

Abstract
The cell adhesion molecule integrin αvβ3 plays a key role in tumor angiogenesis and metastasis. A series of 18F-labeled RGD peptides have been developed for PET of integrin expression based on primary amine-reactive prosthetic groups. In this study we introduced a new method of labeling RGD peptides through a thiol-reactive synthon, N-[2-(4-18F-fluorobenzamido)ethyl]maleimide (18F-FBEM).
Methods
18F-FBEM was synthesized by coupling N-succinimidyl 4-18F-fluorobenzoate (18F-SFB) with N-(2-aminoethyl)maleimide. After high-pressure liquid chromatography purification, it was allowed to react with thiolated RGD peptides, and the resulting tracers were subjected to receptor-binding assay, in vivo metabolic stability assessment, biodistribution, and microPET studies in murine xenograft models.
Results
Conjugation of monomeric and dimeric sulfhydryl-RGD peptides with 18F-FBEM was achieved in high yields (85% ± 5% nondecay-corrected on the basis of 18F-FBEM). The radiochemical purity of the 18F-labeled peptides was >98% and the specific activity was 100~ 150 TBq/mmol. Noninvasive microPET and direct tissue sampling experiments demonstrated that both 18F-FBEM-SRGD (RGD monomer) and 18F-FBEM-SRGD2 (RGD dimer) had integrin-specific tumor uptake in subcutaneous U87MG glioma and orthotopic MDA-MB-435 breast cancer xenografts.
Conclusion
The new tracer 18F-FBEM-SRGD2 was synthesized with high specific activity via 18F-FBEM and the tracer exhibited high receptor-binding affinity, tumor-targeting efficacy, metabolic stability, as well as favorable in vivo pharmacokinetics. The new synthon 18F-FBEM developed in this study will also be useful for radiolabeling of other thiolated biomolecules.
Keywords: thiol-reactive synthon, 18F-FBEM, microPET, 18F labeling, integrin αvβ3
The αvβ3 integrin, which binds to arginine-glycine-aspartic acid (RGD)-containing components of the interstitial matrix, such as vitronectin, fibronectin, and thrombospondin, is significantly upregulated on endothelium during angiogenesis but not in quiescent endothelium (1,2). The special role of integrin αvβ3 in tumor invasion and metastasis arises from its ability to recruit and activate matrix metalloproteinase MMP-2 and plasmin, which degrade components of the basement membrane and interstitial matrix (3). Integrins expressed on endothelial cells modulate cell migration and survival during angiogenesis, whereas integrins expressed on carcinoma cells potentiate metastasis by facilitating invasion and movement across blood vessels (2,4). Antagonists of integrin αvβ3 (antibodies, peptides, and peptidomimetics) can inhibit tumor angiogenesis, tumor growth, and metastasis in vivo (5). The ability to noninvasively visualize and quantify tumor integrin αvβ3 expression level will provide new opportunities to document tumor (tumor cells and sprouting tumor vasculature) integrin expression, to more appropriately select patients for antiintegrin treatment, and to monitor treatment efficacy in integrin-positive patients.
Over the last several years, significant advances have been achieved in developing novel probes for multimodality molecular imaging of tumor integrin expression (6). Small molecules, peptides, peptidomimetic integrin αvβ3 antagonists, and antibodies have been labeled with radioisotopes, superparamagnetic nanoparticles, fluorescent dyes, quantum dots, and microbubbles for PET, SPECT, MRI, near-infrared fluorescence, and ultrasound imaging of small animals, mostly tumor models (6,7). Because of the high sensitivity and adequate spatial and temporal resolution of PET, development of PET probes for integrin expression imaging is currently the most active among all of these modalities.
Cyclic RGD peptide was first labeled with 18F by Haubner et al. and the resulting 18F-galacto-RGD exhibited integrin αvβ3–specific tumor uptake in an integrin-positive M21 melanoma xenograft model (8,9). Initial clinical trials in healthy volunteers and a limited number of cancer patients revealed that this tracer can be administered safely to patients and is capable of delineating certain lesions that are integrin positive with some indication of integrin αvβ3 expression level in vivo (9,10). We labeled a series of mono-, di-, and tetrameric RGD peptides with 18F or 64Cu for integrin-positive tumor targeting (11–19). In particular, the dimeric RGD peptide-based tracer 18F-FRGD2 was found to be able to visualize and quantify the integrin expression level in vivo (11,12).
Radiofluorination of RGD peptides generally uses 18F-synthons such as N-succinimidyl 4-18F-fluorobenzoate (18F-SFB) (20,21) or p-nitrophenyl 18F-fluoropropionate (18F-NFP) (22) to form a stable amide bond by reacting with primary amino groups of RGD peptides. It was also reported that oxoamino derivatives of RGD peptides react with 4-18F-fluorobenzaldehyde (18F-FBA) under acidic condition to form an oxime (23). A few 18F labeled thiol-reactive reagents have been reported in the literature—namely, 1-(4-18F-fluorophenyl)pyrrole-2,5-dione (18F-FPPD) (24), N-[3-(2,5-dioxo-2,5-dihydropyrrol-1-yl)phenyl]-4-18F-fluorobenzamide (18F-DDPFB) (24), 1-[3-(2-(18F-fluoropyridin-3-yloxy)propyl]pyrrole-2, 5-dione (18F-FPyME) (25), and N-[4-[(4-18F-fluorobenzylidene)aminooxyl]butyl]maleimide (18F-FBABM) (26). However, no in vivo microPET data have been reported on tracers synthesized using these prosthetic groups. In this study, we developed a new thiol-reactive synthon, N-[2-(4-18F-fluorobenzamido)ethyl]maleimide (18F-FBEM), for 18F labeling of thiol-containing molecules. Two thiolated RGD peptides were labeled with 18F through 18F-FBEM and tested in murine xenograft models.
MATERIALS AND METHODS
Unless otherwise specified, all chemicals were of analytic grade and commercially available. RGD peptides were synthesized as previously reported (11,12). N-Succinimidyl S-acetylthioacetate (SATA), hydroxylamine·HCl, and tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl) were purchased from Pierce Biotechnology, Inc. N-(2-Aminoethyl)maleimide trifluoroacetate salt, 4-fluorobenzonic acid, N,N,N′,N′-tetramethyl-O-(N-succinimidyl) uronium tetrafluoroborate (TSTU), N-hydroxysuccinimide (NHS), and N,N-diisopropylethylamine (DIPEA) were purchased from Sigma-Aldrich. No-carrier-added 18F-F− was obtained from the in-house PETtrace cyclotron (GE Healthcare). The 18F-F− was provided in a mixture solution of K2CO3 (2 mg/mL in water) and Kryptofix 2.2.2. (Sigma-Aldrich; 10 mg/mL in acetonitrile). The semipreparative reversed-phase high-pressure liquid chromatography (RP-HPLC) system was reported earlier (12). Reversed-phase extraction C18 Sep-Pak cartridges (Waters) were pretreated with methanol and water before use.
The SATA-RGD peptides were prepared following the protocol supplied by the vender. Briefly, c(RGDyK) or E[c(RGDyK)]2 (5 μmol) in 1 mL 50 mmol/L Na2B4O7 buffer (pH 8.5) was mixed with 100 μL SATA solution in dimethyl sulfoxide (DMSO; 6 μmol). After the reaction had gone to completion as shown by analytic RP-HPLC, it was quenched by 100 μL 2% trifluoroacetic acid (TFA) in water. The crude product was lyophilized without purification. The yield of SATA-c(RGDyK) (HPLC retention time [Rt], 12.1 min) was 95% and that of SATA-E[c(RGDyK)]2 (Rt, 13.6 min) was 65% on the basis of analytic RP-HPLC.
The crude SATA-RGD peptides (20 mg) were dissolved in 1 mL water and 100 μL of 0.5 mol/L hydroxylamine solution were added. The pH was adjusted to 6.0. After 2 h, sulfhydryl-c(RGDyK) and sulfhydryl-E[c(RGDyK)]2 (denoted as SRGD and SRGD2, respectively) were purified by semipreparative RP-HPLC. The overall yield was 80% and 50% for SRGD (Rt, 10.7 min) and SRGD2 (Rt, 13.1 min), respectively; little or no dimerization was observed for either peptide when stored under acidic condition (pH 3). MALDI-TOF MS (matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy): SRGD, C29H43N9O9S, calculated 693.3, observed 694.5 ([M+H]+); SRGD2, C61H89N19O19S, calculated 1,423.6, observed 1,422.7 ([M+H]+).
N-(2-Aminoethyl)maleimide (5 μmol) in 200 μL acetonitrile, N-succinimidyl 4-fluorobenzoate (4.5 μmol) in 100 μL acetonitrile, and 500 μL 50 mmol/L Na2B4O7 buffer (pH 8.5) were mixed and reacted at 50°C for 20 min. The reaction was quenched by adding 100 μL 2% TFA in water. HPLC purification gave FBEM in 85% yield. 1H NMR (chloroform-d, 400 MHz): 7.79–7.76 (m, 2H, phenyl o-H); 7.26–7.09 (m, 2H, phenyl m-H); 6.75 (2H, CH=CH); 3.86–3.65 (m, 4H, CH2 − CH2).
The sulfhydryl-RGD peptides (0.5 μmol) were dissolved in 0.5 mL phosphate-buffered saline (PBS, pH 7.4; Invitrogen Corp.). FBEM (0.55 μmol) was dissolved in 200 μL acetonitrile and added to the solution. After 30 min, the reaction mixture was subjected to HPLC purification. FBEM-SRGD (Rt, 14.8 min) and FBEM-SRGD2 (Rt, 15.2 min) were obtained with 80% yield. MALDI-TOF MS: FBEM-SRGD, C42H54FN11O12S, calculated 956.1, observed 956.7 ([M+H]+); FBEM-SRGD2, C73H98FN21O22S, calculated 1,672.5, observed 1,673.5 ([M+H]+).
Radiochemistry
18F-SFB was synthesized as previously reported (12,15) with C18 Sep-Pak cartridge purification. It was dissolved in acetonitrile (300 μL) and 1 mg N-(2-aminoethyl)maleimide in 500 μL acetonitrile and 20 μL DIPEA were added. The reaction mixture was heated to 40°C for 20 min and then quenched by addition of 50 μL TFA. HPLC purification gave 18F-FBEM (Rt, 13.9 min; total reaction time, 150 ± 20 min with nondecay-corrected radio-chemical yield of 5% ± 2% on the basis of 18F-F−; specific activity, 150~ 200 TBq/mmol).
18F-FBEM was dissolved in 600 μL PBS buffer (pH 7.4); 0.2 mg of sulfhydryl-RGD peptide in 50 μL DMSO and 1.0 mg TCEP·HCl in 0.1 mL water were then added. The pH was adjusted to 7.0~ 7.5 using 0.2 mol/L NaOH solution. The reaction mixture was kept at room temperature (r.t.) for 20 min. HPLC purification gave 18F-FBEM-SRGD (Rt, 14.9 min) and 18F-FBEM-SRGD2 (Rt, 15.3 min) in 80% nondecay-corrected yield in both cases. The radiotracers were reconstituted in PBS and passed through a 0.22-μm Millipore filter into a sterile vial for in vivo applications. Nonreacted sulfhydryl-RGD peptides were baseline-separated from the desired products during HPLC and the specific activity of the tracers was 100–150 TBq/mmol.
Eppendorf microcentrifuge tubes containing 500 μL of octanol, 500 μL of normal saline, and ~ 370 kBq of 18F-FBEM-SRGD or 18F-FBEM-SRGD2 were vortexed vigorously for 1 min. Each tube was centrifuged at 14,000 rpm for 5 min and the activities in 20-μL aliquots of both organic and aqueous layers were measured by a γ-counter (GMI, Inc.). The reported octanol/water partition coefficient represents the mean ± SD of 6 measurements.
In Vitro Cell-Binding Assay
Both U87MG and MDA-MB-435 cell lines were purchased from American Type Culture Collection and the culture media were obtained from Invitrogen Co. U87MG glioblastoma cells were grown in Dulbecco’s modified Eagle medium (DMEM, low glucose) and MDA-MB-435 breast cancer carcinoma cells were grown in Leibovitz’s L-15 medium. Both cell lines were cultured in the medium supplemented with 10% (v/v) fetal bovine serum (FBS) at 37°C. In vitro integrin-binding affinities and specificities were assessed via displacement cell-binding assays using 125I-echistatin as the integrin αvβ3–specific radioligand. Both U87MG and MDA-MB-435 cells are integrin αvβ3 positive (12). Cell-binding assay were performed using U87MG cells. The cells were harvested, washed twice with PBS, and resuspended (2 × 106 cells/ mL) in binding buffer (20 mmol/L Tris, pH 7.4, 150 mmol/L NaCl, 2 mmol/L CaCl2, 1 mmol/L MgCl2, 1 mmol/L MnCl2, 0.1% bovine serum albumin). Millipore 96-well filter multiscreen DV plates (pore size, 0.65 μm) were seeded with 105 cells per well and incubated with 125I-echistatin (30,000 cpm/well) in the presence of increasing concentrations of different RGD peptide analogs (0–1,000 nmol/L). The total volume in each well was adjusted to 200 μL. After incubation at r.t. for 2 h, the plates were filtered through a multiscreen vacuum manifold and washed twice with cold binding buffer. The filters were collected and the radioactivity was measured using a γ-counter. The best-fit IC50 (inhibitory concentration of 50%) values for U87MG cells were calculated by fitting the data by nonlinear regression using GraphPad Prism (GraphPad Software, Inc.). Experiments were performed twice with triplicate samples.
Animal Models
All animal experiments were performed under a protocol approved by the Stanford University Administrative Panel on Laboratory Animal Care. The MDA-MB-435 breast cancer model was established by orthotopic injection of 5 × 106 cells into the left mammary fat pad of female athymic nude mice, whereas the U87MG tumor model was obtained by injecting a mixture of 5 × 106 cells suspended in 50 μL medium and 50 μL Matrigel (BD Biosciences) into the right front leg. The mice were used for biodistribution and microPET imaging studies when the tumor volume reached 300–400 mm3 (3–4 wk after inoculation for both U87MG and MDA-MB-435 tumors).
Biodistribution Studies
Female athymic nude mice bearing both U87MG and MDA-MB-435 tumors were injected with 1 MBq of 18F-FBEM-SRGD or 18F-FBEM-SRGD2. The mice were sacrificed and dissected at 10, 30, and 60 min after injection. Blocking experiment was performed by coinjecting radiotracer with a saturating dose of c(RGDyK) (10 mg/ kg mouse body weight) and the mice were sacrificed at 60 min after injection. Blood, tumor, major organs, and tissues were collected and wet weighed (contents in the intestines were removed before weighing). The radioactivity in the tissues was measured using a γ-counter. The results were calculated as percentage injected dose per gram (%ID/g). For each mouse, the radioactivity of the tissue samples was calibrated against a known aliquot of the injectate and normalized to a body weight of 20 g. Values are expressed as mean ± SD for 3 animals per group.
microPET and Image Analysis
PET scans were performed using a microPET R4 rodent model scanner (Concorde Microsystems Inc.). The scanner has a computer-controlled bed and 10.8-cm transaxial and 8-cm axial fields of view (FOVs). It has no septa and operates exclusively in the 3-dimensional list mode. Animals were placed near the center of FOV of the microPET scanner, where the highest image resolution and sensitivity are available. Mice were injected with 3.7 MBq of 18F-FBEM-SRGD or 18F-FBEM-SRGD2 via tail vein under isoflurane anesthesia. The 60-min dynamic (5 × 60 s, 5 × 120 s, 5 × 180 s, 6 × 300 s) microPET data acquisition (total of 21 frames) was started about 3 min after radiotracer injection. Later time-point static images were also acquired as 10-min static images after obtaining a 1-h dynamic scan. The images were reconstructed by a 2-dimensional ordered-subsets expectation maximum algorithm and no correction was applied for attenuation or scatter (27).
For each microPET scan, regions of interests (ROIs) were drawn over each tumor, normal tissue, and major organs by using vendor software ASI Pro 5.2.4.0 on decay-corrected whole-body coronal images. The maximum radioactivity concentration (accumulation) within a tumor or an organ was obtained from mean pixel values within the multiple ROI volume, which were converted to MBq/mL/min by using a conversion factor. Assuming a tissue density of 1 g/mL, the ROIs were converted to MBq/g/min, and then divided by the administered activity to obtain an imaging ROI-derived %ID/g.
Metabolic Stability
Athymic nude mice bearing U87MG tumor were intravenously injected 3.7 MBq of 18F-FBEM-SRGD2. The animals were sacrificed 60 min after tracer injection. Blood, urine, liver, kidneys, and tumor were collected. Blood was immediately centrifuged for 5 min at 13,200 rpm. Organs were homogenized using an IKA Ultra-Turrax T8 homogenizer (IKA Works Inc.), suspended in 1 mL of PBS buffer, and centrifuged for 5 min at 13,200 rpm. After removal of the supernatant, the pellets were washed with 500 μL of PBS. For each sample, supernatants of both centrifugation steps were combined and passed through Sep-Pak C18 cartridges. The urine sample was directly diluted with 1 mL of PBS and then passed through the cartridge. The cartridges were washed with 2 mL of water and eluted with 2 mL of acetonitrile containing 0.1% TFA. After removal of acetonitrile, the residue was redissolved in 1 mL of water and injected onto an analytic HPLC column. Radioactivity was monitored using a solid-state radiation detector. The eluent was also collected using a fraction collector (0.5 min/fraction) and the activity of each fraction was measured by a γ-counter.
Statistical Analysis
Quantitative data are expressed as mean ± SD. Means were compared using 1-way ANOVA and a Student t test. P values < 0.05 were considered significant.
RESULTS
Chemistry
Both monomeric peptide SRGD and the dimeric peptide SRGD2 were synthesized with good overall yield (80% for SRGD and 50% for SRGD2, respectively). FBEM was prepared by reacting SFB with N-(2-aminoethyl)maleimide (90% yield). The conjugation of SRGD or SRGD2 with FBEM was performed at r.t. The resulting conjugates FBEM-SRGD and FBEM-SRGD2 were purified by RP-HPLC and confirmed by MALDI-TOF mass spectrometry.
18F-SFB was synthesized following a previously reported procedure (12,15). 18F-FBEM was obtained by coupling 18F-SFB with N-(2-aminoethyl)maleimide (Fig. 1A). After HPLC purification, the thiol-reactive synthon was allowed to react with SRGD or SRGD2 at r.t. for 20 min. Starting from 18F-F−, the total reaction time including final HPLC purification was about 200 ± 25 min. The overall decay-corrected radiochemical yield was 20% ± 4% (n = 5) for both 18F-FBEM-SRGD (Fig. 1B) and 18F-FBEM-SRGD2 (Fig. 1C). On the basis of 18F-FBEM, both reactions were achieved in high yields (85% ± 5% nondecay corrected), virtually quantitative. The radiochemical purity of the 18F-labeled peptides was >98% according to analytic HPLC. The octanol/water partition coefficient (log P) for 18F-FBEM-SRGD was determined to be 0.93 ± 0.02, indicating the hydrophobic character of this tracer, whereas 18F-FBEM-SRGD2 was hydrophilic (log P = −1.69 ± 0.02).
FIGURE 1.
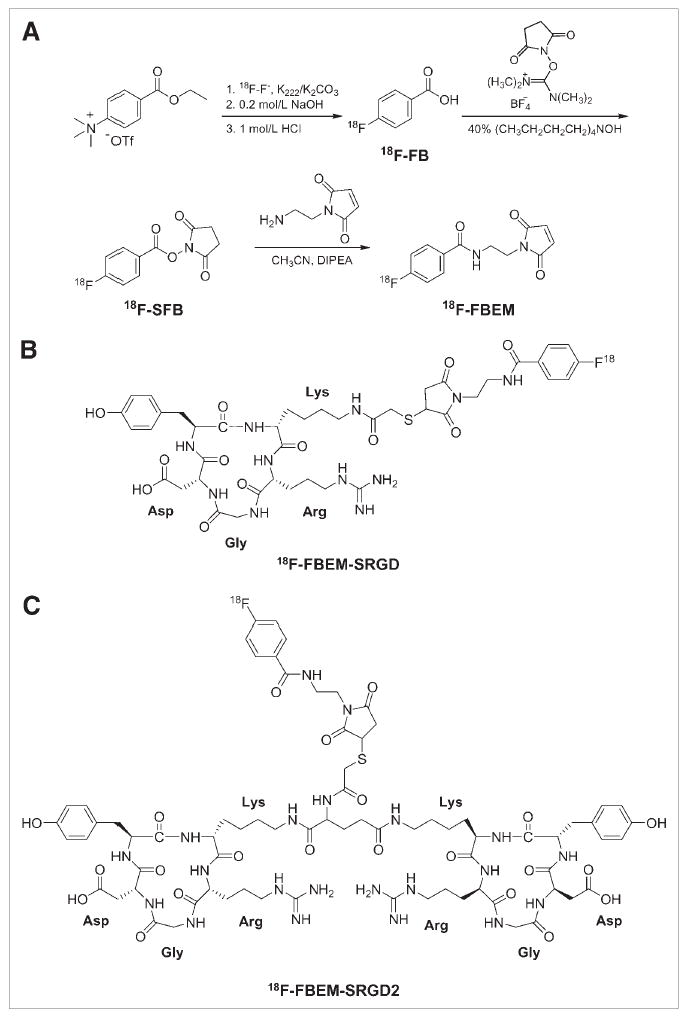
(A) Synthetic route for N-[2-(4-18F-fluorobenzamido)ethyl]maleimide (18F-FBEM). (B) Structure of 18F-FBEM-SRGD. (C) Structure of 18F-FBEM-SRGD2.
In Vitro Cell-Binding Assay
The cell-binding affinity studies of c(RGDyK), E[c(RGDyK)]2, FBEM-SRGD, and FBEM-SRGD2 with U87MG cells are summarized in Figure 2. All 4 peptides inhibited the binding of 125I-echistatin to U87MG cells in a dose-dependent manner. The IC50 values for c(RGDyK), E[c(RGDyK)]2, FBEM-SRGD, and FBEM-SRGD2 were 51.3 ± 4.2, 26.1 ± 3.2, 66.8 ± 5.1, and 55.1 ± 6.5 nmol/L, respectively, indicating that FBEM conjugation had minimal effect on the integrin-binding avidity of the RGD peptides.
FIGURE 2.
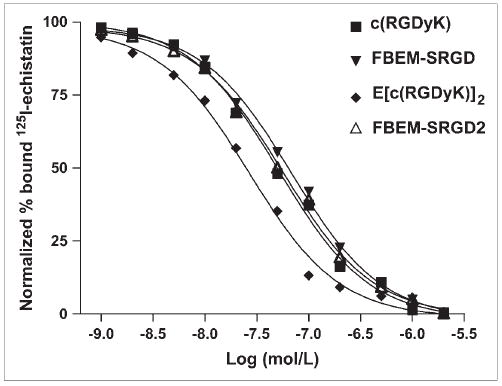
Cell-binding assay of c(RGDyK), E[c(RGDyK)]2, FBEM-SRGD, and FBEM-SRGD2 using U87MG cells (integrin αvβ3–positive human glioblastoma). The cell-binding affinity of the peptides was determined by performing competitive displacement studies with 125I-echistatin. IC50 values for c(RGDyK), E[c(RGDyK)]2, FBEM-SRGD, and FBEM-SRGD2 were 51.3 ± 4.2, 26.1 ± 3.2, 66.8 ± 5.1, and 55.1 ± 6.5 nmol/L, respectively (n = 6).
Biodistribution
Biodistribution of 18F-FBEM-SRGD and 18F-FBEM-SRGD2 was determined in athymic nude mice bearing both U87MG and MDA-MB-435 tumors and the results are shown in Figure 3. For 18F-FBEM-SRGD (Fig. 3A), the U87MG and MDA-MB-435 tumor uptakes were 1.33 ± 0.28 and 1.43 ± 0.11 %ID/g, respectively, at 60 min after injection. When blocked by coinjection of c(RGDyK) at a dose of 10 mg/kg body weight, the tumor uptake decreased to 0.40 ± 0.02 %ID/g for the U87MG tumor (P < 0.05 when compared with the U87MG tumor without blocking) and 0.77 ± 0.04 %ID/g for the MDA-MB-435 tumor (P < 0.05 when compared with the MDA-MB-435 tumor without blocking), respectively. Intestine exhibited a high uptake of 18F-FBEM-SRGD (16.57 ± 0.81 %ID/g when c(RGDyK) was coinjected), most probably due to the relatively hydrophobic nature of the tracer, which is consistent with our previous result for 18F-FB-RGD (18). For 18F-FBEM-SRGD2 (Fig. 3B), the U87MG and MDA-MB-435 tumor uptakes were 2.71 ± 0.19 and 5.25 ± 0.17 %ID/g, respectively, at 60 min after injection (P < 0.01 for U87MG and P < 0.001 for MDA-MB-435 when compared with 18F-FBEM-SRGD). When blocked by coinjection of c(RGDyK) at 10 mg/kg body weight, the tumor uptake of U87MG tumor decreased >5-fold to 0.52 ± 0.26 %ID/g (P < 0.01). Blocking reduced the tumor uptake of both tracers to the background level (due to nonspecific binding in normal organs), clearly indicating integrin-specific binding. Kidney uptake of 18F-FBEM-SRGD2 was high at an early time point (11.40 ± 0.22 %ID/g at 10 min after injection), but the washout was also fast. The 2 tracers exhibited different excretion routes due to the difference in hydrophilicity. 18F-FBEM-SRGD is relatively hydrophobic, therefore exhibiting mainly hepatobiliary excretion, whereas 18F-FBEM-SRGD2 is more hydrophilic and mainly excreted through the kidney. It is worth noting that more radioactivity accumulated in the MDA-MB-435 tumor with time for 18F-FBEM-SRGD2 (Fig. 3B), which might be due to the internalization of the tracer. A similar phenomenon was observed in our previous studies for the 18F-FRGD2 tracer (12). Comparing these 2 radiotracers, 18F-FBEM-SRGD2 has significantly higher kidney and tumor uptake, whereas 18F-FBEM-SRGD has much higher intestine uptake. There was no major difference in the tracer uptake by other organs between the 2 tracers.
FIGURE 3.
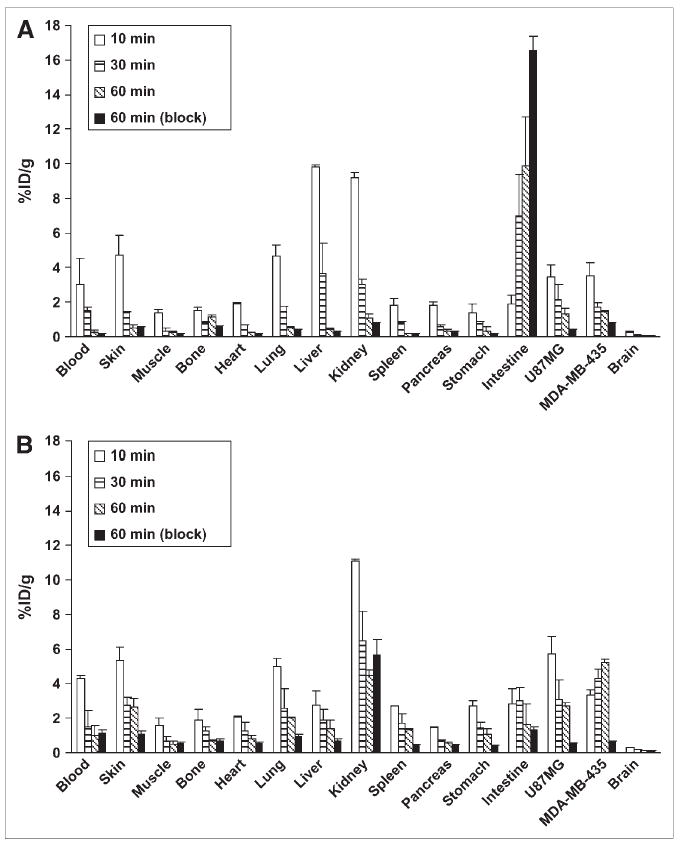
Biodistribution of 18F-FBEM-SRGD (A) and 18F-FBEM-SRGD2 (B) in athymic nude mice bearing both U87MG and MDA-MB-435 tumors at 10, 30, and 60 min after injection (n = 3). Biodistribution of both tracers at 60 min after injection when coinjected with 10 mg/ kg mice body weight of c(RGDyK) is also shown.
microPET
Dynamic microPET scans were performed for both radiotracers. Selected coronal images at different time points after injection in a mouse bearing both subcutaneous U87MG and orthotopic MDA-MB-435 tumors are shown in Figure 4. High tumor activity accumulation was observed as early as 6 min after injection for both tracers. For 18F-FBEM-SRGD, the U87MG and MDA-MB-435 tumor uptakes were 1.27 and 1.04 %ID/g at 60 min after injection, respectively, whereas the liver and kidney uptake were much higher than that of the tumors. Up to 4 h after injection, there is still a fair amount of activity accumulated in the abdomen. 18F-FBEM-SRGD was excreted through both liver and kidneys (Fig. 5A). For 18F-FBEM-SRGD2, most radioactivity in nontargeted tissues was cleared at 60 min after injection. The uptakes in the U87MG, MDA-MB-435, kidneys, liver, and lung at 60 min after injection were 2.14, 2.11, 4.00, 1.46, and 0.58 %ID/g, respectively. Time–activity curves showed that this tracer excreted predominantly through the renal route (Fig. 5B). When the microPET images of these 2 radiotracers were compared, 18F-FBEM-SRGD2 had a much better tumor-to-background contrast and higher tumor uptake, which makes it more suitable for future clinical studies.
FIGURE 4.
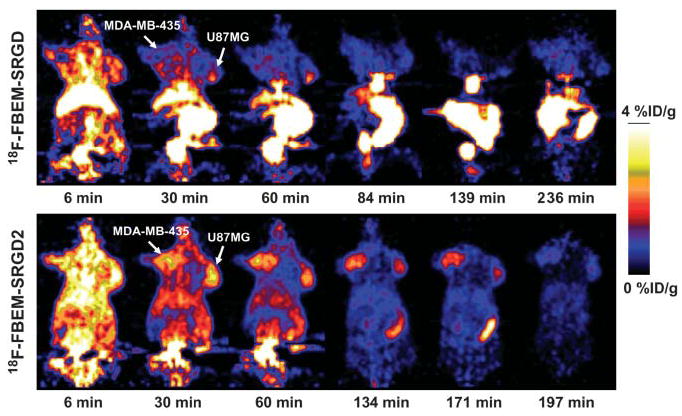
Dynamic microPET scans using both radiotracers at different time points in a mouse bearing both U87MG and MDA-MB-435 tumors. Ten-minute static scans at several later time points were also conducted to complete the tracer kinetic study.
FIGURE 5.
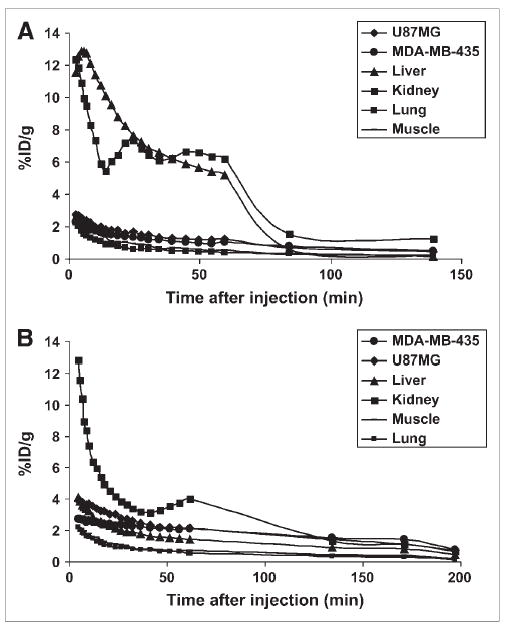
Time–activity curves of 18F-FBEM-SRGD (A) and 18F-FBEM-SRGD2 (B) obtained from microPET scans. The inflection point for tracer clearance is most likely due to the slower metabolism during the dynamic scan when mice were under anesthesia and body temperature was lowered.
Static microPET scans with blocking were then performed for 18F-FBEM-SRGD2 on U87MG tumor-bearing mice (Fig. 6). When coinjected with 10 mg/kg of c(RGDyK), the tracer uptake in the U87MG tumor dropped from 2.21 %ID/g to 0.94 %ID/g at 30 min after injection and from 1.73 %ID/g to 0.44 %ID/g at 60 min after injection, which are essentially at the background level. Successful blocking again confirmed the integrin αvβ3–specific binding of the radiotracer 18F-FBEM-SRGD2.
FIGURE 6.
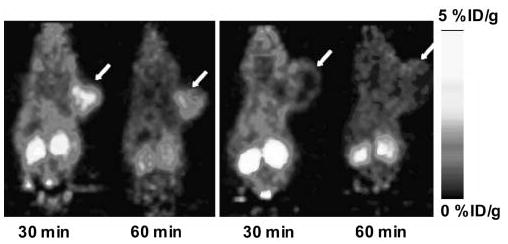
Ten-minute static microPET scans of U87MG tumor-bearing mice (arrows) injected with 3.7 MBq of 18F-FBEM-SRGD2. (Left) Control mouse. (Right) Blocking with 10 mg/kg mouse body weight of c(RGDyK).
Metabolism of 18F-FBEM-SRGD2
The metabolic stability of 18F-FBEM-SRGD2 was also determined in mouse blood and urine samples and in liver, kidneys, and U87MG tumor homogenates at 60 min after tracer injection. The extraction efficiency for all organs was >90% (Table 1). The lowest elution efficiency was found for the U87MG tumor and the liver. HPLC analysis results of the soluble fractions of all samples are shown in Figure 7. The percentage of intact tracer was between 41.7% (urine) and 85.8% (kidney). Although we did not identify the composition of the metabolites, we found that all metabolites came out earlier from the HPLC column than the parent compound. The major metabolite peak was found at about 13 min for almost all organs except the kidney. No defluoridation of 18F-FBEM-SRGD2 was observed, as there is no bone uptake visible in all of the microPET scans.
TABLE 1.
Extraction and Elution Efficiency Data and HPLC Analysis of Soluble Fraction of Tissue Samples at 60 Minutes After Injection of 18F-FBEM-SRGD2
| Organ or tissue | Extraction efficiency (%) | Elution efficiency (%) | Intact fraction (%) |
|---|---|---|---|
| Blood | 91.5 | 94.9 | 80.7 |
| Urine | 100.0 | 99.3 | 41.7 |
| Liver | 91.4 | 66.2 | 59.7 |
| U87MG | 91.0 | 66.2 | 77.5 |
| Kidney | 93.5 | 86.0 | 85.8 |
FIGURE 7.
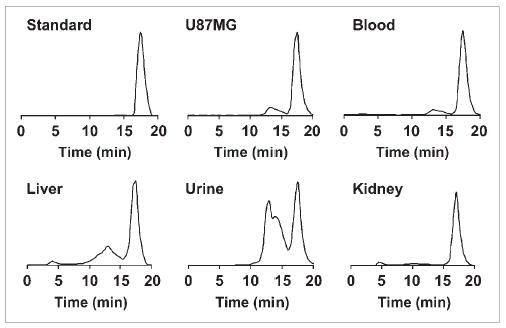
Metabolic stability of 18F-FBEM-SRGD2 in mouse blood and urine samples and in liver, kidneys, and U87MG tumor homogenates 60 min after injection. HPLC profile of tracer itself (Standard) is also shown.
DISCUSSION
18F labeling is generally achieved through 3 types of functional groups: amino group, carboxylic acid group, and sulfhydryl group. Most known 18F-labeling reagents for peptides and protein react with the primary amino groups at the N terminus or the lysine side chain. This can be achieved through active esters, aldehydes, imidates, or azido functionalities. 18F-SFB is probably the most-often-used active ester for 18F labeling via an acylation reaction (12,15,21). Reductive amination using 4-18F-fluorobenzaldehyde (18F-FBA) (28), oxime formation using 18F-FBA (23,29), imidation reaction using 3-18F-fluoro-5-nitrobenzimidate (18F-FNB) (30), photochemical conjugation using 4-azidophenacyl 18F-fluoride (18F-APF) (22), and alkylation reactions using 4-18F-fluorophenacyl bromide (18F-FPB) have been reported earlier (30). The major concern for protein labeling using these reagents is the possible interference with biologic activity: modification of one or more lysines located at or near the active site could reduce the binding affinity through steric hindrance if a bulky group is added. 18F labeling of peptide or protein via the carboxylic acid group at the C terminus or internal glutamic/aspartic acid side chain is less common. Only 2 amines, 1-[4-(18F-fluoromethyl)benzoyl]aminobutane-4-amine (18F-FMBAA) (31) and 4-18F-fluorophenylhydrazine (18F-FPH) (32), have been reported for these amidation reactions.
Thiol-reactive agents have been used to modify peptides and proteins at specific sites, providing high chemoselectivity as compared with amine and carboxylate-reactive reagents (32,33). The disulfide bonds of a protein can be reduced to enable modification using thiol-specific reagents (34–36). More recently, site-directed mutagenesis was used to place cysteine residues on the surface of proteins to provide reactive sulfhydryl groups (37,38). Several thiol-reactive 18F-synthons have been described (24–26), all of which bear a maleimide group allowing for thiol-specific Michael addition reaction. The total synthesis time (100–150 min) and radiochemical yield (10%–20% non-decay corrected) of these synthons are comparable to 18F-FBEM. However, no in vivo data have been reported on tracers synthesized using these prosthetic groups.
We have labeled c(RGDyK) with 18F using 18F-SFB as a prosthetic group (18). The labeling yield was reasonably good for in vivo imaging applications. The resulting 18F-FB-RGD had good tumor-to-blood and tumor-to-muscle ratios but also rapid tumor washout and unfavorable hepatobiliary excretion, making it suitable only for visualizing lesions above the liver (e.g., breast cancer, head and neck cancer, and brain tumor). Because the bent conformation of c(RGDyK) has been optimized to fit into the deep cleft between the α- and β-units of integrin αvβ3, it is unlikely that one can further improve integrin affinity and selectivity of the monomeric RGD peptide by fine tuning the pentapeptide configuration (39). Therefore, the polyvalency effect has been applied to develop dimeric and multimeric RGD peptides, with repeating cyclic pentapeptide units connected by glutamates (11,12,14,19,40). We have found that 18F-FRGD2 (18F-FB-E[c(RGDyK)]2) had predominant renal excretion and almost twice as much tumor uptake in the same animal model as compared with the monomeric tracer 18F-FB-RGD (11,12). The synergistic effect of polyvalency and improved pharmacokinetics may be responsible for the excellent imaging characteristics of 18F-FRGD2. At late time points when most of the nonspecific binding had been cleared, the tumor-to-background ratio also had a linear relationship with the tumor integrin levels, thus making it possible to quantify the tumor integrin expression level in vivo with static PET scans using 18F-FRGD2. We are currently in the process of translating 18F-FRGD2 into clinical trials. In parallel, we developed 18F-FBEM as a prosthetic group for dimeric RGD peptide labeling as well as for protein or engineered antibody labeling through site-specific Michael addition with the sulfhydryl group. The reaction between 18F-FBEM and the thiolated RGD peptides was virtually quantitative. Although 18F-FBEM-SRGD2 demonstrated integrin specificity, as evidenced by effective inhibition of tumor activity accumulation in the presence of a blocking dose of integrin αvβ3 antagonist c(RGDyK), whether it is able to quantify integrin expression in vivo remains to be determined in future studies. It is also worth noting that addition of the thiolated RGD peptides to 18F-FBEM generates a new asymmetric center, resulting in 2 diastereomeric products. Because we only observed one sharp peak in the analytic HPLC for both 18F-FBEM-SRGD and 18F-FBEM-SRGD2, it is very likely that such a small change was not detectable by the HPLC system used.
This study demonstrated that 18F-FBEM could be used to efficiently label peptides through the sulfhydryl group. The major advantage of 18F-FBEM lies in the fact that it can be applied to label a variety of peptides, proteins, or oligonucleotides containing sulfhydryl groups. Because most proteins contain Cys residues, whereas those that do not can be easily engineered to incorporate a Cys residue without compromising the biologic activity, it is expected that 18F-FBEM will have wide applications in the near future for development of novel tracers for in vivo PET.
CONCLUSION
The objective of the present work was to develop a new thiol-reactive 18F-labeling reagent for the prosthetic labeling of peptides and proteins via selective conjugation with a sulfhydryl group. 18F-FBEM was thus incorporated with thiolated monomeric and dimeric RGD peptides via efficient alkylation of the free thiol group though the maleimido function. The advantage of labeling at the sulfhydryl group using 18F-FBEM over labeling at the primary amino group using 18F-SFB was confirmed. The dimeric RGD peptide labeled through the 18F-FBEM strategy showed high integrin affinity in vitro and effective tumor targeting in vivo. The fast tracer clearance, good tumor-to-background contrast, relatively good metabolic stability, and favorable pharmacokinetics of 18F-FBEM-SRGD2 promise further investigation of this tracer in both preclinical and clinical settings for documenting tumor integrin expression (such as the correlation between the tumor uptake and the integrin αvβ3 expression level in vivo). 18F-FBEM also provides a general method of labeling thiol-containing peptides, proteins, antibodies, as well as 5′-thio-functionalized oligonucleotides in high radiochemical yield and high specific activity for successful PET applications.
Acknowledgments
This work was supported, in part, by National Institute of Biomedical Imaging and Bioengineering grant R21 EB001785, Department of Defense (DOD) Breast Cancer Research Program (BCRP) Concept Award DAMD17-03-1-0752, DOD BCRP IDEA Award W81XWH-04-1-0697, DOD Ovarian Cancer Research Program Award OC050120, DOD Prostate Cancer Research Program New Investigator Award DAMD1717-03-1-0143, American Lung Association California, Benedict Cassen Postdoctoral Fellowship from the Education and Research Foundation of the Society of Nuclear Medicine (W. Cai), National Cancer Institute (NCI) Small Animal Imaging Resource Program grant R24 CA93862, and NCI In Vivo Cellular Molecular Imaging Center grant P50 CA114747.
References
- 1.Felding-Habermann B, O’Toole TE, Smith JW, et al. Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci U S A. 2001;98:1853–1858. doi: 10.1073/pnas.98.4.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2:91–100. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- 3.Brooks PC, Stromblad S, Sanders LC, et al. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin αvβ3. Cell. 1996;85:683–693. doi: 10.1016/s0092-8674(00)81235-0. [DOI] [PubMed] [Google Scholar]
- 4.Bogenrieder T, Herlyn M. Axis of evil: molecular mechanisms of cancer metastasis. Oncogene. 2003;22:6524–6536. doi: 10.1038/sj.onc.1206757. [DOI] [PubMed] [Google Scholar]
- 5.Kumar CC. Integrin αvβ3 as a therapeutic target for blocking tumor-induced angiogenesis. Curr Drug Targets. 2003;4:123–131. doi: 10.2174/1389450033346830. [DOI] [PubMed] [Google Scholar]
- 6.Cai W, Gambhir SS, Chen X. Multimodality tumor imaging targeting integrin αvβ3. Biotechniques. 2005;39(suppl):S6–S17. doi: 10.2144/000112091. [DOI] [PubMed] [Google Scholar]
- 7.Haubner RH, Wester HJ, Weber WA, Schwaiger M. Radiotracer-based strategies to image angiogenesis. Q J Nucl Med. 2003;47:189–199. [PubMed] [Google Scholar]
- 8.Haubner R, Wester H-J, Weber WA, et al. Noninvasive imaging of αvβ3 integrin expression using 18F-labeled RGD-containing glycopeptide and positron emission tomography. Cancer Res. 2001;61:1781–1785. [PubMed] [Google Scholar]
- 9.Haubner R, Weber WA, Beer AJ, et al. Noninvasive visualization of the activated αvβ3 integrin in cancer patients by positron emission tomography and [18F]galacto-RGD. PLoS Med [serial online] 2005;2:e70. doi: 10.1371/journal.pmed.0020070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beer AJ, Haubner R, Goebel M, et al. Biodistribution and pharmacokinetics of the αvβ3-selective tracer 18F-galacto-RGD in cancer patients. J Nucl Med. 2005;46:1333–1341. [PubMed] [Google Scholar]
- 11.Chen X, Tohme M, Park R, Hou Y, Bading JR, Conti PS. Micro-PET imaging of αvβ3-integrin expression with 18F-labeled dimeric RGD peptide. Mol Imaging. 2004;3:96–104. doi: 10.1162/15353500200404109. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Xiong Z, Wu X, et al. Quantitative PET imaging of tumor integrin αvβ3 expression with [18F]FRGD2. J Nucl Med. 2006;47:113–121. [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, Hou Y, Tohme M, et al. Pegylated Arg-Gly-Asp peptide: 64Cu labeling and PET imaging of brain tumor αvβ3-integrin expression. J Nucl Med. 2004;45:1776–1783. [PubMed] [Google Scholar]
- 14.Chen X, Liu S, Hou Y, et al. MicroPET imaging of breast cancer αv-integrin expression with 64Cu-labeled dimeric RGD peptides. Mol Imaging Biol. 2004;6:350–359. doi: 10.1016/j.mibio.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Chen X, Park R, Hou Y, et al. MicroPET imaging of brain tumor angiogenesis with 18F-labeled PEGylated RGD peptide. Eur J Nucl Med Mol Imaging. 2004;31:1081–1089. doi: 10.1007/s00259-003-1452-2. [DOI] [PubMed] [Google Scholar]
- 16.Chen X, Park R, Shahinian AH, et al. 18F-Labeled RGD peptide: initial evaluation for imaging brain tumor angiogenesis. Nucl Med Biol. 2004;31:179–189. doi: 10.1016/j.nucmedbio.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Chen X, Sievers E, Hou Y, et al. Integrin αvβ3-targeted imaging of lung cancer. Neoplasia. 2005;7:271–279. doi: 10.1593/neo.04538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, Park R, Tohme M, Shahinian AH, Bading JR, Conti PS. MicroPET and autoradiographic imaging of breast cancer αv-integrin expression using 18F- and 64Cu-labeled RGD peptide. Bioconjug Chem. 2004;15:41–49. doi: 10.1021/bc0300403. [DOI] [PubMed] [Google Scholar]
- 19.Wu Y, Zhang X, Xiong Z, et al. MicroPET imaging of glioma αv-integrin expression using 64Cu-labeled tetrameric RGD peptide. J Nucl Med. 2005;46:1707–1718. [PubMed] [Google Scholar]
- 20.Mading P, Fuchtner F, Wust F. Module-assisted synthesis of the bifunctional labelling agent N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB) Appl Radiat Isot. 2005;63:329–332. doi: 10.1016/j.apradiso.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 21.Vaidyanathan G, Zalutsky MR. Improved synthesis of N-succinimidyl 4-[18F]fluorobenzoate and its application to the labeling of a monoclonal antibody fragment. Bioconjug Chem. 1994;5:352–356. doi: 10.1021/bc00028a012. [DOI] [PubMed] [Google Scholar]
- 22.Wester HJ, Hamacher K, Stoecklin G. A comparative study of n.c.a. fluorine-18 labeling of proteins via acylation and photochemical conjugation. Nucl Med Biol. 1996;23:365–372. doi: 10.1016/0969-8051(96)00017-0. [DOI] [PubMed] [Google Scholar]
- 23.Poethko T, Schottelius M, Thumshirn G, et al. Two-step methodology for high-yield routine radiohalogenation of peptides: 18F-labeled RGD and octreotide analogs. J Nucl Med. 2004;45:892–902. [PubMed] [Google Scholar]
- 24.Shiue CY, Wolf AP, Hainfeld JF. Synthesis of 18F-labelled N-(p-[18F]fluorophenyl)maleimide and its derivatives for labelling monoclonal antibody with 18F. J Labelled Compds Radiopharm. 1998;26:287–289. [Google Scholar]
- 25.de Bruin B, Kuhnast B, Hinnen F, et al. 1-[3-(2-[18F]Fluoropyridin-3-yloxy) propyl]pyrrole-2,5-dione: design, synthesis, and radiosynthesis of a new [18F]fluoropyridine-based maleimide reagent for the labeling of peptides and proteins. Bioconjug Chem. 2005;16:406–420. doi: 10.1021/bc0497463. [DOI] [PubMed] [Google Scholar]
- 26.Toyokuni T, Walsh JC, Dominguez A, et al. Synthesis of a new heterobifunctional linker, N-[4-(aminooxy)butyl]maleimide, for facile access to a thiol-reactive 18F-labeling agent. Bioconjug Chem. 2003;14:1253–1259. doi: 10.1021/bc034107y. [DOI] [PubMed] [Google Scholar]
- 27.Visvikis D, Cheze-LeRest C, Costa DC, Bomanji J, Gacinovic S, Ell PJ. Influence of OSEM and segmented attenuation correction in the calculation of standardised uptake values for [18F]FDG PET. Eur J Nucl Med. 2001;28:1326–1335. [PubMed] [Google Scholar]
- 28.Herman LW, Fischman AJ, Tompkins RG, et al. The use of pentafluorophenyl derivatives for the 18F labelling of proteins. Nucl Med Biol. 1994;21:1005–1010. doi: 10.1016/0969-8051(94)90091-4. [DOI] [PubMed] [Google Scholar]
- 29.Schottelius M, Poethko T, Herz M, et al. First 18F-labeled tracer suitable for routine clinical imaging of sst receptor-expressing tumors using positron emission tomography. Clin Cancer Res. 2004;10:3593–3606. doi: 10.1158/1078-0432.CCR-03-0359. [DOI] [PubMed] [Google Scholar]
- 30.Kilbourn MR, Dence CS, Welch MJ, Mathias CJ. Fluorine-18 labeling of proteins. J Nucl Med. 1987;28:462–470. [PubMed] [Google Scholar]
- 31.Shai Y, Kirk KL, Channing MA, et al. 18F-Labeled insulin: a prosthetic group methodology for incorporation of a positron emitter into peptides and proteins. Biochemistry. 1989;28:4801–4806. doi: 10.1021/bi00437a042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilbur DS. Radiohalogenation of proteins: an overview of radionuclides, labeling methods and reagents for conjugate labeling. Bioconjug Chem. 1992;3:432–470. doi: 10.1021/bc00018a001. [DOI] [PubMed] [Google Scholar]
- 33.Brinkley M. A brief survey of methods for preparing protein conjugates with dyes, haptens, and cross-linking reagents. Bioconjug Chem. 1992;3:2–13. doi: 10.1021/bc00013a001. [DOI] [PubMed] [Google Scholar]
- 34.Iznaga-Escobar N. Direct radiolabeling of monoclonal antibodies with rhenium-188 for radioimmunotherapy of solid tumors: a review of radiolabeling characteristics, quality control and in vitro stability studies. Appl Radiat Isot. 2001;54:399–406. doi: 10.1016/s0969-8043(00)00277-3. [DOI] [PubMed] [Google Scholar]
- 35.Rhodes BA. Direct labeling of proteins with 99mTc. Int J Rad Appl Instrum B. 1991;18:667–676. doi: 10.1016/0883-2897(91)90004-5. [DOI] [PubMed] [Google Scholar]
- 36.Saito G, Swanson JA, Lee KD. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities. Adv Drug Deliv Rev. 2003;55:199–215. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 37.Bragg PD. Site-directed mutagenesis of the proton-pumping pyridine nucleotide transhydrogenase of Escherichia coli. Biochim Biophys Acta. 1998;1365:98–104. doi: 10.1016/s0005-2728(98)00049-8. [DOI] [PubMed] [Google Scholar]
- 38.Chorostowska-Wynimko J, Swiercz R, Skrzypczak-Jankun E, Wojtowicz A, Selman SH, Jankun J. A novel form of the plasminogen activator inhibitor created by cysteine mutations extends its half-life: relevance to cancer and angiogenesis. Mol Cancer Ther. 2003;2:19–28. [PubMed] [Google Scholar]
- 39.Xiong JP, Stehle T, Zhang R, et al. Crystal structure of the extracellular segment of integrin αvβ3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 40.Janssen M, Oyen WJ, Massuger LF, et al. Comparison of a monomeric and dimeric radiolabeled RGD-peptide for tumor targeting. Cancer Biother Radiopharm. 2002;17:641–646. doi: 10.1089/108497802320970244. [DOI] [PubMed] [Google Scholar]