

Abstract
The design, synthesis and unexpected inhibitory activity against S-adenosylhomocysteine hydrolase (SAHase) for a series of truncated carbocyclic pyrimidine nucleoside analogues is presented. Of the four nucleosides obtained, 10 was found to be active with a Ki value of 5.0 μM against SAHase.
Introduction
The significant inhibitory activity exhibited by the carbocyclic nucleosides against S-adenosylhomocysteine hydrolase (SAHase) has, for the most part, been confined to adenosine analogues (Figure 1). 1–3 One notable exception exception4 was the unexpected inhibition exhibited by a split guanosine nucleoside “fleximer”, however to date, few non-adenosine analogues have been reported. Inhibition of SAHase and several other enzymes critical for viral replication have been suggested as strategic targets for the inhibition of the orthopox viruses.5 Given the lack of effective chemotherapeutics against the poxviruses and their potential for use in bioweapons, the search for new and potent inhibitors has increased in intensity.
Figure 1.
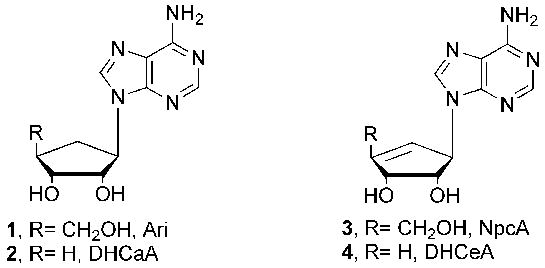
Potent Carbocylic SAHase Inhibitors.
It is well known that inhibition of SAHase also leads to inhibition of S-adenosyl-l-methionine-(SAM) mediated biomethylations.1 As a result, methylation reactions involved in viral replication become attractive targets for inhibition.6 Two of the first naturally occurring carbocyclic nucleosides to exhibit activity were aristeromycin (Ari, 1) and neplanocin A (NpcA, 3).7–9 Unfortunately their close resemblance to adenosine rendered them susceptible to recognition and phosphorylation by cellular kinases, resulting in significant toxicity.10 In response to this, strategic removal of the 4′-hydroxymethyl group of Ari and NpcA to afford the 4′,5′-saturated (2) and 4′,5′-unsaturated (4) derivatives proved to be successful, as both were potent inhibitors of SAHase.11,12 Interestingly, neither served as a substrate for adenosine kinase or adenosine deaminase, thus did not exhibit the toxicity associated with Ari and NpcA.11,12
Related to this, a report report5 of potent activity by two carbocyclic pyrimidine nucleosides (Figure 2) against cytidine triphosphate (CTP) synthetase, in addition to numerous substituted pyrimidines against thymidylate synthase, prompted us to consider the design and synthesis of additional carbocyclic pyrimidine analogues (Figure 3), incorporating the truncated scaffold. In that regard, the first of the pyrimidine targets is described.
Figure 2.
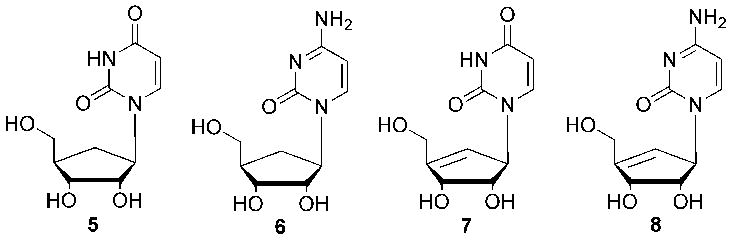
Carbocylic derivatives of uridine and cytidine
Figure 3.
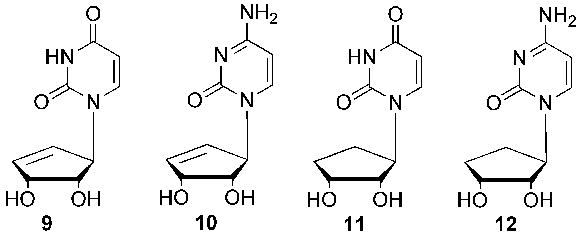
Carbocylic Targets.
Chemistry
Traditional Mitsunobu coupling was envisioned as a logical route to realize the targets. The cyclopentenyl alcohol precursor precursor13,14 was obtained in good yield, and, as shown in Scheme 1, coupled to N3-Bzuracil. The benzoyl protecting group was removed from the coupled product by stirring with a 1% NaOH solution in MeOH to afford analogue 13 in 45% yield for two steps. This precursor is extremely important to this synthesis because from it three different routes can be employed. The first included deprotection of analogue 13 with a mixture of TFA and H2O to result in the first final target 9. Secondly, in order to get the next immediate target, analogue 13 was first reacted with TIPBSCl then followed with mild ammonolysis conditions to give analogue 14 which was then deprotected in a similar fashion as with analogue 9 to give final target 10.
Scheme 1.
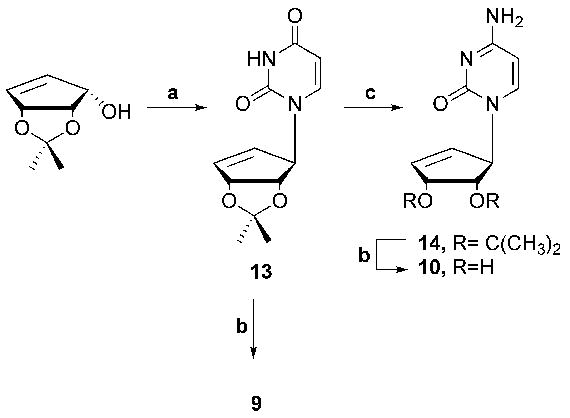
Reagents and comditions: (a) i. N3-Bz-uracil, PPh3, DIAD, CH3CN, 0 °C to rt, 70%, ii. NaOH in MeOH (1%),rt, 15h, 75%: (b) TFA:H2O (2:1), rt, 3h, 95%; (c) i. TIPBSCI, NEt3, DMAP, CH3CN, rt, 4 h; ii. NH4OH, rt, 15 h, 65%.
In order to synthesize the final two targets in this series, it was necessary to reduce the double bond of the cyclopentyl ring system and as a result analogue 13 was reduced using catalytic hydrogenation to give saturated cyclopentyl analogue 15 (Scheme 2). The isopropylidene group of analogue 15 was removed to give final target 11. The oxo group of analogue 15 was also converted to amino 16, which was then deprotected to give final target 12.
Scheme 2.
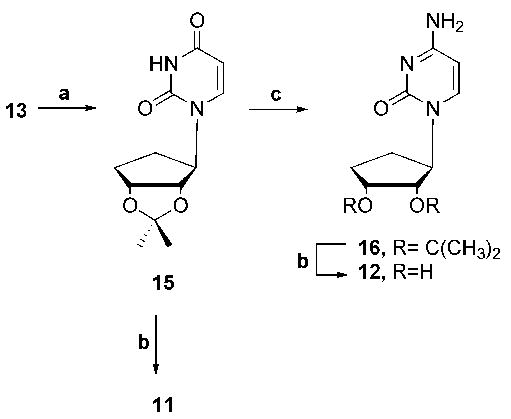
Reagents and conditions: (a) H2 (25 psi), Pd/C, EtOH, rt, 30 min, quant.; (b) TFA/H2O (2:1), rt, 3 h, 95%; (c) i. TIPBSCI, NEt3, DMAP, CH3CN, rt, 4 h; ii. NH4OH, rt, 15 h, 65%.
The four targets were then screened for their ability to inhibit SAHase, DNA methyltransferase (MTase), and CTP synthetase. No activity was seen for any of the analogues against DNA methyltransferase, or CTP synthetase (data not shown). For SAHase, in the first experiment the concentration of inhibitor was varied while the concentration of substrate (SAH) was held constant at 10 μM. Conversion of SAH to adenosine and then to inosine was monitored at 265 nm (Figure 4). In the second experiment the concentration of inhibitor was held constant at 45 μM while the concentration of substrate was varied. Initial rates in the presence and absence of inhibitors were measured (Figure 5). There was no change in absorbance at 265 nm in the absence of SAH. These experimental data best fit a competitive inhibition mechanism. The Ki value for compound 10 was found to be 5.0 ± 0.9 μM against SAHase in the hydrolysis direction, based on a Km value of 7.9 μM for the SAH substrate.15 Ongoing studies suggest that the mechanism may be more complicated than a simple competitive inhibition mechanism, so that the “true” inhibitory form probably binds to the enzyme tighter than SAH. Detailed mechanistic studies will be reported in the future.
Figure 4.
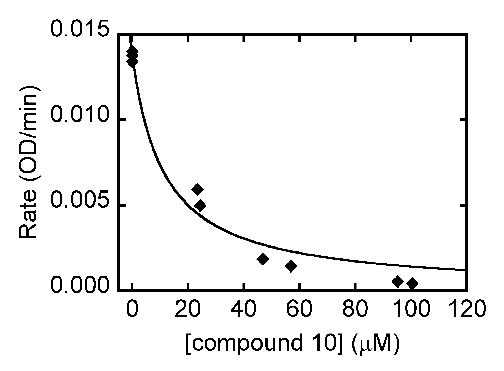
Inhibition of SAHase by 10. Assay solutions contained 50 mM potassium phosphate at pH 7.4, 0.39 units of adenosine deaminase, 132 nM of SAHase, 10 μM of SAH (substrate), and various concentrations of 10. Consumption of substrate was monitored at 265 nm.
Figure 5.
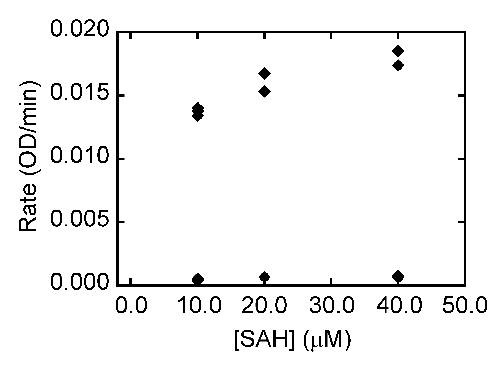
Inhibition of SAHase by 10. In the assay solution, the concentration of 10 was held constant at 45 μM while the concentration of substrate was varied. Rates in the presence (data points at bottom of plot) and absence (data points at top of plot) of inhibitors were measured.
The four compounds were also screened for their ability to inhibit S-adenosylhomocysteine (SAH) nucleosidase (EC 3.2.2.9). SAH nucleosidase irreversibly cleaves SAH and 5′-methylthioadenosine (MTA) to form adenine and the analogous thioribose. This enzyme plays an important role in microbial pathways such as quorum sensing, biological methylation, polyamine synthesis, and methionine recycling. Inhibition of SAH nucleosidase has been shown to lead to a buildup of SAH and MTA, which are feedback inhibitors to various enzymes. This enzyme, encoded by the pfs gene, is presented in about half of all bacterial species, including Staphylococcus aureus, Enterococcus faecalis, Streptococcus pyogenes, Streptococcus pneumoniae, Mycobacterium tuberculosis, Salmonella typhimurium, Haemophilus influenzae, Vibrio cholerae, and Bacillus anthracis; but is not found in mammals.16,17 Knockout of the pfs gene has been found to be lethal or marked impaired in cell growth. All together, SAH nucleosidase represents an attractive target for antimicrobial drug therapy. For SAH nucleosidase inhibition, concentration of inhibitor was varied while the concentration of substrate (SAH) was held constant at 10.8 μM. Consumption of substrate was monitored at 265 nm. Compound 10 inhibited about 50% of the activity at approximately a 13-fold excess (data not shown). Given the limited structural similarity between the SAH substrate and compound 10, the finding was unexpected. Thus in spite of its moderate inhibitory activity, compound 10 may serve as a lead compound for different approach to inhibit this nucleosidase.
EXPERIMENTAL
General
Melting points are uncorrected. 1H and 13C spectra were operated at 300 and 75 MHz, respectively, all referenced to internal tetramethylsilane (TMS) at 0.0 ppm. The spin multiplicities are indicated by the symbols s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet) and b (broad). Reactions were monitored by thin-layer chromatography (TLC) using 0.25 mm Whatman Diamond silica gel 60-F254 precoated plates. Column chromatography was performed on Whatman silica, 200–400 mesh, 60 Å and elution with the indicated solvent system. Yields refer to chromatographically and spectroscopically (1H and 13C NMR) homogeneous materials.
(1′S, 2′R, 3′S)-1-[(2′,3′-O-isopropylidene)-4′-cyclopenten-1′-yl]uracil -(13)
To cyclopenten-ol (6.50 g, 41.67 mmol), N3-Bzuracil (18.02 g, 83.33 mmol) and PPh3 (21.88 g, 83.33 mmol) in anhydrous CH3CN (500 mL) at 0 ºC was added dropwise DIAD (16.85 g, 83.33 mmol, 16.52 mL). After stirring at rt for 15 h the mixture was concentrated and purified by column chromatography (EtOAc:hexanes, 3:1) to afford 9.74 g of 3-benzoyl-1′-(2′,2′-dimethyl-4′,6′-dihydro-3′H-cyclopenta[1′,3′]dioxol-4′-yl)-1H-pyrimidine- 2,4-dione as a white solid. 1H NMR (DMSO-d6): δ 1.23 (3H, s), 1.3 (3H, s), 4.71 (1H, d), 5.27 (1H, s), 5.29 (1H, d), 5.81 (1H, d), 5.84 (1H, dd), 6.23 (1H, dt), 4.71 (1H, d), 5.29 (2H, d), 5.84 (1H, d), 5.85 (1H, d), 6.22 (1H, m), 7.51 (1H, d), 7.57 (2H, t), 7.78 (1H, t), 7.97 (2H, m). 13C NMR (DMSO- d6): δ 24.4, 26.2, 69.1, 83.2, 84.6, 101.1, 111.8, 128.9, 129.0, 130.1, 131.6, 135.0, 139.0, 143.0, 149.9, 163.0, 168.8.
NaOH (10 mL, 1% in MeOH) was added to the coupled product and the mixture was allowed to stir at rt for 12 h. After this time the mixture was neutralized with HCl (1N), the solvent removed and the resulting residue was dissolved in EtOAc (50 mL), washed with H2O (25 mL), dried (MgSO4), filtered, and concentrated to give a solid. This solid was purified by column chromatography (EtOAc:hexanes, 4:1) to afford 13 (7.50 g, 71%) as a white solid. 1H NMR (DMSO-d6): δ 1.22 (3H, s), 1.31, (3H, s), 4.53 (1H, d), 5.27 (2H, m), 5.52 (1H, dd), 5.77 (1H, dd), 6.19 (1H, d), 7.23 (1H, d), 11.31 (1H, s). 13C NMR (CD3OD): δ 24.4, 26.2, 68.4, 83.4, 84.6, 101.2, 111.7, 129.3, 138.5, 142.5, 151.3, 165.0. Anal. Calcd. For C12H14N2O4: C, 57.59; H, 5.64; N, 11.19. Found: C, 57.92; H, 5.75; N, 11.18.
(1′S, 2′R, 3′S)-1-[(2′,3′-Dihydroxy)-4′-cyclopenten-1′-yl]uracil (9)
A mixture of 13 (0.15 g, 0.60 mmol) in TFA/H2O (2:1, 4 mL) was allowed to stir for 12 h at rt upon which time the TFA/H2O was removed by evaporation. The resulting residue was coevaporated with MeOH (3 × 10 mL) to remove trace amounts of TFA. The resulting residue was then purified using C-18 HPLC eluting 90:10, H2O:MeOH → 50:50, H2O:MeOH → 10:90, H2O:MeOH afford 9 (0.106 g, 83 %) as a white solid. 1H NMR (CD3OD): δ 4.10 (1H, t), 4.55–4.60 (1H, dd), 5.45–5.50 (1H, dd), 5.70 (1H, d), 5.90 (1H, d), 6.20–6.23 (1H, dd), 7.40 (1H, dd). 13C NMR (CD3OD): δ 66.6, 73.0, 76.5, 101.6, 132.4, 136.7, 142.3, 151.8, 165.0. HRMS: calcd for C9H10N2O4 (M + H)+, 211.19; found, 211.07.
(1′S, 2′R, 3′S)-1-[(2′,3′-O-isopropylidene)-4′-cyclopenten-1′-yl]cytosine -(14)
To a solution of 13 (0.75 g, 3.00 mmol), DMAP (1.47 g, 12.00 mmol) and Et3N (20 mL) in anhydrous CH3CN (100 mL) at 0 ºC was added portionwise TIPBSCl (3.63 g, 12.00 mmol). After stirring at rt for 3 h NH4OH (50 mL) was added and the mixture continued to stir for 12 h at rt. The solvent was then removed and the resulting residue was purified via column chromatography (EtOAc:acetone:EtOH:H2O, 6:1:1:0.5) to afford 14 (0.58 g, 78%) as a white solid. 1H NMR (CD3OD): δ 1.27 (3H, s) 1.38 (3H, s), 4.53–4.54 (1H, d), 5.32–5.34 (1H, m), 5.43 (1H, m), 5.76–5.78 (1H, m), 5.81–5.83 (1H, d), 6.19–6.21 (1H, m), 7.30–7.32 (1H, m). 13C NMR (CD3OD): δ 24.6, 26.3, 69.3, 83.8, 84.7, 94.8, 111.54, 130, 138.1, 142.8, 157.4, 166.3. Anal. Calcd. For C12H15N3O3 (0.75 H2O): C, 54.88; H, 6.33; N,16.00. Found: C, 54.64; H, 5.83; N, 15.73.
(1′S, 2′R, 3′S)-1-[(2′ 2′,3′-Dihydroxy)-4′-cyclopenten-1′-yl]cytosine, 3′-(10)
See procedure 9 for other reaction details. Compound 14 (0.15 g, 0.60 mmol) and TFA (4 mL) afforded 10 (0.11 g, 87%) as a white solid. 1H NMR (CD3OD): δ 4.03–4.05 (1H, t), 4.58–4.59 (1H, m), 5.47–5.50 (1H, m), 5.90–5.98 (2H, dd), 6.19–6.21 (1H, m), 7.55–7.56 (1H, d). 13C NMR (CD3OD): δ 67.8, 73.7, 76.9, 95.1, 132.6, 136.4, 142.6, 157.9, 166.0. Anal. Calcd. For C C9H11N3O3: C, 51.67, H, 5.30, N, 20.09. Found: C,51.78, H, 5.39, N, 19.70.
(1′S, 2′R, 3′S)-1-[(2′,3′- O-isopropylidene)-cyclopent-1′-yl]uracil -(15)
To 13 (0.25 g, 1 mmol) in MeOH (10 mL) was added Pd/C (10 mol %, 0.025 g). This mixture was hydrogenated at a pressure of 25 psi for 20 min. The Pd/C was removed by filtration and the filtrate was concentrated to afford 0.26 g of 15 in quantitative yield as a white solid. 1H NMR (DMSO-d6): δ 1.20 (3H, s), 1.35 (3H, s), 1.65–1.82 (2H, m), 1.95–2.15 (2H, m), 4.63 (1H, m), 4.64 (1H, m), 4.71 (1H, dd), 5.52 (1H, d), 7.58 (1H, d) and 11.26 (1H, s). 13C NMR (DMSO-d6) δ 24.4, 27.3, 29.1, 31.2, 63.9, 80.3, 84.4, 101.8, 110.0, 143.9, 152.6, 163.8. Anal. Calcd. For C12H16N2O4 (0.25 H2O): C, 56.30; H, 6.50; N, 10.95. Found: C, 56.42; H, 6.44; N,10.88.
(1′S, 2′R, 3′S)-1-[(2′,3′-Dihydroxy)-cyclopent-1′-yl]uracil (11)
See procedure 9 for other reaction details. Compound 15 (0.25 g, 0.99 mmol) and TFA (15 mL) afforded 11 (0.19 g, 91%) as a white solid. 1H NMR (CD3OD): δ 1.69–1.79 (2H, m), 2.06–2.21 (2H, m), 4.06 (1H, m), 4.22–4.25 (1H, dd), 4.61–4.67 (1H, q), 4.86 (1H, s), 5.65–5.67 (1H, d), 7.62–7.64 (1H, d). 13C NMR (CD3OD): δ 19.9, 26.2, 59.5, 77.2, 85.9, 102.4, 141.3, 150.9, 163.6. Anal. Calcd. For C9H12N2O4 (0.6 mol H2O): O): C, 48.49, H, 5.96, N, 12.57. Found: C, 48.79, H, 5.51, N, 12.20.
(1′S, 2′R, 3′S)-1-[(2′,3′-O-isopropylidene)-cyclopent-1′-yl]cytosine -(16)
See procedure 14 for reaction details. Compound 15 (0.42 g, 1.67 mmol), DMAP (0.81 g, 6.66 mmol), Et3N (12 mL) and CH3CN (75 mL) afforded 16 (0.34 g, 81%) as a white solid. 1H NMR (DMSO-d6): δ 1.18 (s, 3H), 1.36 (s, 3H), 1.64–1.80 (m, 2H), 1.94–2.15 (m, 2H), 4.40–4.50 (m, 1H), 4.60–4.68 (dd, 1H), 4.69 (m, 1H), 5.65 (d, 1H), 7.10 (br s, 1H), 7.30 (br s, 1H), 7.50 (d, 1H). 13C NMR (DMSO-d6): δ 22.3, 23.8, 29.5, 30.5, 64.9, 80.6, 84.7, 110.7, 144.9, 163.0.
(1′S, 2′R, 3′S)-1-[(2′,3′-Dihydroxy)-cyclopent-1′-yl]cytosine (12)
See procedure for 9 for other reaction details. Compound 16 (0.10 g, 0.40 mmol) and TFA (5 mL) afforded 12 (0.07 g, 85%) as a white solid. 1H NMR (DMSO-H d6): δ 1.4–1.6 (m, 2H), 1.8–2.1 (m, 2H), 3.85 (s, 1H), 4.05 (s, 1H), 4.3–4.6 (m, 2H), 4.61–4.8 (m, 1H), 5.65 (s, 1H), 6.95 (s, 2H), 7.55 (d, 1H). 13C NMR (DMSO-C d6): δ 39.4, 39.6, 63.1, 71.1, 75.5, 93.8, 144.7, 156.6, 165.8. Anal. Calcd. For C C9H13N3O3 (0.5 mol H H2O): C, 49.08, H, 6.41, N, 19.08. O): Found: C,49.21, H, 6.03, N, 18.73.
SAHase Inhibition Assay
A reported enzyme-coupled continuous assay in the hydrolysis direction was employed. In this assay, SAH is hydrolyzed to homocysteine and adenosine, which is subsequently converted by adenosine deaminase into ammonia and inosine, a process associated with an absorbance decrease around 265 nm. 15 All assays were performed in thermostatted 1 cm quartz cuvettes at 37 °C maintained by a Peltier unit on a Cary 100 ultraviolet-visible photospectrometer. In a total volume of 928 μL, a typical enzyme assay solution contained 50 mM potassium phosphate at pH 7.4, 0.39 units of adenosine deaminase (Worthington Biochemical, catalog number LS009043), 132 nM of SAHase (final concentration; provided by Lynne Howell), 10 μM of SAH (final concentration) and various concentrations of inhibitors. The reactions were initiated by the addition of SAH. Under these conditions, SAH hydrolysis catalyzed SAHase was rate limiting (data not shown). The kinetic data were analyzed using KaleidaGraph 4.0 (Synergy). Based on a competitive inhibition mechanism, the Ki value was determined using the equation, v = kcat × [S] × [E]/{Km × (1 + [I]/Ki) + [S]}; and v, kcat, [S], [E], Km, [I] and Ki stand for the initial reaction rates, rate constant, substrate concentration, enzyme concentration, Michaelis-Menten constant, inhibitor concentration and dissociation constant of the enzyme-inhibitor complex, respectively. The Km value for SAH in the hydrolysis direction was assumed to be 7.9 μM as previously reported.15
SAH Nucleosidase Assay
In this assay assay18, SAH is hydrolyzed into S-ribosylhomocysteine ribosylhomocysteine and adenine, which is subsequently converted by adenine deaminase into ammonia and hypoxanthine, a process associated with a decrease in absorbance around 265 nm. Assays were performed in thermostatted 1-cm quartz cuvettes at 37 °C maintained by a Peltier unit on a Cary 100 ultraviolet-visible photospectrometer. In a total volume of 920 μL, a typical enzyme assay solution contained 50 mM KPi (pH 7.4), 100 μM MnSO4, 30.0 nM adenine deaminase, 4.2 nM SAH nucleosidase, 10.8 μM SAH, and various concentrations of inhibitors. The reaction was initiated by the addition of SAH. Under the assay conditions, SAH hydrolysis catalyzed by the nucleosidase was rate limiting (data not shown). Manganese or other divalent ions (e.g., zinc) are required for adenine deaminase activity.19,20
Acknowledgments
This work was supported by the National Cancer Institute (NCI/NIH RO1 CA 97634 to KSR), the National Institute of Allergy and Infectious Diseases (NIAID/NIH, 1R01AI058146 to ZSZ.), the National Institutes of Health/NIGMS Initiative for Minority Student Development (R25-GM55036 to SLM), and the Herman Frasch Foundation (541-HF02 to ZSZ). We are grateful to Prof. George Carmen of Rutgers University for the CTP synthetase assay and Prof. Moshe Szyf of McGill University for the DNA methyltransferase assays.
References
- 1.De Clercq E. Biochem Pharmacol. 1987;36:2567–2575. doi: 10.1016/0006-2952(87)90533-8. [DOI] [PubMed] [Google Scholar]
- 2.De Clercq E. Nucleosides & Nucleotides. 1994;13:1271–1295. [Google Scholar]
- 3.De Clercq E. Nucleosides & Nucleotides. 1998;17:625–634. doi: 10.1080/07328319808005205. [DOI] [PubMed] [Google Scholar]
- 4.Seley KL, Quirk S, Salim S, Zhang L, Hagos A. Bioorganic Med Chem Lett. 2003;13:1985–1988. doi: 10.1016/s0960-894x(03)00331-7. [DOI] [PubMed] [Google Scholar]
- 5.De Clercq E. Clinical Microbiol Rev. 2001;14:382–397. doi: 10.1128/CMR.14.2.382-397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Razin A, Szyf M. Biochim Biophys Acta. 1984;782:331–342. doi: 10.1016/0167-4781(84)90043-5. [DOI] [PubMed] [Google Scholar]
- 7.Yaginuma S, Muto N, Tsujino M, Sudate Y, Hayashi M, Otani M. J Antibiot. 1981;34:359–366. doi: 10.7164/antibiotics.34.359. [DOI] [PubMed] [Google Scholar]
- 8.Hayashi M, Yaginuma S, Yoshioka H, Nakat Nakatsu su K. J Antibiot. 1981;34:675–680. doi: 10.7164/antibiotics.34.675. [DOI] [PubMed] [Google Scholar]
- 9.Kusaka T, Yamamoto H, Shibata M, Muroi M, Kishi T, Mizuno K. J Antibiot. 1968;21:255–263. doi: 10.7164/antibiotics.21.255. [DOI] [PubMed] [Google Scholar]
- 10.Glazer RI, Knode MC. J Biol Chem. 1984;259:12964–12969. [PubMed] [Google Scholar]
- 11.Hasobe M, McKee JG, Borcherding DR, Borchardt RT. Antimicrob Agents Chemother. 1987;31:1849–1851. doi: 10.1128/aac.31.11.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hasobe M, McKee JG, Borcherding DR, Keller BT, Borchardt RT. Mol Pharmacol. 1988;33:713–720. [PubMed] [Google Scholar]
- 13.Seley KL, Schneller SW, Rattendi D, Lane S, Bacchi CJ. Antimicrob Agents Chemother. 1997;41:1658–1661. doi: 10.1128/aac.41.8.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang M, Ye W, Schneller SW. J Org Chem. 2004;69:3993–3996. doi: 10.1021/jo040119g. [DOI] [PubMed] [Google Scholar]
- 15.Elrod P, Zhang J, Yang X, Yin DH, Hu Y, Borchardt RT, Schowen RL. Biochemistry. 2002;41:8134–42. doi: 10.1021/bi025771p. [DOI] [PubMed] [Google Scholar]
- 16.Lee JE, Cornell KA, Riscoe MK, Howell PL. J Biol Chem. 2003;278:8761–70. doi: 10.1074/jbc.M210836200. [DOI] [PubMed] [Google Scholar]
- 17.Lee JE, Settembre EC, Cornell KA, Riscoe MK, Sufrin JR, Ealick SE, Howell PL. Biochemistry. 2004;43:5159–69. doi: 10.1021/bi035492h. [DOI] [PubMed] [Google Scholar]
- 18.Dorgan KM, Wooderchak WL, Wynn DP, Karschner EL, Alfaro JF, Cui Y, Zhou ZS, Hevel JM. Anal Biochem. 2006;350:249–55. doi: 10.1016/j.ab.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 19.Matsui H, Shimaoka M, Kawasaki H, Takenaka Y, Kurahashi O. Biosci Biotechnol Biochem. 2001;65:1112–8. doi: 10.1271/bbb.65.1112. [DOI] [PubMed] [Google Scholar]
- 20.Dorgan KM, Zhou ZS. unpublished results. [Google Scholar]