in αB crystalline-deficient MCF7 cells (data not shown).
In contrast to cytoplasmic ubiquitination machinery, there may also exist a nuclear, phosphorylation-independent cyclin D1 destruction pathway that is not mediated by the SCFFBX4-αB crystallin complex. Evidence for this pathway comes from the discovery of cyclin D1b, an alternative splice variant of canonical cyclin D1 (Betticher et al., 1995; Howe and Lynas, 2001; Lu et al., 2003; Solomon et al., 2003). Cyclin D1b lacks the last exon of canonical cyclin D1, and therefore Thr286. Since cyclin D1b cannot be phosphorylated at Thr286, it cannot be exported to the cytoplasm. Although the half-life of cyclin D1b is longer than canonical cyclin D1, cyclin D1b degradation remains proteasome-dependent (data not shown) suggesting the existence of phosphorylation-independent machinery that earmarks nuclear cyclin D1b for destruction. Phosphorylation-independent destruction of cyclin D1 has been proposed to occur through the activity of a protein called antizyme (Newman et al., 2004).
Other SCFFBX4 substrates
FBX4 also targets the telomeric DNA repeat binding protein, Pin2/Trf1, for degradation (Lee et al., 2006) potentially contributing to telomere maintenance. In contrast to D1, recognition does not depend upon αB crystallin. At first, it seems somewhat counterintuitive that impairment of FBX4 function could promote both a proliferative signal, via cyclin D1 stabilization, and a growth inhibitory one by inducing telomere shortening via Pin2/Trf1 stabilization. However, the dominant FBX4 downstream effector substrate in vivo may depend on cellular context. For instance, a cancer cell may have acquired the expression of telomerase or additional factors to evade checkpoint responses induced by the effects of Pin2/Trf1 stabilization. In addition, stabilization of one FBX4 substrate may compensate for the effect of stabilizing another. Cyclin D1, for example, can induce the maintenance of telomere length, via a telomerase independent mechanism, (Opitz et al., 2001). Additional work should focus on establishing substrate hierarchy.
Role of FBX4 and αB crystallin in cancer and cell cycle progression
Cyclin D1 is overexpressed in a variety of human tumors that do not exhibit cyclin D1 gene amplification or structural abnormalities of the D1 locus, which suggests that increased cyclin D1 stability is a potential mechanism whereby cyclin D1 overexpression occurs in human cancer. Indeed, mutations of cyclin D1 at Thr286 and Pro-287 have been found in human tumors (Benzeno, 2006), thereby rendering cyclin D1 more stable. Impairment of SCFFBX4-αB crystallin function may also account for cyclin D1 overexpression in a subset of human tumors. In fact, the human αB crystallin locus is located on chromosome 11q22.3-q23.1, a region frequently deleted in human cancer (Dohner et al., 1999).
In a preliminary screen of several human cancers, we have observed that both FBX4 and αB crystallin are down regulated at the protein and mRNA levels, suggesting that SCFFBX4-αB crystalline function might be impaired in human cancer. Our results do not exclude the possibility that variations in FBX4 and αB crystallin levels in the tumors examined is secondary to differences in tumor proliferative fractions. However, re-introduction of αB crystallin in MCF7 cells, a breast cancer cell line that lacks αB crystallin, restored the rate of cyclin D1 proteolysis to that of wild type. Future studies will by necessity address whether mutations in FBX4 or αB crystallin that specifically abolish SCFFBX4-αB crystallin binding to cyclin D1 occur in human cancer in the absence of significant alteration in cyclin D1 mRNA levels. Future studies with higher power will also address whether loss of FBX4 and αB crystallin are mutually exclusive or coordinate events and its relation to cyclin D1 overexpression. Our results suggest that impairment of SCFFBX4-αB crystallin function may contribute to cyclin D1 overexpression in human cancer via decreased ubiquitin-mediated cyclin D1 degradation.
While the function of the SCFFBX4-αB crystallin complex in cancer remains speculative, our data reveal a role of this E3 ubiquitin ligase as regulator of cell cycle progression. Knockdown of either FBX4 or αB crystallin reproducibly accelerated G1 phase progression from a G2/M (nocodazole) block, but not from a G0 (serum starvation) block, suggesting that decreased cyclin D1 proteolysis increases the rate of cell cycle progression. The failure of FBX4 and αB crystallin knockdown to accelerate the G0 to S phase transition likely reflects the fact that the G0 to S transition heavily relies on the induction of cyclin D1 transcription (Matsushime et al., 1991). Indeed our results are consistent with the observation that αB crystallin−/− lens epithelial cells exhibit hyperproliferation and genomic instability (Bai et al., 2003). Future efforts are needed to establish the potential tumor suppressive action of both FBX4 and αB crystallin in relation to cyclin D1 as a substrate.
METHODS
Cell Culture Conditions and Transfections
NIH3T3, 293T and U20S cells were maintained in DMEM medium containing glutamine, antibiotics and 10% FBS (Gemini). NIH3T3 cell derivatives engineered to overexpress Flag-FBX4 and Flag-(ΔF) FBX4 were generated by transfection using LipofectAMINE Plus (Invitrogen) with pcDNA3 vectors encoding the appropriate cDNA. Transfected cell lines were selected and maintained in 400 μg/mL G418. Stable NIH3T3 cell lines expressing cyclin D1 have been described previously(Alt et al., 2000). Insect Sf9 cells were grown have been described previously(Summers, 1987). Transient expression of vectors encoding respective cDNAs was achieved by using LipofectAMINE Plus according to the instructions from the manufacturer. Metabolic labeling and protein half-life measurements were performed as previously described (Diehl and Sherr, 1997; Rimerman et al., 2000).
Gene silencing
pSM2c shRNA vectors targeting mouse FBX4 and mouse αB crystallin were purchased from Open Biosystems CSHL library; (Mouse αB crystallin target sequence: TATTAGCTTAATAATCTGGGCC); (Mouse FBX4 target sequence: ATTCATCTTGCCATTCATGACGT). NIH3T3cells were infected with retroviruses encoding αB crystallin or FBX4 shRNAs; pools and clones of infected cells were selected with puromycin. Knockdown of human FBX4 was achieved by transfection with shRNA vectors(Paddison et al., 2002; Paddison and Hannon, 2002) containing either of the following target sequences: GCCGGTACAGTGTGATTCCACAGATTCAA; GGCATTGAGTGGATTCTTGAAGAAGTGGA.
Construction of FBX4 mutants
pcDNA3 Flag-tagged FBX4 was a gift from Michelle Pagano. Flag-(ΔF)FBX4 mutant was generated using the following primers by mutagenesis using the Quick Change Mutagenesis kit (Stratagene); Forward: 5-GAGGAGGTGGATGAGAGGGATCTTCCTTCTTGG-3; Reverse: 5- CCAAGAAGGAAGATCCCTCTCATCCACCTCCTC-3.
Immunoprecipitation and immunoblotting
NIH3T3 cells were treated with 10 μM MG132 or 50 μM LLnL for 4 hrs. Cells were lysed in buffer containing 50 mM HEPES (pH 8.0), 150 mM NaCl, 2.5 mM EGTA, 1 mM EDTA, 0.1% Tween 20, protease and phosphatase inhibitors (1mM PMSF, 20 U/ml aprotinin, 5 mg/ml leupeptin, 1 mM DTT, 0.4 mM NaF, 10 mM β-Glycerophosphate, 100 nM Okadaic Acid). Cyclin D1 was precipitated with the mouse monoclonal antibody D1-72-13G. Alternatively, FBX4 was precipitated with a rabbit polyclonal antibody generated by immunizing rabbits with FBX4 derived peptides. Immune complexes were captured with protein A Sepharose, and resolved by SDS-PAGE. The following antibodies were used for westerns: cyclin D1 (72-13G), αB crystallin (SPA 223) (Stressgen), HA (12CA5), SKP1 (Transduction Labs), β-actin (AC15) (Sigma). FBX4 antiserum was generated by immunizing rabbits with peptides generated based on the N or C-terminus of FBX4 (Rockland Immunochemicals Inc, PA).
In vitro peptide pull down
293T cells transfected as indicated were lysed in Tween 20 buffer plus 1mM PMSF, 20 U/ml aprotinin, 5 mg/ml leupeptin, 1 mM DTT, 0.4 mM NaF, 10 mM β-Glycerophosphate, 100 nM Okadaic Acid. Whole cell extracts were incubated with 100 μg of peptides biotinylated peptides (synthesized by the Baylor College of Medicine Protein Chemistry Core ) and bound proteins were captured on avidin agarose beads. Beads were boiled in SDS sample buffer prior to separation by SDS-PAGE.
Mass Spectrophotometry
Cyclin D1 complexes were separated by SDS-PAGE and visualized with Coomassie Brilliant Blue staining. Excised bands were destained with 50% acetonitrile (v/v)-25mM NH4HCO3, dehydrated with 100% acetonitrile, dried under vacuum and incubated with bovine trypsin (1ug/ml, Sigma) overnight at 37°C. Peptides were extracted with 50% acetonitrile and 5% trifluoroacetic acid, dried under vacuum and reconstituted in 50% acetonitrile and 0.1% trifluoroacetic acid. Peptide solutions were mixed with an equal volume of a-Cyano-4-hydroxycinnamic acid matrix and spotted onto MALDI plates. Peptides were calibrated with Sequazyme peptide mass standards kit (PE Biosystem) and analyzed by MALDI-TOF mass spectrometry (Voyager DE Pro, Applied Biosystems). Identification of proteins was performed using MS-Fit software (http://prospector.ucsf.edu/ucsfhtml4.0/msfit.htm).
In vivo ubiquitination
Cell expressing HA-ubiquitin were lysed in buffer containing 50 mM Tri-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 1% deoxycholate, 1 mM PMSF, 20 U/ml of aprotinin, 5 mg/ml of leupeptin, 1 mM DTT, 0.4 mM NaF, 5 mM NEM and 20 μM MG132. Cyclin D1 or Flag-D1 derivatives were precipitated with either the cyclin D1 antibody or with the M2 monoclonal antibody. Proteins were separated by SDS-PAGE, and ubiquitinated intermediates were detected by immunoblot.
In vitro ubiquitination
293T cells were transfected with vectors encoding Flag-FBX4, αB-crystallin, Cul1, Skp1 and ROC1. The SCF complexes were purified from the lysates of these cells using M2 agarose (Sigma) and incubated with in vitro translated 35S-labeled cyclin D1 and unlabeled CDK4 in the presence of recombinant GSK3β (5U), E1, E2 (UbcH5A), ATP and ubiquitin for 10 min at 30°C followed by incubation for another 50 min at 37°C. Proteins were resolved on 10% SDS-PAGE and visualized by autoradiography.
Subcellular fractionation
Cells were collected by centrifugation at 300g for 4 min at 4°C and suspended in 400 μL buffer A {10 mM HEPES (pH 7.5), 10 mM KCl, 1 mM DTT, 1 mM PMSF, 20 U/ml aprotinin, 5 mg/ml leupeptin, 0.4 mM NaF, 10 mM β-Glycerophosphate}. After incubation on ice for 15 min, 12.5 μL of 10% NP-40 was added and incubated on ice for 10 min. Nuclei were pelleted at 1500g for 5 min, and the supernatant (cytoplasmic extracts) was centrifuged at 13,000g. Nuclei were washed with 1 mL of buffer A twice, and suspended in 50 μL of buffer B {20 mM HEPES (pH 7.5), 0.4 M NaCl, 1 mM DTT, 1 mM PMSF, 20 U/ml aprotinin, 5 mg/ml leupeptin, 0.4 mM NaF, 10 mM β-Glycerophosphate}. Nuclear extract was collected by centrifugation at 18,000g for 5 min.
Immunofluorescence
Cells were fixed in 70% methanol, 30% acetone for 15 min at −20°C, blocked for 1hr in 4%BSA/PBS followed by incubation in primary antibody (6702, 1:100; affinity purified 7099, 1:2) for 1 hr at room temperature. Cells were washed in PBS and incubated in secondary FITC conjugated antibody (1:200) for 30 min at room temperature. Cells were washed with PBS and counterstained with Hoechst (1:500).
Tumor vs. normal mRNA array
mRNA array representing the matched malignant/normal tissues was purchased from Biochain Institute, Inc (Hayward, CA). Hybridizations with cDNA probes specific for FBX4 and αB crystallin performed according to instructions. mRNA levels were normalized to β-actin.
Supplementary Material
Acknowledgments
We thank M. Pagano for the FBX4, FBW2, CUL1 and SKP1 constructs, P. Sicinski for immortalized D1−/− fibroblasts and M. Lessie for technical assistance. We thank members of the Diehl, Fuchs, Bassing and Kushner labs for critical insight. This work was supported by grants from the NIH CA93237, CA111360; a Leukemia & Lymphoma Scholar award (JAD) and the Abramson Cancer Center (JAD and SYF).
References
- Alt JR, Cleveland JL, Hannink M, Diehl JA. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 2000;14:3102–3114. doi: 10.1101/gad.854900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt JR, Gladden AB, Diehl JA. p21(Cip1) Promotes cyclin D1 nuclear accumulation via direct inhibition of nuclear export. J Biol Chem. 2002;277:8517–8523. doi: 10.1074/jbc.M108867200. [DOI] [PubMed] [Google Scholar]
- Bai F, Xi JH, Wawrousek EF, Fleming TP, Andley UP. Hyperproliferation and p53 status of lens epithelial cells derived from alphaB-crystallin knockout mice. J Biol Chem. 2003;278:36876–36886. doi: 10.1074/jbc.M304010200. [DOI] [PubMed] [Google Scholar]
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature. 2004;428:190–193. doi: 10.1038/nature02330. [DOI] [PubMed] [Google Scholar]
- Bax B, Carter PS, Lewis C, Guy AR, Bridges A, Tanner R, Pettman G, Mannix C, Culbert AA, Brown MJ, et al. The structure of phosphorylated GSK-3beta complexed with a peptide, FRATtide, that inhibits beta-catenin phosphorylation. Structure (Camb) 2001;9:1143–1152. doi: 10.1016/s0969-2126(01)00679-7. [DOI] [PubMed] [Google Scholar]
- Benzeno S, Lu F, Guo M, Barbash O, Zhang F, Herman JG, Klein PS, Rustgi A, Diehl JA. Identification of mutations that disrupt phosphorylation-dependent nuclear export of cyclin D1. Oncogene . 2006 doi: 10.1038/sj.onc.1209644. In Press. [DOI] [PubMed] [Google Scholar]
- Betticher DC, Thatcher N, Altermatt HJ, Hoban P, Ryder WD, Heighway J. Alternate splicing produces a novel cyclin D1 transcript. Oncogene. 1995;11:1005–1011. [PubMed] [Google Scholar]
- Bornstein G, Bloom J, Sitry-Shevah D, Nakayama K, Pagano M, Hershko A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem. 2003;278:25752–25757. doi: 10.1074/jbc.M301774200. [DOI] [PubMed] [Google Scholar]
- Burton JL, Tsakraklides V, Solomon MJ. Assembly of an APC-Cdh1-substrate complex is stimulated by engagement of a destruction box. Mol Cell. 2005;18:533–542. doi: 10.1016/j.molcel.2005.04.022. [DOI] [PubMed] [Google Scholar]
- Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. The p21(Cip1) and p27(Kip1) CDK 'inhibitors' are essential activators of cyclin D-dependent kinases in murine fibroblasts. Embo J. 1999;18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman ML, Marshall CJ, Olson MF. Ras promotes p21(Waf1/Cip1) protein stability via a cyclin D1-imposed block in proteasome-mediated degradation. Embo J. 2003;22:2036–2046. doi: 10.1093/emboj/cdg189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Engelsman J, Bennink EJ, Doerwald L, Onnekink C, Wunderink L, Andley UP, Kato K, de Jong WW, Boelens WC. Mimicking phosphorylation of the small heat-shock protein alphaB-crystallin recruits the F-box protein FBX4 to nuclear SC35 speckles. Eur J Biochem. 2004;271:4195–4203. doi: 10.1111/j.1432-1033.2004.04359.x. [DOI] [PubMed] [Google Scholar]
- den Engelsman J, Keijsers V, de Jong WW, Boelens WC. The small heat-shock protein alpha B-crystallin promotes FBX4-dependent ubiquitination. J Biol Chem. 2003;278:4699–4704. doi: 10.1074/jbc.M211403200. [DOI] [PubMed] [Google Scholar]
- Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA, Sherr CJ. A dominant-negative cyclin D1 mutant prevents nuclear import of cyclin-dependent kinase 4 (CDK4) and its phosphorylation by CDK-activating kinase. Mol Cell Biol. 1997;17:7362–7374. doi: 10.1128/mcb.17.12.7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:957–972. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- Dohner H, Stilgenbauer S, Dohner K, Bentz M, Lichter P. Chromosome aberrations in B-cell chronic lymphocytic leukemia: reassessment based on molecular cytogenetic analysis. J Mol Med. 1999;77:266–281. doi: 10.1007/s001090050350. [DOI] [PubMed] [Google Scholar]
- Ganiatsas S, Dow R, Thompson A, Schulman B, Germain D. A splice variant of Skp2 is retained in the cytoplasm and fails to direct cyclin D1 ubiquitination in the uterine cancer cell line SK-UT. Oncogene. 2001;20:3641–3650. doi: 10.1038/sj.onc.1204501. [DOI] [PubMed] [Google Scholar]
- Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M, Hershko A. The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat Cell Biol. 2001;3:321–324. doi: 10.1038/35060126. [DOI] [PubMed] [Google Scholar]
- Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, Pavletich NP. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol Cell. 2005;20:9–19. doi: 10.1016/j.molcel.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Harper JW, Burton JL, Solomon MJ. The anaphase-promoting complex: it's not just for mitosis any more. Genes Dev. 2002;16:2179–2206. doi: 10.1101/gad.1013102. [DOI] [PubMed] [Google Scholar]
- Howe D, Lynas C. The cyclin D1 alternative transcripts [a] and [b] are expressed in normal and malignant lymphocytes and their relative levels are influenced by the polymorphism at codon 241. Haematologica. 2001;86:563–569. [PubMed] [Google Scholar]
- Jin J, Shirogane T, Xu L, Nalepa G, Qin J, Elledge SJ, Harper JW. SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003;17:3062–3074. doi: 10.1101/gad.1157503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294:173–177. doi: 10.1126/science.1065203. [DOI] [PubMed] [Google Scholar]
- Kraft C, Vodermaier HC, Maurer-Stroh S, Eisenhaber F, Peters JM. The WD40 propeller domain of Cdh1 functions as a destruction box receptor for APC/C substrates. Mol Cell. 2005;18:543–553. doi: 10.1016/j.molcel.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Latres E, Chiaur DS, Pagano M. The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene. 1999;18:849–854. doi: 10.1038/sj.onc.1202653. [DOI] [PubMed] [Google Scholar]
- Lee TH, Perrem K, Harper JW, Lu KP, Zhou XZ. The F-box protein FBX4 targets PIN2/TRF1 for ubiquitin-mediated degradation and regulates telomere maintenance. J Biol Chem. 2006;281:759–768. doi: 10.1074/jbc.M509855200. [DOI] [PubMed] [Google Scholar]
- Lu F, Gladden AB, Diehl JA. An alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear oncogene. Cancer Res. 2003;63:7056–7061. [PubMed] [Google Scholar]
- Marshall C. How do small GTPase signal transduction pathways regulate cell cycle entry? Curr Opin Cell Biol. 1999;11:732–736. doi: 10.1016/s0955-0674(99)00044-7. [DOI] [PubMed] [Google Scholar]
- Matsushime H, Roussel MF, Ashmun RA, Sherr CJ. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 1991;65:701–713. doi: 10.1016/0092-8674(91)90101-4. [DOI] [PubMed] [Google Scholar]
- Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A, Pagano M. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 1999;13:1181–1189. doi: 10.1101/gad.13.9.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman RM, Mobascher A, Mangold U, Koike C, Diah S, Schmidt M, Finley D, Zetter BR. Antizyme targets cyclin D1 for degradation. A novel mechanism for cell growth repression. J Biol Chem. 2004;279:41504–41511. doi: 10.1074/jbc.M407349200. [DOI] [PubMed] [Google Scholar]
- Ohtsubo M, Roberts JM. Cyclin-dependent regulation of G1 in mammalian fibroblasts. Science. 1993;259:1908–1912. doi: 10.1126/science.8384376. [DOI] [PubMed] [Google Scholar]
- Opitz OG, Suliman Y, Hahn WC, Harada H, Blum HE, Rustgi AK. Cyclin D1 overexpression and p53 inactivation immortalize primary oral keratinocytes by a telomerase-independent mechanism. J Clin Invest. 2001;108:725–732. doi: 10.1172/JCI11909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddison PJ, Caudy AA, Hannon GJ. Stable suppression of gene expression by RNAi in mammalian cells. Proc Natl Acad Sci U S A. 2002;99:1443–1448. doi: 10.1073/pnas.032652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddison PJ, Hannon GJ. RNA interference: the new somatic cell genetics? Cancer Cell. 2002;2:17–23. doi: 10.1016/s1535-6108(02)00092-2. [DOI] [PubMed] [Google Scholar]
- Pfleger CM, Lee E, Kirschner MW. Substrate recognition by the Cdc20 and Cdh1 components of the anaphase-promoting complex. Genes Dev. 2001;15:2396–2407. doi: 10.1101/gad.918201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quelle DE, Ashmun RA, Shurtleff SA, Kato JY, Bar-Sagi D, Roussel MF, Sherr CJ. Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev. 1993;7:1559–1571. doi: 10.1101/gad.7.8.1559. [DOI] [PubMed] [Google Scholar]
- Resnitzky D, Gossen M, Bujard H, Reed SI. Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol Cell Biol. 1994;14:1669–1679. doi: 10.1128/mcb.14.3.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimerman RA, Gellert-Randleman A, Diehl JA. Wnt1 and MEK1 cooperate to promote cyclin D1 accumulation and cellular transformation. J Biol Chem. 2000;275:14736–14742. doi: 10.1074/jbc.m910241199. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Solomon DA, Wang Y, Fox SR, Lambeck TC, Giesting S, Lan Z, Senderowicz AM, Knudsen ES. Cyclin D1 splice variants. Differential effects on localization, RB phosphorylation, and cellular transformation. J Biol Chem. 2003;278:30339–30347. doi: 10.1074/jbc.M303969200. [DOI] [PubMed] [Google Scholar]
- Spruck C, Strohmaier H, Watson M, Smith AP, Ryan A, Krek TW, Reed SI. A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol Cell. 2001;7:639–650. doi: 10.1016/s1097-2765(01)00210-6. [DOI] [PubMed] [Google Scholar]
- Summers MDaSGE. A manual of methods for baculovirus vectors and insect culture procedures. Tex Agric Exp St Bull. 1987:1555. [Google Scholar]
- Yu ZK, Gervais JL, Zhang H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc Natl Acad Sci U S A. 1998;95:11324–11329. doi: 10.1073/pnas.95.19.11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.