

Abstract
We previously described a filament-based antibody recognition assay (FARA) that generates ELISA-like sandwich structures immobilized on a filament. FARA allows the coupling of antibodies to precise locations along a filament, on-line fluorescence detection of captured pathogen, and feedback-directed filament motion. These properties suggest that this approach might be useful as an automated means to rapidly classify unknown pathogens. In this report, we describe validation of the novel decision tree aspect of this technology using mammalian reovirus. Based on available antibodies, we developed a decision tree algorithm to detect virus with increasing specificity at each level of the tree. Using three strains of reovirus and a bacteriophage control, our system correctly classified the reovirus strains at a concentration of 2 × 1012 virions ml−1 and M13K07 phage at 3 × 1011 virions ml−1. Classification of reovirus strain type 3 Dearing (T3D) required three levels of testing: general reovirus classification in level 1, serotype 3 classification in level 2, and final T3D strain classification in level 3. Strain T3SA + also required three levels of testing before a final classification was returned in level 3. Classification of strain type 1 Lang (T1L) required two levels of testing. M13K07 phage detection required only one level of testing for classification. These results indicate that automated pathogen classification using FARA is feasible. Furthermore, the simplicity of the design could be exploited for development of more complex sub-classification networks with additional levels and branches.
Keywords: Virus detection, Virus classification, Fluorescence detection, Pathogen detection
Introduction
Identification of specific pathogens is essential for the selection of pathogen-specific treatments, minimizing the spread of infection, and monitoring for long-term complications. Currently, specific pathogen identification is achieved through available RT-PCR and antibody-based strategies. In the clinic, these tests are usually applied consecutively to evaluate for the presence of the most likely to least likely pathogen based on patient findings. An automated classification strategy that is less dependent on clinical knowledge but achieves rapid accurate identification of a single pathogen from among a group of possible pathogens is currently unavailable.
Our previously described filament-based antibody recognition assay (FARA) employs antibodies immobilized at known locations along a filament to detect specific pathogens.9,10 The filament is pulled through a series of small reaction chambers, and pathogens, if present, are captured by filament-bound antibodies. Detection of pathogen binding is achieved by using a fluorescently labeled second antibody specific for the pathogen.
FARA was first reported using immobilized anti-M13K07 antibody to detect M13K07 phage.10 This virus and antibody pair provided a well-characterized test system to demonstrate the feasibility of a filament-based, pathogen-detection platform. However, in this first generation approach, the filaments were removed from the system for fluorescence scanning. Subsequent improvements to FARA include an integrated fluorescence detector and a feedback algorithm to control filament position.9 The integrated detector enables adaptive pathogen detection in which regions of interest along the filament can be reincubated in the appropriate reaction chambers to increase filament fluorescence when the initial signal is low.
In this report, we describe a small-scale test of the use of a simple classification tree together with feedback-controlled FARA to identify four viruses. The feedback feature of online FARA is used to guide the selection of subsequent tests within the classification tree. Unnecessary tests are not performed, and each subsequent test becomes more specific for a single pathogen. These results establish FARA as a robust platform for classification of diverse pathogens.
Materials and Methods
Cells and Viruses
Murine L929 (L) cells were cultured in suspension in Joklik’s modified Eagle’s medium supplemented to contain 5% fetal bovine serum, 2 mM l-glutamine, 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, and 0.25 g mL−1 amphotericin-B. Reovirus strains T1L and T3D are laboratory stocks. T3SA + is a monoreassortant virus isolated from L cells co-infected with T1L and T3C44MA.1 T3SA + contains the S1 gene segment from the type 3 parental strain and all other gene segments from T1L.1 Reovirus particles were purified as previously described.1,3,6 L cells were inoculated with second-passage L-cell lysate stocks of twice plaque-purified reovirus at a multiplicity of infection of 10 plaque-forming units per cell. Virus was purified from infected cells by freon extraction and CsCl-gradient centrifugation. Purified M13K07 virus was obtained from the Vanderbilt Molecular Recognition Core.
Antibodies
Mouse monoclonal antibodies 4F2, 5C6, 8H6, and 9BG5 specific for reovirus proteins2,12,13 (Table 1) were purified from mouse hybridoma supernatants using Protein A column chromatography. Anti-M13K07 monoclonal antibody was purchased from Amersham Biosciences (Piscataway, NJ).
Table 1.
Antibody specificity.
| Antibody | Protein specificity | Reovirus strain | ||
|---|---|---|---|---|
| T1L | T3D | T3SA+ | ||
| 5C6 | T1 σ1 | + | − | − |
| 9BG5 | T3 σ1 | − | + | + |
| 8H6 | T1 and T3 μ1 | + | + | + |
| 4F2 | T3D σ3 | − | + | − |
Antibodies 4F2 and 8H6 were used for fluorescence detection of virus (step 4, Table 2) and were labeled with Alexa Fluor 555 or Alexa Fluor 647 (AF555 or AF647, Molecular Probes, Eugene, OR), respectively, according to the manufacturer’s instructions (Molecular Probes). Labeled antibodies were purified using PD-10 size-exclusion chromatography (Amersham Biosciences). Antibody concentration and number of fluores per labeled antibody were determined by using absorbance measurements at 280 nm and the peak absorbance wavelength of each label. Aliquots of both labeled and unlabeled antibodies were stored at −20°C, and working solutions were kept at 4°C. Final antibody concentration was adjusted immediately prior to experiments.
Table 2.
The five reaction chambers in FARA processing
| Chamber | Description | Solution | Incubation time | Chamber ID / Volume (mm / μl) |
|---|---|---|---|---|
| 1 | Block/Wash Filament | PBS-Ta | 15 min | 2/235 |
| 2 | Virus Incubation | Unknown Virus | 40 min | 1/60 |
| 3 | Wash | PBS-T | 1 min | 2/235 |
| 4 | Detecting Antibody Incubation | Fluorescently Labeled Detecting Antibody | 5 min | 1/60 |
| 5 | Wash | PBS-T | 1 min | 2/235 |
aTween-20 (0.1%) was added to PBS as a blocking agent.
Filament Preparation
Capture antibodies were passively adsorbed to the filament surface in groups of three by placing the filament across the concave teeth of a PhastGel sample applicator (Amersham Biosciences) (Fig. 1). Three capture-antibody regions, corresponding to the three levels of testing, were prepared along each filament using three applicator combs glued end to end. Antibody solution pipetted onto the filament without the comb spread unimpeded along the filament. Surface tension within the teeth of the comb overcame this tendency and produced a small, distinct circumferential band of immobilized antibody. Antibody was spotted in a volume of 0.75 μL and allowed to passively adsorb to the filament for 45 min in a humidified box. Following incubation at 25°C, filaments were rinsed in phosphate buffered saline with 0.1% Tween 20 (PBS-T) and threaded through the reaction chambers for virus detection experiments. Preliminary experiments were performed to determine the optimal concentration of each capture antibody.
Figure 1.
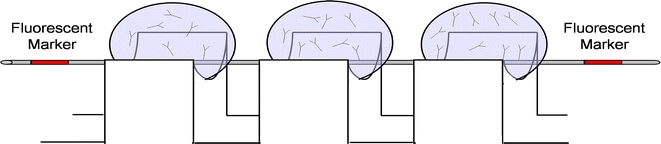
Schematic of a device for adsorption of capture antibodies to the filament. The filament is placed within the concave teeth of a PhastGel applicator to localize the spotted antibody to a small circumferential band around the filament.
Red fingernail polish (Poisonberry, Noxel Corporartion, Hunt Valley, MO) was used as a visible and fluorescent fiducial marker to identify the leading and trailing edges of capture-antibody regions during experiments and during laser scanning. The polish was applied by pipette between the teeth of the comb flanking the antibody region. A simple bar code system was used to identify each of the three capture-antibody regions. The first and second regions were preceded by a single fiducial marker, and the third region was preceded by two fiducial markers.
Micro-reaction Chambers
Glass microreaction chambers were fashioned from 0.25 inch stock tubing into 75 mm lengths. The ends were flared outward to facilitate movement of the filament through the chambers. Interior diameters of the chambers were 1 or 2 mm depending on the required reaction volume (Table 2). Chambers were carefully positioned in a straight line on the top of a horizontal aluminum stage using machined aluminum mounts.9 Fine adjustment of chamber position was achieved by using oblong mounting holes on each mount. PBS-T was used in all wash chambers and for virus and labeled antibody solutions. Reovirus was used at a concentration of 2 × 1012 virions mL−1, and phage M13K07 was used at a concentration of 3 × 1011 virions mL−1. Detecting antibodies were present in the detecting-antibody chamber at a concentration of approximately 40 μg mL−1 for each antibody in the three antibody mixture. Solutions were added to appropriate chambers at the initiation of experiments and used for all three levels of testing (Table 2). If testing proceeded to level three, fluid loss from the chambers was monitored and replenished if necessary.
Filament Control
Movement of the filament and, therefore, the antibody bands through the chambers, was achieved by using a rotary stage to wind or unwind the filament around a spindle. A small weight was attached to the opposite end of the filament to maintain a constant tension. Filament positioning to within several microns was achieved by using a rotary stage encoder from Yaskawa Instruments (Waukegan, IL) and a custom control algorithm written as a LabView Virtual Instrument (VI) (National Instruments, Austin, TX). Parameters including filament speed and residence times within chambers were controlled by using the LabView software interface.
Between chambers, the speed of the filament was 1 cm sec−1. Within each chamber, the capture-antibody region undergoing processing was oscillated 2.5 cm back and forth at a speed of 1 cm sec−1 to increase interactions between immobilized molecular structures attached to the filament and the molecular species in solution. Within the detector, filament speed was 4 cm sec−1.
Lasers and PMTs
Filament fluorescence was measured by passing the filament between two diode lasers. The two laser excitation sources were attached to either side of a detection chamber (Fig. 2). Laser 1 (638 nm, 25 mW diode laser; Coherent, Santa Clara, CA) was used to excite the antibody tag AF647. Laser 2 (532 nm, 20 mW diode-pumped, solid state laser; B&W Tek, Inc., Newark, DE) was used to excite the antibody tag AF555. The effective power of laser 1 was reduced to approximately 5 mW by using a polarizer and an excitation slit. Laser 2 was not attenuated. Two Hamamatsu R928 photomultiplier tubes (PMT) were attached to the top and bottom of the sample chamber and powered by 850 and 800 V signals for the AF647 and AF555 channels, respectively. Current from the PMT was converted to voltage by using transimpedance amplifiers that amplified the signal by a factor of 106 for AF647 and 105 for AF555. Voltage was sampled at a rate of 800 samples sec−1 by using a digital acquisition board from National Instruments (DaqPAD 6020E). Fluorescence signal was acquired as a function of filament position by using LabView. Following fluorescence scanning for most experiments, filament regions were cut and scanned again in a microarray flatbed scanner (GenePix 4000B, Axon Instruments, Union City, CA).
Figure 2.
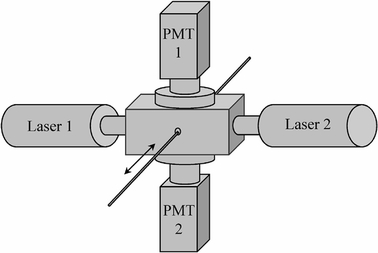
Schematic of lasers and photomultipliers (PMT) to detect online fluorescence of the filament. The filament is moved (arrow) through a rectangular sample chamber with a laser excitation source attached to either side. Photomultiplier tubes are attached to the top and bottom of the chamber.
Filters
Filter sets were placed in the light path between the sample chamber and the photomultipliers (Fig. 2). Long-pass filters with cutoffs at 685 nm (Chroma, Rockingham, VT) and 665 nm (Melles Griot, Rochester, NY) were combined to reduce reflected laser light from the AF647 laser. For the AF555 channel, two long-pass filters (570 nm cutoff, Melles Griot) were combined with a bandpass filter centered at 565 nm (30 nm bandwidth, Chroma) to reduce reflected light.
Classification Algorithm
The LabView program coordinated all filament movement, filament scanning, and feedback control. Elements of the feedback control are described by the nodes of the decision tree used to classify the viruses shown in Fig. 3. The fluorescent fiducial markers on either side of the capture-antibody region produced characteristically sharp emission peaks. Since the physical location of the immobilized antibody positions between the markers was known, experimental conclusions were based on the distance of the first detected peak from the initial fluorescent marker.
Figure 3.
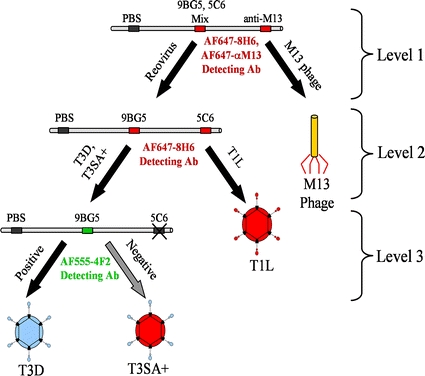
Decision-tree algorithm to classify virus strains. The filament control program enters the decision tree from the top at level 1 and, based on the type of virus found at each level, follows different branches of the decision tree. When reovirus is detected at level one, the filament is moved forward to the next antibody region for level 2 testing. If a serotype 3 reovirus strain is detected (T3D or T3SA+ ) at level 2, testing continues to level 3 to distinguish between these strains.
The filament-control program was designed to find each peak, calculate its location along the filament, and make a decision about additional tests. Spatially localized fluorescence from the filament was measured as a 0–14 V signal from the transimpedance amplifier. Voltage data from the scan were used as input data by a peak-detection function in LabView. The LabView peak detector fit a quadratic polynomial to sequential sets of points depending on a width parameter entered by the user. Data were then compared to a threshold parameter, also entered by the user, to identify peaks. A binary decision to stop or continue testing was based on the location and number of peaks found. Peak-detection threshold parameters were defined as 0.3 V (approximately three times background) with a width of 30 data points, which corresponded to a width slightly larger than the physical width of the comb tooth.
A common set of parameters was used in all experiments based on previous work with M13K07.10 A filament region containing three capture antibodies and two fiducial markers was incubated within each chamber for the times shown in Table 2. Each of the capture-antibody regions contained a PBS negative control in the first position. In level one the second position was a mixture of 9BG5 and 5C6 (0.25 mg mL−1, 0.2 mg mL−1). The third position was anti-M13K07 (0.5 mg mL−1). In levels two and three, the negative control position was followed by 9BG5 (mg mL−1) in the second position and 5C6 (0.2 mg mL−1) in the third position.
The logic encoded in the decision tree shown in Fig. 3 was followed. In level 1, if fluorescence was detected from the anti-M13K07 position of the first region of the filament, a classification of M13K07 virus was made, and no further testing was conducted. If fluorescence was detected in the 9BG5/5C6 region, a classification of reovirus was made and the filament was advanced to the appropriate region for level 2 testing, where a more specific test for reovirus was performed. In level 2 testing, fluorescence from the 5C6 region indicated a serotype 1 reovirus, which in this scheme corresponded to reovirus T1L. A classification of T1L represented an endpoint in the decision tree, and testing was discontinued. Fluorescence from the 9BG5 region indicated the presence of a serotype 3 reovirus, and the program advanced the filament for further subtyping in level 3. Level 3 testing used AF555 conjugated 4F2 antibody as the detecting antibody and a second detection channel with a green excitation laser. If fluorescence was found in the 9BG5 region using this channel, a classification of reovirus T3D was made and testing along that branch ended. If no fluorescence was detected from the second channel for that region, the program ended with a classification of reovirus T3SA+ .
If no peaks were found between the fiducial markers, a message was generated indicating that no virus was found. Similarly, if a peak was detected in the negative control region of the filament, a warning message was generated. Although theoretically possible, this situation did not arise during testing.
Results
FARA utilizes a polyester filament with circumferential bands of immobilized antibody that is passed through a series of five glass micro-reaction chambers that are similar to the five major steps of an ELISA (Table 2). In the first reaction chamber of the FARA approach employed in this study, capture antibody coupled to the filament was rehydrated. In this chamber the filament also was blocked to prevent non-specific binding of virus. In the second chamber, the immobilized capture antibody was incubated with virus solution. In this chamber, if virus in solution bound to antibody coupled to the filament, then the virus became coupled to the filament via this interaction. In the third chamber, non-specifically bound virus was removed by washing before incubation with a fluorescently labeled detecting antibody in the fourth chamber. A final wash in the fifth chamber removed non-specifically bound detecting antibody. The capture antibody region was then passed through an integrated fluorescence detector.
In these experiments, the test virus was classified with greater specificity at each level of a decision tree (Fig. 3). Representative fluorescence signals in volts obtained during testing for phage M13K07 and reoviruses T1L, T3D, and T3SA+ are shown in Fig. 4. Labels for the PBS control position and the antibodies in each capture antibody position are shown adjacent to the filament. The specificity of each test antibody for its corresponding virus was high, with little or no cross-reactivity. No signal was detected in the PBS negative-control position.
Figure 4.
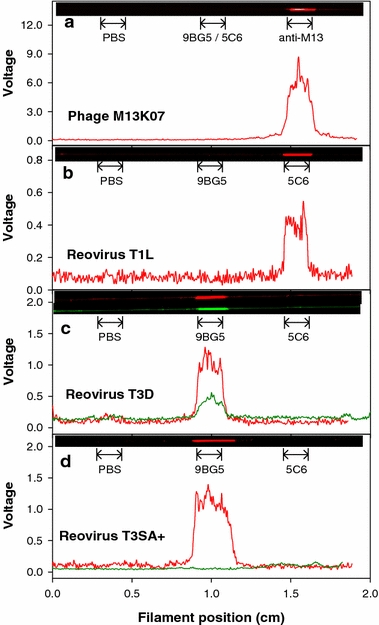
Fluorescence intensity in volts as a function of filament position for M13K07 phage (A) and reoviruses T1L (B), T3D (C), and T3SA+ (D). The immobilized capture antibody positions on the filament are labeled within each panel. Captured virus was detected by a fluorescently labeled second antibody (anti-M13K07 [red], 8H6 [red], or 4F2 [green]). Corresponding images of the scanned filaments are shown at the top of each graph. Each panel shows a representative experiment of three performed.
Detection of M13K07 was achieved by level 1 testing. Strong fluorescence was observed in the anti-M13K07 position but not in the 9BG5/5C6 or PBS positions (Fig. 4a). This finding indicated capture of the phage M13K07 by the anti-M13K07 capture antibody. For detection of M13K07, AF647 conjugated anti-M13K07 was used. Because a classification of M13K07 was made, the other two capture regions of the filament were not evaluated.
Detection of the reovirus strains employed in this study required analysis beyond level 1. Level 1 testing indicated the presence of a reovirus for all three strains with a peak in the second position corresponding to virus capture by the 9BG5/5C6 antibody mixture. In level 2 testing, strain T1L was detected in the 5C6 antibody position but not in the 9BG5 or PBS positions (Fig. 4b). In this case, the capture and detecting antibodies differed. Because a reovirus T1L classification was made, the third capture region of the filament was not evaluated.
For reovirus T3D, level 1 testing indicated the presence of a reovirus with a peak in the second position corresponding to virus capture by 9BG5/5C6 (data not shown). Level 2 testing showed a distinct peak in the 9BG5 antibody position (Fig. 4c, red curve). In this case, 8H6 antibody labeled with AF647 was the detecting antibody indicating a type 3 reovirus. Level 3 testing showed a distinct peak in the 9BG5 antibody position (Fig. 4c, green curve). In this case, 4F2 antibody labeled with AF555 was the detecting antibody. Neither curve showed cross reactivity of reovirus T3D with the immobilized 5C6 region or the negative-control PBS region.
For reovirus T3SA+ , level 1 testing indicated the presence of a reovirus with a peak in the second position corresponding to virus capture by 9BG5/5C6 (data not shown). Level 2 testing yielded a distinct peak in the 9BG5 antibody position (Fig. 4d, red curve), like the result obtained with T3D. However, level 3 testing with 4F2 did not yield a positive signal in the 9BG5 antibody position, indicating that the captured virus was not T3D. No detectable cross reactivity was found in the 5C6 antibody region or the negative-control PBS region. Thus, the virus was classified as T3SA+.
Discussion
In this study, we demonstrated the feasibility of a FARA pathogen-classification approach using a well-characterized virus system. Three reovirus strains and one phage were successfully classified by performing sequential antibody-binding assays directed by the decision tree. The structure of the decision tree was based on published characteristics of each virus strain and the known specificity of each antibody. Because the capture antibodies were arrayed at known locations along the filament, and the fluorescence of the detection antibodies was measured as a function of filament location, an increase in filament fluorescence at a particular location indicated antibody interaction with the virus. For each virus strain tested, the expected fluorescence peaks were observed, and the correct classification was made. Moreover, successful transit of all decision tree branches was demonstrated.
The biochemical components of FARA are similar to those used in standard ELISAs. Both assays create a dual-antibody sandwich that results in similar detection limits for M13K07 for FARA and ELISA.10 One of the virus-specific antibodies acts as the capture antibody, and the second acts as the detecting antibody. The main difference in the biochemistry of the two assays is that, in the current implementation of FARA, enzyme amplification is not utilized. Like ELISA, FARA utilizes antibodies adsorbed to the surface of a substrate to capture virus from solution. The capacity of bound antibody to retain its antigen-binding activity is essential to the success of both assays. Each of the immobilized test antibodies in our system retained functionality and bound its corresponding virus (Fig. 4). The absence of peaks in the negative-control positions and in positions occupied by antibodies not specific to the test virus demonstrates minimal cross reactivity in the FARA platform employed in this study.
We chose reovirus as a test system to show clinical relevance and to avoid potential safety concerns in the laboratory. Reovirus has been used as a model to study mechanisms of viral pathogenesis in mice.14 Although it is a human pathogen, it is rarely associated with human disease.11 Many reovirus field-isolate strains have been characterized,4,7,8 and a broad array of antibodies are available that recognize reovirus with varying specificity.12
We found that not every antibody tested was suitable for use as an immobilized capture antibody. Neither antibody 8H6 nor 4F2 bound virus when immobilized on the filament, even after virus incubation time was increased to greater than 100 min (data not shown). It is possible that these antibodies undergo conformational changes when passively adsorbed to a solid substrate, rendering them inactive. Alternatively, these antibodies may be incapable of antigen binding when immobilized as a consequence of steric hindrance. Antibodies 8H6 and 4F2 recognize the μ1 and σ3 proteins, respectively,12 which form the bulk of the viral outer capsid.5 It is possible that extension of the viral attachment protein σ1 may shield the μ1 and σ3 proteins from binding to the 8H6 and 4F2 antibodies on the filament surface. This conclusion also is consistent with our observation that 9BG5 and 5C6 were both excellent capture antibodies, since they recognize the σ1 protein, which extends farthest from the viral capsid. The failure of the 8H6 and 4F2 antibodies to function as effective capture antibodies complicated our virus detection scheme. Although 8H6 is not serotype-specific and should bind all reovirus strains, we could not use this antibody as the immobilized capture antibody for level 1 testing. Antibodies 5C6 and 9BG5 are specific for serotype 1 and serotype 3 reovirus strains, respectively;12 therefore, we immobilized a mixture of these antibodies for detection of all reovirus isolates in level 1. Although 4F2 recognizes most type 3 strains, T3SA+ is a reassortant virus that contains a σ3-encoding gene segment from T1L.1 As a result, 4F2 does not recognize T3SA+ and is specific only for T3D in our scheme. Since 4F2 could not be used as a capture antibody in level 3, we incorporated a fluorescently labeled 4F2 antibody as a detecting antibody in solution.
Although the nature of the biochemical interactions is similar in FARA and ELISA, the capture antibody employed in a standard ELISA is static, and solutions are changed in the well-plate in a fixed sequence. In FARA, the capture antibody is attached to a mobile substrate, and solutions are changed by positioning the filament in different solution chambers. As we have shown previously, this gives FARA a sensitivity limit similar to ELISA on the order of 107 virus particles.10 A major advantage of FARA is that it is dynamic and allows modification of processing in response to results.9 Virus incubation time with the capture antibody can be reduced to as low as one minute depending on the virus concentration and antibody affinity, reducing the overall assay duration. On the other hand, the incubation time can be increased in order to increase sensitivity.9 Moreover, because the filament is mobile, the capture antibody is brought to the virus solution, thus enabling use of capture antibodies with different specificities to interrogate a single aliquot of virus solution.
Attachment of the capture antibody to a mobile filament in FARA makes accurate positioning of the filament and correct identification of the capture antibody regions essential for virus strain classification. These parameters are particularly important in multilevel testing of the type used in our study. A simple bar code system based on fluorescent fiducial markers was used to identify the leading edge of each of the capture antibody regions and position the filament in the reaction chambers. The pattern of the fluorescent marks also was used to identify the level of testing. Since our simple test system involved three levels, and testing always began in level 1, only searches for patterns containing one or two fluorescent marks corresponding to levels two and three were required. However, this simple approach allows for the incorporation of more complex bar codes using additional spots or patterns of spots.
This study demonstrates the feasibility of an automated system for diagnosing specific virus strains. Although the scheme we report is a relatively simple implementation of this system, more complex designs are possible. For example, each antibody capture region could contain additional antibodies. In addition, there is essentially no limit to the overall length of the filament, which would allow the incorporation of many additional testing regions. The sensitivity of FARA could also be increased by the use of quantum-dot labeled detection antibodies as well as a more sensitive fluorescence detection system. The most important requirement is the availability of a wide range of antibodies to the specific pathogens of interest. Such a strategy may have applications for detecting specific pathogens from complex mixtures. This approach would have both clinical and environmental applications.
Acknowledgments
We thank members of our laboratories for many useful discussions and Dr. Ray Mernaugh for assistance with the experimental designs. Antibody 8H6 was provided by Dr. Greg Wilson (Vanderbilt University).
This research was supported in part by Public Health Service awards R21 EB003516 (F.R.H.) and R01 AI38296 (J.D.W. and T.S.D.) and the Elizabeth B. Lamb Center for Pediatric Research. Additional support was provided by Public Health Service awards CA68485 for the Vanderbilt-Ingram Cancer Center and DK20593 for the Vanderbilt Diabetes Research and Training Center.
References
- 1.Barton E. S., Connolly J. L., Forrest J. C., Chappell J. D., Dermody T. S. 2001 Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J. Biol. Chem. 276: 2200–2211 [DOI] [PubMed]
- 2.Burstin S. J., Spriggs D. R., Fields B. N. 1982 Evidence for functional domains on the reovirus type 3 hemagglutinin. Virology 117: 146–155 [DOI] [PubMed]
- 3.Chappell J. D., Duong J. L., Wright B. W., Dermody T. S. 2000 Identification of carbohydrate-binding domains in the attachment proteins of type 1 and type 3 reoviruses. J. Virol. 74: 8472–8479 [DOI] [PMC free article] [PubMed]
- 4.Dermody T. S., Nibert M. L., Bassel-Duby R., Fields B. N. 1990 Sequence diversity in S1 genes and S1 translation products of 11 serotype 3 reovirus strains. J. Virol. 64: 4842–4850 [DOI] [PMC free article] [PubMed]
- 5.Dryden K. A., Wang G., Yeager M., Nibert M. L., Coombs K. M., Furlong D. B., Fields B. N., Baker T. S. 1993 Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J. Cell. Biol. 122: 1023–1041 [DOI] [PMC free article] [PubMed]
- 6.Furlong D. B., Nibert M. L., Fields B. N. 1988 Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J. Virol. 62: 246–256 [DOI] [PMC free article] [PubMed]
- 7.Goral M. I., Mochow-Grundy M., Dermody T. S. 1996 Sequence diversity within the reovirus S3 gene: reoviruses evolve independently of host species, geographic locale, and date of isolation. Virology 216: 265–271 [DOI] [PubMed]
- 8.Leary T. P., Erker J. C., Chalmers M.L., Cruz A. T., Wetzel J. D., Desai S. M., Mushahwar I. K., Dermody T. S. 2002 Detection of mammalian reovirus RNA by using reverse transcription-PCR: sequence diversity within the lambda3-encoding L1 gene. J. Clin. Microbiol. 40: 1368–1375 [DOI] [PMC free article] [PubMed]
- 9.Stone G. P., Lin K. S., Haselton F. R. 2006 Adaptive virus detection using filament-coupled antibodies. J. Biomed. Opt. 11: 34012, 2006. [DOI] [PMC free article] [PubMed]
- 10.Stone G. P., Mernaugh R., Haselton F. R. 2005 Virus detection using filament-coupled antibodies. Biotechnol. Bioeng. 91: 699–706 [DOI] [PMC free article] [PubMed]
- 11.Tyler, K. L., and B. N. Fields. Reoviruses. In: Fields Virology 4th edition, edited by B. N. Fields, D. M. Knipe, and P. M. Howley. Philadelphia: Lippincott, Williams & Wilkins, 2001, pp. 1729–1745
- 12.Virgin H. W., Mann M. A., Fields B. N., Tyler K. L. 1991 Monoclonal antibodies to reovirus reveal structure/function relationships between capsid proteins and genetics of susceptibility to antibody action. J. Virol. 65: 6772–6781 [DOI] [PMC free article] [PubMed]
- 13.Virgin H. W., Tyler K. L. 1991 Role of immune cells in protection against and control of reovirus infection in neonatal mice. J. Virol. 65: 5157–5164 [DOI] [PMC free article] [PubMed]
- 14.Virgin,W. H., K. L. Tyler, and T. S. Dermody. Reovirus. In: Viral Pathogenesis, edited by N. Nathanson. Philadelphia: Lippincott-Raven, 1997, pp. 669–699