

Abstract
Analytical methods for the analysis of polychlorinated biphenyls (PCBs) and organochlorine pesticides (OCPs) are widely available and are the result of a vast amount of environmental analytical method development and research on persistent organic pollutants (POPs) over the past 30–40 years. This review summarizes procedures and examines new approaches for extraction, isolation, identification and quantification of individual congeners/isomers of the PCBs and OCPs. Critical to the successful application of this methodology is the collection, preparation, and storage of samples, as well as specific quality control and reporting criteria, and therefore these are also discussed. With the signing of the Stockholm convention on POPs and the development of global monitoring programs, there is an increased need for laboratories in developing countries to determine PCBs and OCPs. Thus, while this review attempts to summarize the current best practices for analysis of PCBs and OCPs, a major focus is the need for low-cost methods that can be easily implemented in developing countries. A “performance based” process is described whereby individual laboratories can adapt methods best suited to their situations. Access to modern capillary gas chromatography (GC) equipment with either electron capture or low-resolution mass spectrometry (MS) detection to separate and quantify OCP/PCBs is essential. However, screening of samples, especially in areas of known use of OCPs or PCBs, could be accomplished with bioanalytical methods such as specific commercially available enzyme-linked immunoabsorbent assays and thus this topic is also reviewed. New analytical techniques such two-dimensional GC (2D-GC) and “fast GC” using GC–ECD may be well-suited for broader use in routine PCB/OCP analysis in the near future given their relatively low costs and ability to provide high-resolution separations of PCB/OCPs. Procedures with low environmental impact (SPME, microscale, low solvent use, etc.) are increasingly being used and may be particularly suited to developing countries.
Electronic supplementary material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s00216-006-0765-y and is accessible for authorized users.
Keywords: PCBs, Organochlorines, Pesticides, POPs, Review, Analysis, GC–ECD, GC–MS, Extraction, Sample preparation and QA/QC
Introduction
Scope of the review
Polychlorinated biphenyls (PCBs) and organochlorine pesticides (OCPs; aldrin, chlordane, DDT, dieldrin, endrin, heptachlor, mirex, toxaphene, hexachlorobenzene (HCB)) constitute ten of the twelve chemical substances/groups currently defined under the Stockholm Convention on Persistent Organic Pollutants (POPs). While bans and phase-outs of these chemicals occurred during the 1970s and 1980s in most developed countries, they were not in place in many developing countries. Article 7 of the Stockholm Convention requires National Implementation Plans (NIPs) to be developed by signatory countries [1]. For countries where PCBs and OCPs are still in use or loosely regulated, the NIPs will eventually lead to the phase-out of OCPs for agricultural use and reduced use for disease vector control. However, many countries will be faced with the legacy of past use of these persistent chemicals. Residues in soils, sediments and biota will pose a risk for wildlife and human consumers. The problems of destroying existing stockpiles, especially of OCPs and PCBs in electrical equipment, as well as preventing migration of contaminants from storage areas, will need to be solved. There is a need to monitor levels and trends now and into the future in order to assess compliance with the POPs Convention within each country as well as on a regional and global basis.
Methodology is available for PCBs and OCPs as a result of a vast amount of environmental analytical chemistry research and development over the past 30 to 40 years. However, the establishment of an analytical laboratory and the application of this methodology at currently acceptable international standards is a relatively expensive undertaking. Furthermore, the current trend to use isotope-labeled analytical standards and high-resolution mass spectrometry for routine POPs analysis is particularly expensive. These costs limit participation of scientists in developing countries and this is clear from the relative lack of publications and information on POPs from countries in Africa, south Asia and South/Central America. Thus, this review will summarize the best practices in developed countries and new advances in PCB/OCP analysis, while bearing in mind the need for low-cost methods easily implemented in developing countries.
This review will also mainly focus on methodology for the analysis of PCB/OCPs in biological samples (fish, aquatic and terrestrial mammals and birds) as well as soils, sediment and passive samplers such as semipermeable membrane devices (SPMDs). These matrices and environmental compartments have higher concentrations of PCBs and most OCPs than water or air, making them more suitable for routine monitoring and more relevant in the context of exposure of humans and wildlife. Milk and blood are important matrices for POPs monitoring in humans and, in the case of blood, are increasingly used in programs involving live-captured marine mammals and birds. Thus analytical methods for milk and blood are briefly considered here. The advantages and disadvantages of various bioindicator species are discussed in detail by Tanabe and Subramanian [2].
Substances to be analyzed
PCBs (except non-ortho-substituted congeners; no-PCB) and OCPs can be considered together because they are extracted and analyzed together in most cases, as discussed below. In practice, most laboratories determine about 30 or more individual PCB congeners, and 10–20 individual OCPs and their metabolites, regardless of the sample matrix. Ongoing POPs monitoring programs vary in their analyte lists. For example, the Integrated Atmospheric Deposition Network (IADN) in the Great Lakes region of North America includes over 100 PCB congeners [3], while the UNEP/World Bank/GEF project on Persistent Organic Pollutants, Food Security, and Indigenous Peoples in Arctic Russia included 15 PCB congeners [4]. The Arctic Monitoring and Assessment Program recommended 30 ortho-substituted PCBs [5].
The UNEP Global POPs monitoring workshop categorized individual PCB/OCPs as “essential” and “recommended,” taking into consideration the need for various levels of analytical capacity building in developing countries in order to conduct the monitoring (Electronic Supplementary Material, Table 1) [6]. For PCBs, a minimum of seven PCBs (28/31, 52, 101/90, 118, 138, 153 and 180) were regarded as essential for routine monitoring of fish and food products around the world. Using only seven congeners severely underestimates total PCB concentrations in some matrices—particularly in abiotic samples such as air, soil, and sediment. On the other hand, these seven are robustly determined, having been the subject of numerous interlab comparisons (e.g., QUASIMEME [7]). UNEP [6] recommended a suite of 30 ortho-substituted congeners for more comprehensive monitoring.
Toxaphene and chlordane are multicomponent mixtures and analytical standards are available for some, but not all, major components, which makes them the most difficult to analyze among the eight OCPs on the POPs list. Current programs in Europe are determining three toxaphene congeners, Parlar (P) 26, 50 and 62, and interlaboratory comparisons have shown reasonable agreement for these congeners among laboratories [8]. However, P26 is interfered with by coeluting chlorobornanes in some matrices, and P62 can vary widely in its instrumental response. Furthermore, in North America, most datasets for toxaphene are still based on quantification with technical toxaphene, which yields a single value and no congener-specific information. Capacity for determining toxaphene outside of the USA, Canada and western Europe is very limited judging from the relatively large number of measurements from these countries and the dearth of measurements elsewhere.
The situation for chlordane is better because analytical standards are available for the major components (cis- and trans-chlordane; cis- and trans-nonachlor, heptachlor). Nevertheless, other octachloro- and nonachlorochlordane components are important [9] and are often found in top predators [10, 11], and analysts need to be aware of possible interferences from these compounds in seabird and marine mammal extracts.
The chlorinated insecticides endosulfan, lindane and methoxychlor are also candidates for inclusion in analytical methods where impacts of OCPs are being investigated. Global use of these insecticides, particularly endosulfan, is very significant [12, 13] These current-use pesticides can generally be isolated and quantified along with other OCPs.
The no-PCBs need to be considered separately because their analytical methodology is normally quite different from that used for ortho-substituted PCBs and OCPs (see Reiner et al. [14] in this issue).
In summary, the minimum number of PCB/OCPs to be considered will vary with the objectives of a given study. A long list of analytes would be used for initial surveys where no information was available. Chemical structures and physical and chemical properties of selected PCBs and OCPs are listed in Electronic Supplementary Material Table 2 [143, 144].
Analytical standards
The availability of accurate analytical standards is a fundamental requirement of an analytical program designed to quantify PCB/OCPs. Standards are available from commercial chemical supply companies as well as from agencies involved in certification of reference materials, e.g., the National Institute of Standards and Technology (NIST) or from the Bureau Communautaire de Référence (BCR) via the Sigma-Aldrich company. Interlaboratory studies have shown that incorrect analytical standards and calibration errors can be a major source of error in POPs analysis [15, 16]. These authors recommended that multilevel calibration at 4–6 levels should be used to define the response profile, that calibration solutions should be checked against independent solutions, either from another reliable laboratory or by preparing duplicate solutions, and that calibrants should be ampouled (in sealed glass vials) for long-term storage. They noted that refrigerator storage can loosen the caps on screw-top vials. Thus, storage of calibrant solutions in this manner should only be temporary.
Quantification of PCBs can be carried out using two approaches; by Aroclor mixture or individual congener standard. The former method employs a standard that was available as a commercial product (e.g., Aroclor 1254 used in transformer oils) to quantitate the congeners contained in the Aroclor mixture against those matching in the sample. There are several mixtures available, with varying degrees of mean chlorination. Percent ratios of the congeners in various commercial mixtures have been reported [17]. The limitation of this method is the varying congener ratios within a given commercial mixture due to batch-to-batch production variability which, in turn, may lead to congener calculation error [18]. Also, a significant bias resulting in an overestimation of PCB measurements may occur if the PCB pattern in the sample does not match that of the standards. Sather et al. [19] suggested that accurate PCB determinations can be made if the chromatographic pattern of both the standards and the samples are similar. However, if the pattern differs, the study reported a bias doubling the values determined by the individual congener standard. The method of choice is congener determination using standard mixtures comprising composite individual congeners added at known quantities which were purchased individually. This provides an improved and known level of accuracy not possible with the commercial PCB mixture approach [19].
Commonly used analytical methods for PCB/OCP monitoring and surveillance
Overview
Numerous methods have been published over the past 30 years related to specific analytical techniques for the determination of PCBs and OCPs in food and environmental matrices. Laboratory standard operating procedures (SOPs) are available from the National Environmental Methods Index in the USA (which includes US EPA and US Geological Survey methods) (http://www.nemi.gov) and the Japan Environment Agency (http://www.env.go.jp/en/index.html), the National Oceanographic and Atmospheric Administration in the USA [20] and the US Food and Drug Administration [21]. Useful information may also be available from the International Committee for Exploration of the Sea [22], the Oslo–Paris Commission [23] (http://www.ospar.org), the Helsinki Commission (HELCOM) (http://www.helcom.fi), the International Organization for Standardization (http://www.iso.org), the Association of Official Analytical Chemists International (http://www.aoac.org), and Gosstandart of the Russian Federation (http://www.krohne.ru/russia_en/downloads/certificates/cis/russia/gosstandard/).
Not all of these sources provide analytical SOPs for all environmental media. Alternatively, numerous books are available that summarize methods. For example, Keith [24] summarized US EPA methods for PCBs and OCP in sediment and biological materials. Wells and Hess [25, 26] have reviewed and recommended methods for the separation, isolation and recoveries of OCPs and PCBs from soils, sediment and biological matrices. De Boer and Law [27] provide a useful overview of current analytical methodology for OCP/PCBs.
The UNEP POPs workshop on global monitoring (UNEP 2003) noted that, given the broad range of technical expertise for analysis of PCBs and OCPs, as evident from the extensive international participation in interlaboratory calibration projects for these compounds, no single, detailed, step-by-step analytical method can be recommended. Instead laboratories would use methods best-suited to their situation and take part in international interlaboratory comparisons to verify their work. This performance-based approach has also been adopted by the US EPA in an effort to introduce flexibility in conducting environmental monitoring. This approach also encourages analytical innovation by allowing the use of new and alternate methods provided that predetermined performance criteria are met [28].
Sampling
Acquisition of suitably representative samples is fundamental to any environmental measurement program and should not be overlooked. Contamination during field collection can occur, particularly from PCBs in electrical equipment and building products (e.g., marine paints, joint sealants) [29, 30], or due to the ongoing use of OCPs. “Clean” techniques need to be adopted, such as the use of special clothing and disposable gloves for sampling, sealed shipping containers, and field blanks. ICES [22], NOAA [20] and OSPAR [23] provide useful guidelines for sampling of fish/crustaceans, mussels, and sediment, respectively. Azcue and Mudroch [31] provide a detailed overview of sampling methods for lakes and river sediments.
Sample containers should be supplied by the laboratory and checked for contamination. Appropriate sample containers for biological tissue that is to be analyzed for POPs are glass jars or glass vials with screw caps. Screw caps should be lined with solvent-rinsed aluminum foil or Teflon cap inserts. Clear polyethylene bags, and polypropylene jars, are also appropriate for temporary storage but may not be suitable for long-term storage because of the possibility of migration of plasticizers (such as phthalates) into the tissue, especially for samples with high lipid contents. Freezing and storage of multiple small samples suitable for analysis, rather than larger masses, is recommended to avoid multiple freezing and thawing of tissue [25] and to reduce sample handling, which in turn reduces the potential for contamination. Samples can be wrapped in aluminum foil and then inserted into plastic bags.
Blood samples should be collected in ethylenediamine tetraacetic acid (EDTA) vials or vacutainers, centrifuged, and the plasma transferred to vials washed with hexane. Field blanks, consisting of sample containers taken to the lab and returned with other samples, should be included.
Air sampling for POPs is normally conducted using polyurethane foam (PUF) or XAD resin [3]. From 300 to 10,000 m3 of air are passed through the sampling medium [3, 32]. Sampling media are then transferred to clean, wide-mouthed glass jars, sealed and stored at 4 °C prior to extraction. Some sampling cartridges can be sealed and transported and the sampling media transferred to Soxhlet or pressurized liquid extraction systems in a clean laboratory environment.
Water sampling is particularly prone to contamination by PCBs due to ubiquitous contamination, especially on older ships and buildings, and to the very low sample concentrations likely to be encountered [33]. US EPA analytical methods for PCBs in water assume that drinking water is being analyzed [34, 35] and do not place much emphasis on sampling because the drinking water quality guideline is 0.5 ug/L and only small <1 L samples need to be collected to assess compliance. Solid-phase extraction (SPE) cartridges have been widely used to extract relatively small volumes (1–5 L) for OCP analysis [36]. For sampling of natural waters where PCB concentrations are in the low pg/L range, much larger samples are required. For example, Achman et al. [37] pumped lake water (∼100 L) directly into large-diameter filters (0.7 μm nominal pore size) and then through XAD-2 resin columns. Surrogate standards were added to the resin column prior to extraction. Similar solid-phase extraction approaches are used for sampling and extracting wet precipitation [38, 39]. Another large-volume application uses liquid:liquid extraction in a continuous flow approach [40]. This involves a one pass-through extraction unit where the sample is metered into a mixing chamber which contains a solvent (i.e., dichloromethane) and flows out to an overflow exit. The drawback to this method is the significant potential for contamination due to exposure to the surrounding air. Petrick et al. [41] describe an in situ sampler for the collection of very large volumes (∼500 L) of seawater on polyurethane foam for the determination of PCBs at low pg/L concentrations. The studies by Petrick et al. [41] and Sobek and Gustafsson [42] illustrate the great care that is needed to properly determine PCBs at the low pg/L level (femtograms/L of individual congeners) that are found in ocean waters.
SPMDs offer potential for relatively inexpensive sampling of water and air for POPs and potential for broad geographic coverage [43, 44]. For water sampling, lipid-based SPMDs (consisting of triolein inside polyethylene tubing) have generally been used to estimate bioavailable water concentrations. SPMD use in the aquatic environment has been validated in a series of studies [45]. The use of performance reference compounds (PRCs) added to the triolein prior to deployment has been shown to enhance the use of SPMDs by accounting for site-specific variables [45]. The PRCs are gradually lost from the sampler and thus permit an exposure adjustment factor or depuration rate to be calculated, which adjusts for changes in sampling rate.
Lipid-based passive air samplers have also been developed and deployed for air sampling of PCBs and OCPs [46]; however, more recently designs based on polyurethane foam (PUF) and XAD resin have become more widely applied [47, 48]. Care must be taken to prepare and transport the samplers under clean conditions. Following deployment, which is usually for several months for lipid-based and PUF samplers [49] for up to one year for XAD [44], samplers are placed in transport containers and returned to the lab. PRCs are also often added to the passive air samplers [47].
Sample storage and handling
A basic requirement for analytical laboratories involved in the measurement of PCBs and OCPs is the availability of freezer and refrigerator capacity for sample storage and archiving. Wells and Hess [25] and ICES [22] provide recommendations for storage of biological and sediment samples. Kiriluk et al. [50] found no significant differences in concentrations of PCBs in whole fish homogenates stored at −20 or −80 °C over four years. Norstrom and Won [51] observed dehydrochlorination of DDT in long-term-archived egg homogenates, which they attributed to reactions occurring during freeze-drying. De Boer and Smedes [52] found no significant changes in the concentrations of PCBs and DDT components in fish tissue stored at −20 to −70 °C in the dark for up to 24 months. However, they did find changes in lipid extractability caused by oxidation of triglycerides to free fatty acids. The authors pointed out that this could affect calculations of lipid-normalized concentrations. To minimize any effects, they recommended long-term (>2 year) storage at −70 °C.
Sample preparation
Wells et al. [53] noted that sample preparation affected the magnitude of the variance in the analysis of PCB congeners in mussel tissue. They found that freeze-drying samples reduced recoveries of OCPs and PCBs, probably due to tighter binding and occlusion of residues in the dried material. Volatilization losses might also occur during freeze-drying of volatile compounds such as HCB, and there is a greater potential for contamination in the lab [54]. Although freeze-drying has been recommended by some authors for sediments and soils [25], lower recoveries of PCBs from freeze-dried sediments have also been reported [55]. Karl [56] noted that loss of water led to an increase in OCP concentrations in fish products during processing. Thus, in general, maintaining environmental sample tissues and sediments in their original wet state is regarded as the most appropriate approach for preparing samples for PCB/OCP analysis. Furthermore, avoiding a drying step minimizes potential contamination from lab air, particularly in the case of PCBs that are ubiquitous contaminants of older (pre-1980s) buildings [30, 57]. Using wet material also avoids possible volatilization losses. Instead, homogenized samples should be mixed with a desiccant such as sodium sulfate, Celite or Hydromatrix to bind water. The desiccant must be certified to be free of analyates, e.g., by heating at high temperature in the case of sodium sulfate or pre-extraction (Celite; Hydromatrix).
Tables 1 and 2 provide general guidance for various preparation/extraction and isolation/QA steps in the analysis of PCBs and OCPs and sources of information. Starting with sample preparation, the basic approach is to assure that the sample is prepared for extraction in a room that is free from significant contamination. Ideally this would involve a well-ventilated lab with air prefiltered through HEPA and carbon filters, but any clean chemical laboratory facility should be adequate for most work on PCBs and OCPs in most matrices except water or soils and sediments from remote locations. Laboratory buildings built after about 1990 are likely to be free of PCB-containing materials (electrical equipment/paints/sealants) or OCPs used as insecticides (or termiticides in foundations) because of the effects of bans on the use of these substances promulgated during the 1970s and 1980s.
Table 1.
Extraction techniques used for solid environmental samples (from Lopez-Avila [64]; Wells and Hess [25])
| Technique | Overview | Method reference |
|---|---|---|
| Conventional Soxhlet | Sample + desiccant mixture in glass or paper thimble is leached with warm (condensed) solvent for 4–12 hrs. Solvents are, e.g., diethyl ether, DCM, hexane | EPA 3540 [65] |
| Automated Soxhlet (e.g., “Soxtec”) | Extraction thimble is immersed in boiling solvent (30–60 min) then raised for Soxhlet extraction. Solvent can also be evaporated. | EPA 3541 [65] |
| Supercritical fluid extraction (SFE) | Sample (usually +desiccant) placed in high-pressure cartridge and carbon dioxide at 150–450 atm at temp of 40–150 °C passed through. After depressurization, analytes are collected in solvent trap | EPA 3560–3562 |
| High-speed blending | Useful for high water content samples such as plant material. Homogenizes sample with acetone and NaCl. | [21, 66] |
| Column extraction | Sample (+desiccant) placed in large column with filter and stopcock. Eluted with large volume of extraction solvent, e.g., hexane:DCM; hexane | [67] |
| Sonication-assisted extraction | Sample in open or closed vessel immersed in solvent and heated with ultrasonic radiation using ultrasonic bath or probe. | Method 3550C [65] |
| Microwave-assisted extraction (MAE) | Sample in open or closed vessel immersed in solvent and heated with microwave energy. | EPA 3546 [65] |
| Pressurized liquid extraction (PLE) | Sample (usually +desiccant) placed in extraction cartridge and solvent (heated, pressurized) passed through then dispensed in extraction vial. | EPA 3545 [61, 65] |
Table 2.
Guidance for various preparation, extraction and isolation steps in the analysis of PCBs and OCPs
| Environmental matrix | Analytical steps | General procedures | EPA or other method |
|---|---|---|---|
| Soil and sediment | Preparation | Prepare in a PCB- and pesticide-free room. | [35, 86] |
| Avoid air-drying. Wet sieve if necessary to remove large particles. Centrifuge sediment to remove excess water. | |||
| Mix soils/sediments with desiccant such as Na2SO4. | |||
| Separate determination of dry mass by oven drying. For sediments total organic carbon should be determined. | |||
| QA | One blank, soil CRM every ten samples; spike all samples with recovery surrogate standards. Bake glassware overnight at 200 °C or higher. | ||
| Extraction | Soxhlet, PLE, sonication, or MAE with acetone: hexane or DCM | ||
| Solvent evaporation, transfer to hexane. | |||
| Sulfur removal with activated copper turnings required for sediment. | |||
| Isolation/cleanup | Alumina, silica or Florisil elutions: non-polar (hexane) and polar (DCM:hexane or equivalent) | ||
| Vegetation | Preparation | Homogenize using food chopper or blender. Cryoblending using liquid nitrogen or dry ice is useful. Mix with dessicant. Separate determination of dry mass by oven-drying. | [21, 35] |
| QA | Same as soil. Use vegetation CRM if possible | ||
| Extraction | Same as soil. | ||
| Isolation/cleanup | Same as soil. | ||
| Aquatic biota | Preparation | Select muscle or liver depending on species. For mussels and crustaceans use soft tissue. Select tissue that has not been in contact with the sample container. Homogenize using food chopper or blender. Cryoblending is useful. | [21, 35] |
| Mix with drying agent. Separate determination of lipid content. | |||
| QA | Same as soil. Use fish or mussel SRMs. | ||
| Extraction | Soxhlet, pressurized liquid extraction, or column extraction. Use acetone:hexane or DCM. | ||
| Isolation/cleanup | Remove lipid using gel permeation chromatography if possible or by repeated washing of the extract with sulfuric acid. Follow with fractionation on silica or Florisil columns as described for soil. | ||
| Marine mammal blubber | Preparation | Select blubber that has not been in contact with the sample container. Blend or hand-mix with drying agent. Separate determination of lipid content. | [10, 87] |
| QA | Same as soil. Use fish oil or marine mammal SRMs. | ||
| Isolation/cleanup | Same as for fish extracts. | ||
| Air (high volume) | Extraction,QA and cleanup | Assuming that air is collected on polyurethane foams or XAD resin, these would be extracted in a Soxhlet or pressurized liquid extractor. Other steps as for soil or sediments | [32] |
| Semi-permeable membrane devices (SPMD) | Preparation | SPMDs would be removed from their transport cases and rinsed with precleaned water to remove accumulated dust (air-borne samplers) or periphyton (water samplers). | [46] |
| QA | Use PRCs | ||
| Extraction, and cleanup | Assuming that the SPMD is lipid-based, extraction of POPs by “dialysis” into hexane would be achieved in a large glass cylinder. | ||
| Water (including melted snow, ice and wet precipitation) | Extraction | Liquid–liquid, SPE (e.g., C18) extraction for small (<1 L) samples; XAD-2 or modified “Speedisk” for >1 L. | US EPA 508.1 [34] and 525.2 [36, 37, 78, 88] |
| QA and cleanup | Pre-spike XAD columns with surrogates. | ||
| Blood plasma | Extraction | Extract blood plasma with ammonium sulfate/ ethanol/hexane (1:1:3) or C18 SPE extraction. | [72, 73] |
| Determine lipid content. | |||
| QA | Same as fish. Use NIST 1589a SRM. | ||
| Isolation and cleanup | Sulfuric acid partitioning to remove lipids. | ||
| Acid–base silica for additional lipid removal. | |||
| Milk | Extraction | Liquid–liquid partitioning with acetone:hexane or C18 SPE extraction. Determine lipid content. | [75, 77] |
| QA | BCR SRM 284 & 533 milk powder. | ||
| Isolation and cleanup | As with plasma. |
Recommended extraction and isolation techniques for PCBs and OCP
Recovery surrogates/internal standards
A standard QA step in the analysis of PCB/OCPs is to include surrogate recovery standards in each sample. These are generally one or two PCB congeners (e.g., CB30 and CB204) and OCP-related, unlabeled compounds such as pentachloronitrobenzene, endrin ketone, 1,3,5-tribromobenzene, and other compounds that are readily separated from individual OCPs and not commonly found in the environment. If GC–MS is being used as the detection system then 13C-labeled or deuterated surrogates should be used. These standards are carefully added by pipetting the appropriate volume of a standard solution onto the sample matrix prior to extraction. Surrogates do not define absolute recoveries because they are not incorporated into the matrix; however, they do provide information on analyte recovery throughout the isolation process and can also provide sample-to-sample extraction efficiency comparability. If bioanalytical techniques are being used to screen extracts, it may not be possible to include recovery standards, particularly 13C-surrogates, since they may respond similarly to the native compounds.
Extraction techniques
The appropriately prepared sample can then be extracted by any one of a number of techniques (Table 1). The main points to consider are to allow adequate exposure time for the solvent system in the sample matrix and to limit sample handing steps, i.e., avoid filtration steps by using Soxhlet (sample in a glass thimble), extraction columns (sample matrix eluted after soaking in solvent) or semi-automated systems (e.g., pressurized liquid extractors, PLEs). Cross-contamination from residues left behind by high levels in other samples is a concern at this stage and equipment must be thoroughly cleaned and checked from batch to batch. Purity of extraction solvents is a major consideration here. Only high-purity glass-distilled solvents should be used, because later evaporation steps will concentrate any contaminants. PLE is increasingly being used to replace Soxhlet and column extraction methods [58–60]. PLE uses much less solvent than Soxhlet or column elution. However, it suffers the disadvantage of initial high cost and the need for a stable power supply to avoid premature instrument shutdown. Also, operation of an automated PLE requires regular scheduled maintenance by trained service personnel. Preparation of a homogeneous dry sample from wet tissue or sediment for PLE can be a challenge due to the limited size of PLE extraction vials, currently set at 100 ml. A US EPA method (3545) using PLE for solid waste extraction is available [61]. This method recommends acetone/hexane (1:1, v /v) or acetone/dichloromethane (DCM) for extraction of PCBs from solid waste, and these solvent systems appear to be the most commonly used [59]. There has been a recent trend toward not using chlorinated (potentially toxic) solvents such as chloroform and DCM, due to concerns over exposure to lab personnel. Binary mixtures such as hexane/acetone (1:1) are regarded as equally effective for the extraction of POPs from solid samples [62]. In general, extraction with a polar:apolar binary mixture has been found to be more efficient for recovering OCPs and PCBs from fish tissues of low lipid content than an apolar solvent [63].
The time- and solvent-consuming nature of Soxhlet extraction (or related techniques involving percolation of a solvent through the sample) is generally thought to be related to the slow diffusion and desorption of the analytes from the sample matrix. Semivolatile compounds such as naphthalenes can also be lost from Soxhlet apparatus via volatilization [68]. The use of microwave, sonication, supercritical fluids, or elevated temperatures and pressure (as in PLE) increases the rates of diffusion and desorption and thus speeds up extraction [58, 64, 69]. Pressurized hot water extraction has even been used to extract PCBs from sediment and soil. Under pressure, the dielectric constant of the water can be manipulated to facilitate the extraction of nonpolar analytes [70]. Sonication, microwave-assisted extraction (MAE) and supercritical fluid (SFE) extraction have all been successfully employed to recover PCBs and OCPs from solid samples (Table 1). Soxhlet is regarded as more reliable than sonication, SFE, or shake flask-type methods and equivalent to (but not as rapid as) MAE and PLE for a wide range of environmental matrices [25].
Blood plasma lipids can be rapidly extracted using conventional solvent partitioning using a mixture of ammonium sulfate/ethanol/hexane (1:1:3) [71, 72]. Solid-phase extraction using C18 cartridges is also a proven technique [73]. Needham et al. [74] have reviewed methods for the analysis of POPs in human milk. PCB/OCPs can be extracted from milk samples using conventional lipid extraction techniques involving liquid–liquid extraction with hexane/acetone [75]. Blending with acetonitrile and potassium oxalate [76] followed by C18-solid phase extraction has also been used in recent studies [77].
Water sampling devices such as solid-phase extraction (SPE) cartridges, XAD or SPMDs are extracted by elution or dialysis (with hexane for lipid-based SPMDs). The elution of reverse-phase or XAD resin water sampler cartridges generally involves the use of a water miscible solvent first to remove water followed by a solvent of intermediate polarity such as DCM, methyl t-butyl ether or ethyl acetate. Combined extracts are then partitioned into hexane. Usenko et al. [78] compared six different SPE devices for the recovery of PCBs and OCPs from natural waters. These included XAD-2 resin, C18-filter disks and various C18 - and divinyl benzene-based adsorbents. They found that C18-based products, particularly “Speedisk,” a modified divinylbenzene solid-phase extraction device, gave the best overall recoveries.
Another water sampling device, solid-phase microextraction (SPME), involves immersing a polydimethylsiloxane-coated syringe into 3–5 ml samples. Hydrophobic compounds are adsorbed onto the coated fiber and the syringe then placed into a hot injection liner which desorbs these compounds into the GC [79]. The benefit of this approach is that it requires no solvent or multistepped cleanup/concentration procedures. A drawback is that the adsorption efficiency can be affected by complex water matrices and may not be suitable for all water sample types [80]. Particulate phases collected by filtration on glass fiber filters are treated like sediments and should be extracted by Soxhlet or pressurized liquid extraction.
Determination of lipid content
Lipid contents of biological samples should be determined during the analysis of POPs. Most studies have determined total extractable lipid gravimetrically by drying a fraction of the sample extract to constant weight [25]. However, results can vary widely among laboratories due to different extraction efficiencies of various combinations of solvent and extraction apparatus [81]. The benchmark method for total lipid is that of Bligh and Dyer [82]. A modified version of this method has improved sample handling and method precision [83]. Smedes [84] demonstrated that propanol:cyclohexane:water (8:10:11) was an effective substitute for the Bligh and Dyer chloroform:methanol:water. The Smedes method gave more consistent results for extractable lipids in fish tissue with low lipid content (∼1% lipid). Samples with very low fractions of lipid, such as blood plasma, are best analyzed using a colorimetric method [85].
Isolation of analytes from coextractives
There are two aspects to this phase of POPs analysis. The analytes must be separated from nonvolatile materials which affect the performance of GC columns, such as pigments, inorganic sulfur, and triglycerides. Also, there is a need to separate the OCPs and PCBs from each other as much as possible prior to GC analysis in order to limit coelution problems. These isolation steps are also useful, and sometimes essential, for the application of bioanalytical methods to PCBs/OCPs and dioxin-like activity (See “Bioanalytical methods for the quantification of OCPs and PCBs”).
Adsorption “cleanup” columns
Separation of PCB/OCPs from coextractives can be relatively straightforward for low-lipid samples such as soils, sediments and vegetation (Table 2). Generally, small silica gel or Florisil columns (either prepared in the lab or pre-purchased) should suffice. The purpose of this step is to remove coextractive pigments and to separate nonpolar PCBs (plus p,p′-DDE) from more polar OCPs (HCH, most chlordanes, dieldrin/endrin). This is achieved by applying the extract in a small volume of apolar solvent to fractionation by eluting with hexane followed by one or two other elutions of increasing polarity. Polar compounds are retained on the column. Basic alumina and silica gel columns have been effective for the separation of OCPs from fish lipids [89], although there is the possibility of minor losses due to dehydrochlorination of some OCPs, e.g., p,p′-DDT, on the alumina. The effectiveness of these adsorption columns depends on the mass and the water content of the adsorbent together with the polarity of the solvent. In general 3–8 g of absorbent is used in a 0.5–1.0 cm-diameter column with silica gel or Florisil deactivated with a low percentage of water (0–5%) [51, 90]. Typically the sample is applied to the top of the column in hexane and then a first elution solely of hexane separates PCBs from many OCPs. Subsequent elutions with hexane–diethyl ether or hexane–DCM recover the OCPs (p,p′-DDT, dieldrin, toxaphene) [21, 76]. Alumina and Florisil have the capacity to retain about 100 mg lipid per 10 g of adsorbent [26].
Size-exclusion columns
For high-lipid samples (>10% lipid), such as some fish tissues (e.g., salmonid muscle, cod livers) and marine mammal blubber, a lipid removal step must be included. This can be achieved using size exclusion or gel permeation chromatography (GPC), either in automated systems, using HPLC columns or by gravity flow columns. A 60 g bed of Bio-Beads SX3, a neutral porous styrene divinylbenzene resin (BioRad Laboratories, Hercules, CA, USA) can accommodate up to 1 g of lipid and achieves the separation of fish lipids from most PCBs and OCPs with a size exclusion of ∼400 Da [67]. Equivalent HPLC size-exclusion columns can be used [91]. The advantages of GPC are that it is nondestructive and that columns can be reused. A disadvantage is a requirement for large volumes of purified solvent (low-pressure or gravity systems) or expensive columns (HPLC). Automated GPC units are commercially available and allow for unsupervised elution of 12–24 samples. Due to required rinses of the unit sample lines, a portion of the sample is lost and must be accounted for when calculating final analyte concentrations. GPC eluates generally require an adsorption column fractionation step on silica or Florisil to remove remaining low molecular weight lipids, waxes and pigments that are not completely separated from the PCB/OCPs.
Lipid destruction
Lipid removal using sulfuric acid washing or elution of the extract through sulfuric acid (50%) in silica or KOH-treated silica columns is also effective for most PCB/OCPs, but does result in loss of some analytes such as dieldrin [92, 93]. Ethanolic–KOH treatment of Soxhlet extracts is also suitable for lipid removal and works well for PCBs [87], but may degrade chlorinated aliphatics such as DDT, which is converted to DDE. The acid- or base-treated extracts containing the PCBs and OCPs are then subjected to an adsorption column fractionation step on silica or Florisil.
Sulfur removal
Sulfur is coextracted with PCBs and OCPs and presents a particular problem for GC–ECD analysis of sediment extracts because of its strong response in this detector. It is also cytotoxic and needs to be eliminated prior to use with in vitro bioassays [94]. Sulfur can be removed by GPC but can also be removed using activated copper turnings (washed with concentrated HCl and held in an apolar solvent) or using mercury. The latter method removes sulfur more efficiently but is not recommended due to potential for contamination of the lab and lab effluent. Shaking with tetrabutylammonium sulfide has also been used to remove sulfur [95].
Evaporation steps
Solvent evaporation is generally used several times within all analytical methods for PCB/OCPs and successful analyte recoveries critically depend on minimizing losses during this step. Surrogate standards may be added prior to evaporation to monitor compound losses [96]. The Pesticide Analytical Manual of the US FDA [21] includes a discussion of solvent evaporation and an overview of the most widely used equipment, rotary evaporators, Kuderna–Danish apparatus and TurboVap equipment. The US FDA [21] overview favors the rotary evaporator because of greater range of use, e.g., for temperature-sensitive and semivolatile analytes, but notes that it is applicable to only one sample while both the TurboVap and K–D apparatus can be set-up for multiple samples.
Preparation for GC analysis
Following fractionation on silica or Florisil, final extracts are prepared in GC vials for analysis. Addition of an internal standard to check solvent volume is recommended at this stage. Careful evaporation is required at this step, and only high-purity compressed gas (usually nitrogen) should be used. This can be done using a stream of regulated gas via a disposable glass pipet and heating block or via multineedle devices (e.g., “N-Evap”).
Quantification methods
Overview
Numerous analytical approaches are available for quantifying PCBs and OCPs. As with extraction/isolation steps, only general comments are provided here for GC analysis of ortho-substituted PCBs and OCPs. However, a major consideration is that the laboratory will have access to modern capillary GC equipment and either electron capture or mass spectrometry detection. Some general guidance on the application of gas chromatographic analysis of ortho-substituted PCBs and OCPs is provided in Table 4. Cochran and Frame [97], in their review of the GC analysis of PCBs, noted that separation requires attention to (1) proper injection to minimize analyte band-broadening; (2) choice of carrier gas; (3) optimized carrier gas velocity; (4) GC oven programming; (5) column dimensions such as length, inside diameter, film thickness, number of plates; and (6) the type of column stationary phase.
Table 4.
General guidance on GC analysis and data reporting for PCBs and OCPs
| GC detector | Analytes | Configuration | Advantages/disadvantages | Detection limitsa |
|---|---|---|---|---|
| Capillary GC - with electron capture detection | All ortho-subsituted PCBs & all OCPs on the POPs list except toxaphene | 30 or 60 m×0.25 mm id. Column with H2 carrier gas. Dual column nonpolar (DB-1) and intermediate polarity columns (DB-5). | Relatively inexpensive and easy to operate. Similar response factors for most OCs. | DDT/DDE ∼ 1 pg |
| Good sensitivity for all POPs. Adequate for routine tasks. High potential for misidentification of some POPs due to coeluting peaks. | HCB ∼0.5 pg | |||
| Quadrupole mass spectrometry in electron ionization (EI) mode | All PCBs & all OCPs on the POPs list except toxaphene | 30 m×0.25 mm i.d. low-bleed columns with He carrier gas. Selected ion mode for target POPs. | Moderately expensive and more complex to operate and maintain. Newer instruments (post 1997) have adequate sensitivity for routine POPs monitoring at low pg/μL concentrations. Much less potential for misidentification than with ECD. | DDT/DDE ∼1–10 pg |
| HCB ∼1–10 pg | ||||
| Dieldrin ∼25 pg | ||||
| Toxaphene ∼500 pg (as tech mixture) | ||||
| Quadrupole mass spectrometry in electron capture negative ionization (ECNIMS) mode | Toxaphene and other highly chlorinated OCPs and PCB with >4 chlorines | 30 m×0.25 mm i.d. low-bleed columns with He carrier gas. Selected ion mode for target POPs. | Comparable sensitivity in ECNIMS mode to ECD in SIM mode for some POPs. Much less potential for misidentification than with ECD. | DDT/DDE ∼0.1 pg |
| HCB ∼0.1 pg | ||||
| Dieldrin ∼1 pg | ||||
| Toxaphene ∼10 pg (as tech mixture) | ||||
| Ion trap mass spectrometry using MS/MS mode | All PCBs, All OCPs on the POPs list | 30 m×0.25 mm i.d. low-bleed columns with He carrier gas. Same columns as quadrupole MS. | Comparable sensitivity to ECD in MS/MS mode for some POPs. Much less potential for misidentification than with ECD. | DDT/DDE ∼1 pg |
| HCB ∼1 pg | ||||
| Dieldrin ∼5 pg | ||||
| Toxaphene ∼100 pg (as tech mixture) | ||||
| High-resolution magnetic sector mass spectrometry in electron ionization (EI) mode | All PCBs, all OCPs on the POPs list except toxaphene | 30 m×0.25 mm i.d. low-bleed columns with He carrier gas. Selected ion mode for target POPs at 10,000× resolution. | Comparable sensitivity to ECD in SIM mode. Highly reliable identification at low pg/μL levels. | DDT/DDE ∼0.05 pg |
| HCB ∼0.05 pg | ||||
| Dieldrin ∼0.1–0.5 pg | ||||
| Toxaphene ∼10 pg (as tech mixture) |
aInstrumental detection limits at S/N of ∼10
Bioanalytical methods based on immunoassays, or in vitro bioassays for dioxin-like activity, have become widely available over the past ten years for the screening of sample extracts for POPs. These methods and selected applications are briefly reviewed in this section.
GC injection ports
Optimization of injection conditions is critical to proper GC analysis [98, 99]. In the analysis of POPs and OCPs, problems often occur with nonvolatile coextractives such as triglycerides and pigments that, despite various isolation procedures, are still present in final extracts. Most GC applications for PCB and OCP analysis have employed split/splitless injection systems, although on-column injection has also been used. Although on-column injection avoids artifacts associated with heated split/splitless systems (i.e., degradation of labile compounds), it requires highly clean extracts to avoid matrix effects [99]. Retention gaps consisting of an empty capillary column have been used to reduce these effects and permit larger on-column injection volumes. Pressure programming allows improved control over carrier gas flow, resulting in improved separation of PCB and OCPs.
GC columns
Fused silica open tubular capillary columns, generally coated with nonpolar or medium-polarity chemically bonded liquid phases are almost universally used for GC separation of PCBs and OCPs [99]. The use of capillary columns revolutionized PCB analysis, allowing identification of the individual congeners. This improved the comparability of the analytical data from different sources and helped to establish a basis for the understanding of geochemical cycles and toxicological implications. The basic technology for separation of PCB congeners described by Mullins et al. [100] has not changed greatly over the years. These authors used a 5% phenyl methyl silicone phase and a long temperature program (100 min) to achieve high-resolution separation of PCB congeners. Improved routine separations of PCBs have been achieved using 60 m×0.25 mm i.d. columns with hydrogen carrier gas. PCBs within a homolog group elute according to their number of ortho chlorines: 4<3<2<1<0. Coelution of congeners remains a problem for routine analysis by GC–ECD. However, several modified polydimethylsiloxane phases with n-octyl or n-octyldecyl substituents, e.g., DB-XLB (J&W Scientific, Folsom, CA, USA) and HT 8 (SGE Inc., Austin, TX, USA) can resolve all but four pairs of significant congeners and five pairs of minor congeners [18, 101] using mass spectrometry detection. The coelution of PCB congeners on the more commonly used 30 m DB-5 columns is outlined in Table 3. Coelutions are thus important even for routinely monitored congeners such as CB 28, 31, 105 and 153. However, separation of these congeners can be routinely achieved on 60 m 5% phenyl methyl silicone phase capillary columns using H2 carrier gas and on n-octyl phases as well [18, 101]. Use of dual-column GC, in which the sample is injected simultaneously into nonpolar and medium-polar phase columns, is recommended if a suitable instrument is available. Alternatively, confirmation of peak identity in a subset of samples, using a second column of different polarity, can be done instead. Although other carrier gases such as He can be used, the best peak resolution and efficiency is achieved by using H2.
Table 3.
Significant PCB congener coelutions on 5% phenyl phases [97]
| Classificationa | Coeluting PCBs | Number of chlorines |
|---|---|---|
| A | 4, 10 | 1, 2 |
| A | 9, 7 | 2, 2 |
| A | 12, 13 | 2, 2 |
| A | 17, 15 | 2, 3 |
| A | 27, 24 | 3, 3 |
| A | 32, 16 | 3, 3 |
| A | 28, 31 | 3, 3 |
| A | 33, 20, 53 | 3, 3, 4 |
| A | 43, 49 | 4, 4 |
| A | 47, 75, 48 | 4, 4, 4 |
| A | 44, 59 | 4, 4 |
| A | 37, 42 | 4, 4 |
| A | 71, 41, 64 | 4, 4, 4 |
| A | 66, 95 | 4, 5 |
| A | 56, 60 | 4, 4 |
| A | 84, 89, 101, 90 | 5, 5, 5, 5 |
| A | 117, 87, 115 | 5, 5, 5 |
| A | 77, 110 | 4, 5 |
| A | 135, 144, 124 | 6, 6, 5 |
| A | 147, 109 | 6, 5 |
| A | 123, 139, 149, 118 | 5, 5, 6, 5 |
| A | 114, 133 | 5, 6 |
| A | 131, 122 | 6, 5 |
| A | 153, 132, 105 | 6, 6, 5 |
| A | 176, 130 | 7, 6 |
| A | 164, 163, 138 | 6, 6, 6 |
| A | 158, 129 | 6, 6 |
| A | 175, 166 | 7, 6 |
| A | 173, 157, 201 | 7, 6, 8 |
| A | 170, 190 | 7, 7 |
| A | 198, 199 | 8, 8 |
| A | 203, 196 | 8, 8 |
aCapital A indicates a major Aroclor congener
Coelution of major OCPs with each other or with PCBs is also a problem in GC–ECD analysis, despite preseparation by adsorption chromatography. For example, p,p′-DDD can coelute with cis-nonachlor, CB99 with a trans-nonachlor isomer [10], p,p′-DDE with dieldrin and CB85, and toxaphene congener Parlar 50 with CB128. Recently, the coelution of CB180 and brominated diphenyl ether congener 47 has also been noted [102].
Chiral GC separation of OCPs and PCBs
Most OCPs on the POPs list have optically active or chiral isomers (e.g., α-HCH, o,p′-DDT, the main constituents of technical chlordane, cis-/trans-chlordane, heptachlor, as well as chlorobornanes in toxaphene) [103]. There are also 19 PCB atropisomers which are also chiral as a result of restricted rotation at the central biphenyl bond. While PCBs and OCPs are racemic mixtures when manufactured, microbial degradation in soils and water as well as biotransformation reactions in biota can result in nonracemic patterns in environmental samples. Crucial for chiral analysis is the availability of chiral capillary GC columns such as those with various cyclodextrins chemically bonded to a polysiloxane. These phases are relatively heat-stable and have low bleed [104]. Current methods range from the simple use of 30 m chiral columns to a two-dimensional “heart-cutting” technique. This involves a redirection of a targeted portion of the sample after elution from a methyl/phenyl polysiloxane column into a chiral column via a computer-controlled pneumatic valve. This provides higher peak capacity and generally further separation of chiral compounds. While use of chiral GC separations is not part of routine OCP analysis, it is a well-developed technology that is relatively easy to implement in existing GC–ECD and GC–MS instruments. This technique has useful applications for tracking sources of OCPs and transformations of PCBs. For example, Jantunen et al. [105]) and Leone et al. [106] have shown that enantiomer fractions of chlordane isomers can be used to identify emissions of this pesticide from soils. Wong et al. [107] calculated minimum transformation rates for PCB atrophisomers CB95 and 136 in the Lake Superior food web using enantiomer ratios in predators and prey species.
GC–ECD
Since the 1960s, POPs have been determined by GC with electron capture detection (ECD), initially using packed columns. Capillary GC–ECD began to be routinely applied by the early 1980s. Use of GC–ECD is recommended for routine analysis of OCPs, except for toxaphene, as well as for ortho-PCBs (Table 4) but not for non-ortho-PCBs [14]. This instrumentation is widely available at relatively low cost from at least four instrument manufacturers. A substantial knowledge base exists on the use of this 40-year-old technology. GC–ECD is capable of determining PCB/OCPs at low ng/g levels or higher in environmental matrices. Although at one time tritium-based ECDs were once available, the 63Ni detector is now universally used. This detector is operated at high temperatures, 300–350 °C, which makes it relatively unaffected by column bleed. ECD suffers from the potential for false positives due to interferences such as those from sulfur, phthalate esters, and negative peaks generated by hydrocarbons. ECDs are normally operated with N2 or argon/methane gas, which combines with the flow from the GC column (He or H2 carrier gas). Gases used for GC–ECD must be ultrapure to protect both the GC column (which can be oxidized by trace oxygen or siloxanes hydrolyzed by trace water) and the ECD itself. Recent refinements in ECD technology include the use of microcells which have greater linear range than older detector cells [108] and can provide greater sensitivity.
GC–MS
Capillary GC coupled with mass spectrometry (GC–MS) revolutionized environmental organic analysis in the 1980s [109], particularly with the advent of bench-top instruments. Current GC–low-resolution (quadrupole) mass spectrometry (LRMS) instrumentation is capable of determining most PCB/OCPs at low pg concentrations using electron ionization (EI) in selected ion mode. Electron capture negative ion ionization (ECNI) is capable of detection of low femtogram amounts of highly chlorinated OCPs such as chlordane and toxaphene congeners. In this mode, a reagent gas, usually methane, is introduced into the ion source and negative ions are monitored. The ECNI mode also has a more limited linear range than the EI mode, is more difficult to operate routinely due to greater sensitivity to temperature variations, and needs more frequent cleaning due to the reaction of the reagent gas with surfaces in the ion source.
GC–ion trap MS (ITMS) in MS/MS mode offers an equivalent sensitivity to quadrupole ITMS in selected ion mode and improved specificity by examining product ions of major mass fragments. ITMS has been successfully applied to detect mono-ortho-PCBs at subpicogram levels in a range of environmental samples [110] and to determine pg levels of toxaphene congeners [90]. In general, both quadrupole and ion trap LRMS can suffer from false positives due to unit mass resolution. Thus experienced analysts are needed to interpret results using confirmatory information such as full-scan analyses, fragmentation patterns and ion ratios.
GC coupled to HRMS provides much higher specificity for individual PCB/OCPs due to its ability to provide millimass resolution and can also be used for the determination of all ortho-substituted PCBs (e.g., EPA Method 1668 [35]) and OCPs too, and provides a very high level of confidence in the results compared to GC–ECD and LRMS.
The use of GC–MS allows for the application of isotope-dilution techniques and these have increasingly come into routine use for PCB/OCP analysis in environmental samples due to the sensitivities of the latest generation of quadrupole and ion-trap MS systems [111, 112]. A wide range of PCBs and OCPs are available as 13C-labeled compounds from several chemical supply companies, which, when added at the extraction step, increase precision and reproducibility for the native analytes.
Bioanalytical methods for the quantification of OCPs and PCBs
Enzyme-linked immunoabsorbent assays (ELISA)
ELISAs have been used to quantify most DDT/DDE, HCH isomers, toxaphene and cyclodiene OCPs as well as PCBs in environmental samples [113, 114]. ELISAs are based on competitive binding in which the binder molecule, an excess amount of labeled analyte or coating antigen, and the target analyte are allow to approach equilibrium. The sample antigen competes with the coated antigen for binding sites on the labeled antibody; after a wash step, detection is performed by adding substrate and chromophore. Quantification is generally performed via spectrophotometric measurements and the amount of analyte in the sample is interpolated from a calibration curve. Although widely used to screen for current-use herbicides and insecticides as well as their polar metabolites [115], the development of competitive immunoassays for neutral hydrophobic OCPs has lagged [114]. This is in part due to the need for low detection limits. Also, ELISA analysis for some OCPs such as HCH (and lindane) has been challenging due to the small size of the HCH molecule, its structural symmetry, and possibly most importantly, its lack of aromatic structures or other atomic groups capable of supporting delocalized electron networks [116]. The development of a sensitive ELISA for DDT required synthesis of a ligand with Cl replaced by b-alanine carboxamide and lindane after degradation to trichlorobenzenes [117]. Commercial ELISA kits for detection of PCBs and most OCPs are available from Millipore Corp. (Billerica, MA, USA) and Strategic Diagnostics (Newark, DE, USA). Detection limits for PCBs and OCPs are typically in the low μg/L range in water or 0.1–1 μg/g range in soil and plant extracts. These kits are meant to be used with relatively little sample preparation and, although semiquantitative, are ideal for screening samples and complement more elaborate techniques involving GC analysis (Fig. 1).
Fig. 1.
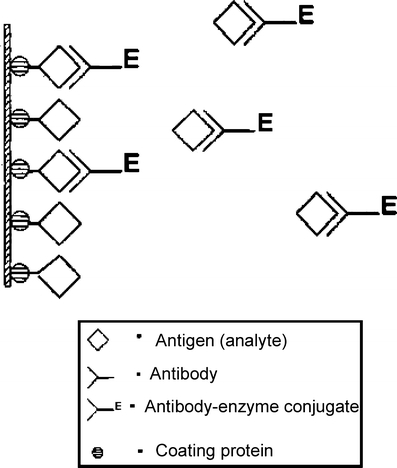
Illustration of the basic components of an ELISA for detection of OCPs and PCBs in environmental samples or extracts. Sample antigen (analyte) competes with antigen for binding sites on coating protein; after a wash step, detection is performed by adding substrate and chromophore
Shivaramaiah et al. [118] used an ELISA specific for DDE to survey DDT residues in surface soils in three river valleys of northern New South Wales, Australia. The technique allowed the analysis of large numbers of samples, thus permitting the areas of contamination to be mapped. Galloway et al. [119] used an ELISA to detect PCBs in extracts of mussels from New Bedford Harbor (USA). The ELISA and GC–ECD results were highly correlated, although the latter were about 20% lower than quantitation by GC. Samples were extracted and lipid partially removed by chromatography on Florisil prior to exchanging the sample into a phosphate buffer/methanol solution for the immunoassay. The use of the ELISA saved additional isolation steps and GC analysis. In general, fatty coextractive materials must be removed prior to application of ELISAs for OCPs. Skerritt et al. [120] examined the application of ELISAs for DDT and cyclodiene insecticides, heptachlor and endosulfan in plant-derived foods. They found that cleanup was necessary for foods that yielded highly colored extracts such as coffee and spinach and for oily products such as cottonseed.
Quality assurance issues for PCBs and OCPs
Ancillary data
A significant amount of ancillary data is required in order for the PCB/OCPs measurements to be properly interpreted in terms of temporal and/or spatial trends and exposure of wildlife and humans. Tanabe and Subramanian [2] have extensively reviewed the factors to be considered for the selection of bioindicator species, including human tissues. Analytical lab reports need to include quality assurance data so that results and lab performance can be assessed independently [6], as outlined in Table 5. The objective here would be to have a record of the entire processing of the sample from preparation through to reporting concentrations that can be evaluated independently. Therefore, the individual labs should report concentrations for analytes, blanks and reference materials. Data reports should also include instrument calibration results and recoveries. This would enable method detection limits (MDLs) to be calculated independently of the lab if necessary.
Table 5.
Minimum reporting dataset for POPs analysis
| Information | Details |
|---|---|
| Sampling protocols | Method, number, size and representativeness |
| Storage temperature and location | |
| Sample tracking information | Date received, date analyzed, lab batch number or other unique identified |
| Published analytical method | e.g., EPA method |
| Limit of detection/quantification | See “QA procedures” |
| Blanks | Reagents and also field blanks if possible |
| Recoveries | |
| Duplicates | |
| Calibration | Source of standards; date stocks prepared |
| Surrogate and internal standards | |
| QA of cofactors | Such as lipid, organic carbon and moisture content |
| Confirmatory tests | e.g., Use of second GC column or other detection system |
| Data manipulations | Blank subtraction, recovery correction |
Field and lab blanks
Determination of PCBs and OCPs requires the analysis of blank samples because of the ubiquitous nature of these contaminants, as discussed previously. For air and water, the sampling media (i.e., pre-extracted water, or the XAD resin, C18 cartridges, or SPMDs) can serve as blanks. Ideally these media should be transported to the field and exposed briefly to the sampling equipment, thus serving as a field blank. Additional samples of the media should be archived in the preparation lab for use as lab blanks. For soils and fatty samples, US EPA Method 1668 for PCBs recommends sand and corn oil, respectively, as lab reference blanks [35]. Clean facilities, ideally with HEPA and carbon-filtered air are recommended, but are not essential, for the determination of PCBs, except for ultralow levels in water. Contamination of lab air can be checked by use of SPMDs or by exposing C18-disks in the lab for a specified period.
QA procedures
Key elements in QA/QC for POPs analysis are the use of reference materials, the use of quality charts, participation in interlaboratory studies, and the use of guidelines for sampling and analysis [121].
Recovery studies in which individual analytes are spiked into the sample matrix prior to extraction are useful for determining the performance of the analytical method. However, they do not provide much information on the true extractability of analytes from naturally contaminated matrices. This can only be accomplished by extracting certified reference materials (CRMs) or, if none are available for a particular type of sample, some other well-characterized material that has been analyzed by an experienced reference lab [25, 74]. Recovery studies can be also useful for determining method detection limits if conducted at concentrations within 3–5 times the estimated limit of detection determined from the instrumental response or from blanks [122].
As a routine measure, spiking surrogate recovery standards into each sample provides useful information on losses of analyte from the extraction step onwards. However, no single PCB or OCP can be representative of all of the organochlorines being determined, and thus recovery correction should be performed with caution. Isotopically labeled surrogates are ideal for analyses of PCBs and OCPs that are being performed by LRMS and HRMS, and isotope dilution techniques correct for the recoveries of these surrogates.
If blanks are significant (for example, averaging greater than 10% of the average level of total PCBs), then blank correction should be carried out. This should only be done with a robust method blank based on at least seven samples or a blank known to be representative of a batch of samples. Ferrario et al. [29] subtracted an MDL value based on the blank+2×σb on the rationale that this ensured a low probability (5%) of false positives compared to subtraction of the average blank only.
Detection limits
Detection limits for PCB/OCPs depend on the analytical method but also on the sample size and QA considerations, e.g., information available from blank or control samples and recovery studies. The selection of detection limits also depends on the goals of the program and how much emphasis is placed on the reliability of the results versus the need to achieve broad geographic coverage, to meet or exceed regulatory or risk assessment guidelines (Electronic Supplementary Material, Table 3 [145–152]). and to avoid reporting “less thans” for a high proportion of samples. A recent US EPA report has reviewed the approaches to calculating detection limits in the analysis of POPs and other environmental contaminants [122].
Detection limits are usually expressed as a concentration, i.e., they are based on the average weight of the sample analyzed, as summarized in Table 6. The recommended method for determining the MDL [122] is to analyze a series of samples, as free from the target analyte as possible (a minimum of seven replicates), that have a concentration of the analyte that is at least equal to, or in the same concentration range as (within a factor of five of), the estimated detection limit. All of the replicate aliquots are processed through the entire analytical method. The standard deviation (σ) of the replicate measurements is determined and the MDL is then determined by multiplying the standard deviation by the Student’s t-value at a 99% percentile for n−1 degrees of freedom. The MDL is thus an estimate of the measured concentration at which there is 99% confidence that a given analyte is present in a given sample matrix. The MDL is defined by the analytical laboratory and is likely to vary between labs because of different blanks and instrumental conditions.
Table 6.
Detection limits defined by various organizations
| Organization | Terminology | Calculation |
|---|---|---|
| US EPA | Method detection limit (MDL) | 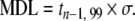 |
| Minimum level of quantitation (ML) | 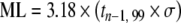 |
|
| American Chemical Society | Limit of detection (LOD) | 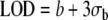 |
| Limit of quantitation (LOQ) | 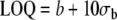 |
|
| International Organization for Standardization / International Union of Pure and Applied Chemistry (ISO/IUPAC) | Critical value (CRV) minimum detectable value (MDV) | 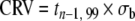 |
| Limit of quantitation. (LOQ ISO/IUPAC) | 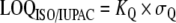 |
The MDL is analogous to the “CRV” described by Currie [123] and reported by ISO/IUPAC, which is tn−1, 95×σb of a sample blank. Note that the t-value is based on the 95th percentile. Thus the MDL and CRV differ in the multiplier used. There may be practical difficulties with measuring some analytes if there no blank value, however the σ of a low-concentration standard can be used. The MDV is similar to the CRV but measured at the detection limit.
Limit of detection (LOD) described by the American Chemical Society [124–126] is equal to the field blank b plus three times the standard deviation of the blank (σb). 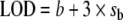 is numerically equivalent to the MDL as b approaches zero or when results are blank-subtracted. This definition has been widely used because of its publication in the peer-reviewed literature. It should be noted that the US EPA “MDL” and the ISO/IUPAC “CRV” implicitly assume that results are adjusted for interferences or blank values.
is numerically equivalent to the MDL as b approaches zero or when results are blank-subtracted. This definition has been widely used because of its publication in the peer-reviewed literature. It should be noted that the US EPA “MDL” and the ISO/IUPAC “CRV” implicitly assume that results are adjusted for interferences or blank values.
The US EPA [122] and other organizations have also defined various lowest levels of quantitation which give a higher degree of confidence in the results. The “minimum level of quantitation” is the lowest level at which the entire analytical system must give a recognizable signal and an acceptable calibration point for the analyte. It is “equivalent to the concentration of the lowest calibration standard, assuming that all method-specified sample weights, volumes, and clean up procedures have been employed. The ML is calculated by multiplying the MDL by 3.18 and rounding the results to the number nearest to (1, 2, or 5) ×10n, where n is an integer.” [122]. The ACS LOQ is defined as being b+10×σb above the gross blank. The ISO/IUPAC LOQ is defined as “the signal or analyte (true) value that will produce estimates having a specified relative standard deviation (RSD), commonly 10%.” [123]. It is calculated as: 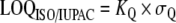 where σQ is the standard deviation of the blank, and where KQ=10.
where σQ is the standard deviation of the blank, and where KQ=10.
Table 7 presents some approximate values for MDLs that should be achievable assuming typical responses of ECD and MS systems, as well as three types of bioanalytical methods, to various OCPs/PCBs and negligible blanks for the individual PCBs and OCPs in a 10 g sample.
Table 7.
Estimated MDLs for individual PCBs and OCPs (ng/g wet wt), after Liem [127]
| Analyte | ELISA (soil/fly ash) | ECDa | MSb (low resolution; EI-SIM) | MSc (high resolution; EI-SIM) |
|---|---|---|---|---|
| PCB 28 | – | 0.05 | 0.1 | 0.01 |
| PCB 52 | – | 0.05 | 0.1 | 0.01 |
| PCB153 | – | 0.05 | 0.05 | 0.005 |
| PCB180 | – | 0.05 | 0.02 | 0.005 |
| p,p′-DDE | 50 | 0.05 | 0.05 | 0.01 |
| Toxaphene | 500 | 0.05 | 0.02 | 0.005 |
| Lindane | 400 | 0.01 | 0.04 | 0.01 |
| HCB | – | 0.01 | 0.02 | 0.005 |
| Dieldrin | 100–500 | 0.01 | 0.02 | 0.01 |
| cis-Chlordane | 100–500 | 0.03 | 0.05 | 0.01 |
| Total PCB | 100–500 | 0.1–1.0 | 0.1–1.0 | 0.01–0.1 |
aAssumes MDL=3×SD of low-level spike (US EPA 2003), assuming 10 g sample and a sample volume of 0.5 mL
bAssumes use of isotope dilution (13C) surrogates; electron ionization, selected ion monitoring mode with 10 g sample, and a sample volume of 0.5 mL
cAssumes use of isotope dilution (13C) surrogates; electron ionization, selected ion monitoring mode with 10 g sample, and a sample final volume of 0.05 mL
Reference materials
Certified reference materials (CRMs; also known as standard reference material or SRM) are essential for determining the accuracy of analytical methods for most PCBs and OCPs [128–130]. The CRMs currently available are shown in Table 8. While no single CRM is currently certified for all POPs, there are published uncertified values available for a broad array of individual PCBs, DDT isomers and toxaphene congeners, especially for NIST 1588a cod liver [131]. Sediment CRMs are freeze-dried and thus potentially not representative of sediment materials normally extracted. Some CRMs, notably SRM 1945a, cannot be shipped outside of the USA without CITES permits.
Table 8.
CRMs for PCBs/OCPs in biota and sediment (adapted from de Boer and McGovern [128])
| CRM | Source | Tissue/species | cis-CHL | trans-CHL | Dieldrin | DDT | HCB | Mirex | PCB congeners |
|---|---|---|---|---|---|---|---|---|---|
| SRM1974b | NIST | mussel | X | X | X | X | |||
| SRM1976 | NIST | lake trout | X | X | X | X | |||
| SRM1588a | NIST | cod liver | X | X | X | X | X | ||
| SRM1945 | NIST | whale bl | X | X | X | X | X | X | |
| SRM2974 | NIST | mussel | X | X | X | ||||
| SRM2977 | NIST | mussel | X | X | X | X | X | ||
| SRM2978 | NIST | mussel | X | X | X | X | |||
| 140/OC | IAEA | plant | X | X | X | ||||
| BCR598 | BCR | cod liver | X | X | X | X | X | X | |
| CARP-1 | NRCC | carp | X | ||||||
| BCR349 | BCR | cod liver | X | ||||||
| BCR350 | BCR | mackerel | X | ||||||
| BCR682 | BCR | mussel | X | ||||||
| BCR618 | BCR | herring | X | ||||||
| EDF 2525 | CIL | lake trout | X | ||||||
| EDF 2514 | CIL | soil | X | ||||||
| SRM1944 | NIST | sediment | X | X | X | X | |||
| SRM1939a | NIST | sediment | X | X | X | ||||
| IAEA383 | IAEA | sediment | X | ||||||
| IAEA408 | IAEA | sediment | X | X | X | ||||
| HS-1 | NRCC | sediment | X | ||||||
| HS-2 | NRCC | sediment | X | ||||||
| BCR536 | BCR | sediment | X | ||||||
| DX-1 | BCR | sediment | |||||||
| DX-1 | BCR | sediment |
Criteria for evaluating the desirability and efficacy of different analytical methods in environmental monitoring and surveillance
Overview
The data generated by chemical analytical and bioanalytical methods for PCBs/OCPs are often used to assess spatial and temporal trends of contamination in environmental media on a national, regional and international basis. The data will also be used by regulators and risk assessors to determine if concentrations exceed guidelines. Thus methods must give accurate results, which are comparable among labs, and at concentrations which are at or below action limits for food or guidelines on wildlife protection (see Electronic Supplementary Material Table 3 [145–152]).
Comparison of detection limits for widely used instrumentation for PCBs and OCPs in Table 7 with guideline limits (Electronic Supplementary Material Table 3) suggests that conventional chemical analytical methodology and bioanalytical methods can meet and exceed these limits in some cases by orders of magnitude. Sample size, lack of coextractive interferences, and final volume are important considerations.
The QA programs discussed above can assure laboratory personnel that accurate and reproducible results are being generated and can be validated by participating in regional, national or international interlaboratory comparisons using samples with assigned values [132]. In the context of implementation of the Stockholm POPs Convention, global comparability is the goal and thus successful participation in international programs should be a requirement of any participating lab. The challenge for future POPs monitoring, especially in developing countries, will be to assess the quality of data and to provide guidance for best laboratory practices.
Interlaboratory comparisons
Interlaboratory comparisons of POPs were conducted, along with the first environmental measurements, in the late 1960s, and became widely adopted in the 1990s [27]. The major international programs related to PCBs and OCPs are the QUASIMEME (Quality Assurance of Information for Marine Environmental Monitoring in Europe) program [53] and the determination of OCPs in food organized in 1993 and 1994 by the Global Environmental Monitoring Scheme (GEMS) of the World Health Organisation (WHO) [133]. Other important ongoing programs are the determination of OCPs/PCBs in human milk under the auspices of AMAP [134] and the intercomparisons operated by the National Institute of Standards and Technology and the Marine Mammal Health and the National Oceanic Atmospheric Administration, National Marine Fishery Service. These programs have been a major stimulus for improved analysis of POPs globally in the past ten years.
The results from interlab programs indicate that training and specific guidance on key aspects of the analytical methods, such as preparation of calibration solutions, are important [26].
PCB/OCP method accuracy
How accurate should analytical methods be? Obviously, they should be accurate enough to identify changes in temporal or spatial trends in contaminant concentrations [121]. Using the approach of Nicholson [135], accuracy is defined as being 95% certainty that the measured values will fall within upper and lower limits defined by:
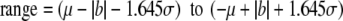 |
where μ is the true concentration, σ is the precision, ±b is the bias, where 1.645=tn−1, 0.95, and where the dataset is normally distributed. Both σ and ∣b∣ can be calculated from multiple analyses of a CRM, where σ is the standard deviation of the analyte and ∣b∣ is the relative percent deviation from the certified value (μ). As a percentage of the true value, the accuracy is given as:
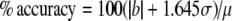 |
Between-laboratory accuracies of 15–20% have been achieved for PCB congeners among experienced labs participating in QUASIMEME [27].
The European Commission [136] has defined the acceptability of an analytical method based on repeated analysis of PCB/OCPs in a CRM as follows:
−50% to +20% for analytes in the range of <1 μg/kg
−30% to +10% for analytes >1 μg/kg to 10 μg/kg
−20% to +10% for analytes >10 μg/kg.
These percentages are also recommended acceptability guidelines for samples spiked with PCBs/OCPs where no CRM is available.
Emerging issues in analytical methods and future directions
Analytical methods for trace organic analysis are constantly evolving and improving, as can be seen from recent reviews of environmental analysis [58, 137, 138]. Although PCB/OCP analysis is a mature area within environmental analytical chemistry, with basic methods that have remained unchanged for the past 20 years, there have been many new technologies that have been gradually adopted and undoubtedly more will emerge in the future. Some emerging procedures with low environmental impact (SPME, microscale glassware, low solvent use, etc.) may be particularly suited for developing countries where analytical budgets are small and product delivery times are lengthy. Thus, strategies must be considered that will allow improved techniques to be adopted by such labs.
A major development in the analysis of POPs has been the introduction of semi-automated extraction instruments for PLE (Accelerated Solvent Extraction; Dionex Instruments, Sunnyvale, CA, USA). The PLE extraction can be combined with solid-phase adsorbents to extract and isolate analytes of interest, for example to remove lipids during the extraction of POPs from fatty samples [139]. In general, automated and semi-automated systems are available for cleanup and isolation of samples for PCB/OCP using disposable solid-phase cartridges, as well as high-performance/pressure LC columns.
Advances in GC that are potentially applicable to PCB/OCP analysis in developing countries are the commercial availability of multidimensional GC (2D-GC) and “fast GC.” In GCxGC, or comprehensive two-dimensional GC, all of the analyte mass is transferred to a second column, and thus resolving power is increased by an order of magnitude [140]. When applied to PCB separations using GCxGC ECD, three- to four-fold lower LODs were obtained compared to one-dimensional GC, and dioxin-like no- and mo-PCBs could be separated from other congeners [141]. Rapid GC separation of PCB congeners by “fast GC” were obtained on narrow-bore (0.1 mm) columns which reduced peak widths and shortened total run times for the full PCB congener mix to minutes [142]. Both techniques can be run with a micro-ECD and are thus relatively inexpensive to implement.
Conclusions
Regulators and managers require data on PCBs and OCPs concentrations in environmental media to assess water quality, food contamination, and to infer possible biological effects. Indeed, the global assessment of the success of the Stockholm POPs convention assumes a chemical measurement capability in all UNEP regions which will deliver data on concentrations in key media [6].
The analytical methodologies discussed here refer to an “analytical system” encompassing information on the collection and storage of samples, the procedures used to extract, isolate, concentrate, separate, identify, and quantify POPs residues in samples, as well as specific quality control and reporting criteria. All aspects of this system must be in operation for POPs to be analyzed and reported.
The chemical analytical methodology for the determination of PCBs and OCPs is a mature area within environmental analytical chemistry as a result of research and development over the past 30–40 years. Basic steps in the quantification of OCPs and PCB congeners have hardly changed in the past 20 years. Analytical methods for the determination of OCP/PCBs in foods, soils, sediments, fish, birds, mammals (including human milk and blood) are available and could be implemented at relatively low cost in developing countries. However, access to modern capillary GC equipment with either electron capture or mass spectrometry (MS) detection to separate and quantify PCBs/OCPs is required in order to conduct the analysis and to take part in regional and international intercomparsons.
In general, ELISAs are very useful tools for the rapid assessment of PCB/OCPs contamination, especially in areas of former heavy use. They are particularly well-suited to laboratories in developing countries which may have access to spectrophotometric equipment but not to GC instrumentation.
Existing analytical methods for PCB/OCPs can determine over 100 individual components at low ng/g concentrations in many environmental media using high-resolution capillary GC–ECD. However, the number of certified values for OCP/PCB congeners in certified reference materials is more limited (approximately 23 PCB congeners and 15 OCPs in NIST 1588a cod liver). At a minimum, the OCP/PCBs for which there are certified values in readily available CRMs should be determined (approximately 38). With this number of analytes, the information would be useful for both regulatory actions as well as for source identification using multivariate analysis or other “fingerprinting” methods.
Interlab comparisons of POPs analysis over the past ten years have shown that availability of accurate analytical standards is a fundamental requirement of an analytical program designed to quantify trace organic contaminants such as POPs. Agencies such as GEF and UNEP Chemicals should give top priority to ensuring that certified analytical standards are available to all labs on a continuing basis.
Quality assurance programs are critically important for demonstrating the performance of analytical methods for POPs within a lab and between labs. QA requirements for PCBs/OCP analysis are well known and include the use of certified reference materials, field and laboratory blanks, the use of quality control charts to monitor long-term lab performance, participation in interlaboratory studies, and the use of guidelines for sampling and analysis.
Determination of PCBs/OCPs requires the analysis of blank samples because of the ubiquitous nature of these contaminants. If blanks are significant (for example, averaging greater than 10% of the average level of total PCBs), then blank correction should be carried out.
As a routine measure, spiking surrogate recovery standards into each sample provides useful information on losses of analyte from the extraction step onwards. However, no single PCB or OCP can be representative of all the organochlorines being determined, and thus recovery correction should be performed with caution. Isotopically labeled surrogates are ideal for the quantification of PCBs and many OCPs via LRMS and HRMS; the application of isotope dilution techniques can correct for the recoveries of these surrogates.
Detection limits depend not only on the analytical method used but also on the sample size and QA considerations, e.g., on information available from blank or control samples and recovery studies. Detection limits should be calculated as described by US EPA or by IUPAC/ISO methodology. Comparison of detection limits for widely used instrumentation for POPs with action limits for POPs in food and tissue residue guidelines suggests that current GC–ECD and GC-MS analytical methodology for PCB/OCPs can meet and exceed these limits, in some cases by orders of magnitude.
Some emerging new analytical techniques, such 2D-GC and “fast GC” using GC–ECD, may be well-suited for use in developing countries in the near future given their relatively low cost and their ability to provide high-resolution separations of OCP/PCBs. Procedures with low environmental impacts (microscale, low solvent use, etc.) may be particularly well-suited to developing countries where analytical budgets are small and product delivery times are lengthy. Thus, strategies must be considered that will allow improved techniques to be adopted by such labs.
Electronic supplementary materials
Below is the link to the electronic supplementary material.
(DOC 4 kb)
(DOC 13 kb)
(DOC 6 kb)
Acknowledgements
The authors would like to thank the Scientific and Technical Advisory Panel (STAP) and the Global Environment Facility (GEF) for permission to use text from a review of PCB and PCDD/F analyses coauthored with Dr. M. Morita (National Institute for Environmental Studies, Tsukuba, Ibaraki, Japan), that was prepared for the STAP Workshop on “The Use of Bioindicators, Biomarkers and Analytical Methods for the Analysis of POPs in Developing Countries” (December 10–12, 2003).
Footnotes
Electronic supplementary material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s00216-006-0765-y and is accessible for authorized users.
References
- 1.UNEP (2001) Final act of the conference of plenipotentiaries on the Stockholm convention on persistent organic pollutants. United Nations Environment Programme, Geneva, Switzerland, p 44
- 2.Tanabe S, Subramanian A (2005) Bioindicators suitable for POPs monitoring in developing countries. Kyoto University Press, Kyoto, Japan
- 3.Integrated Atmospheric Deposition Network (IADN) (1994) Integrated atmospheric deposition network quality assurance program plan. US Environmental Protection Agency, Great Lakes National Program Office, Chicago, IL, p 143
- 4.RAIPON/AMAP/GEF Project (2001) Persistent toxic substances (PTS), Food security and indigenous peoples of the Russian north. In: Kimstach V (ed) Arctic monitoring and assessment program. Olso, Norway
- 5.Arctic Monitoring and Assessment Programme (AMAP) (2000) AMAP trends and effects programme, section B, trend monitoring programme, Table B2. Arctic Monitoring and Assessment Programme, Oslo, Norway
- 6.United Nations Environment Program (UNEP) (2003) Proceedings of the UNEP workshop to develop a global POPs monitoring programme to support the effectiveness evaluation of the Stockholm convention, 24–27 March 2003. UNEP Chemicals, Geneva, Switzerland, p 260
- 7.QUASIMEME (Quality Assurance of Information for Marine Environmental Monitoring in Europe) (2002) Laboratory performance studies. Round 30 exercise protocols FRS marine laboratory, Aberdeen, UK, p 24
- 8.de Boer J, Oehme M, Smith K, Wells DE (2000) Chemosphere 41:493–497 [DOI] [PubMed]
- 9.Dearth MA, Hites RA (1991) Environ Sci Technol 25:245–254 [DOI]
- 10.Muir DCG, Norstrom RJ, Simon M (1988) Environ Sci Technol 22:1071–1079 [DOI] [PubMed]
- 11.Muir DC, Jones PD, Karlsson H, Koczansky K, Stern GA, Kannan K, Ludwig JP, Reid H, Robertson CJ, Giesy JP (2002) Environ Toxicol Chem 21:413–423 [DOI] [PubMed]
- 12.Li YF, Macdonald RW (2005) Sci Total Environ 342:87–106 [DOI] [PubMed]
- 13.United Nations Economic Commission for Europe (UNECE) (2004) Technical review report on Lindane. Reports on substances scheduled for re-assessments under the UNECE POPs protocol. UNECE, Geneva, Switzerland, p 38
- 14.Reiner EJ, Clement RE, Okey AB, Marvin CH (2006) Anal Bioanal Chem DOI 10.1007/s00216-006-0479-1 [DOI] [PMC free article] [PubMed]
- 15.Wells DE, Maier EA, Griepink B (1992) Int J Environ Anal Chem 46:255–264
- 16.Wells DE, de Boer J (1994) Mar Poll Bull 29:174–184 [DOI]
- 17.Swackhamer DL (1988) Quality assurance plan: green bay mass balance study, I. PCBs and Dieldrin. US EPA Great Lakes National Program Office, Chicago, IL
- 18.Frame GM (1999) J High Res Chromatogr 22:533–540 [DOI]
- 19.Sather PJ, Newman JW, Ikonomou MG (2003) Environ Sci Technol 24:5678–5686 [DOI] [PubMed]
- 20.National Oceanic and Atmospheric Administration (NOAA) (1998) Sampling and analytical methods of the national status and trend program, Mussel watch projects: 1993–1996 update. NOAA, Silver Spring, MD
- 21.US FDA (1999) Pesticide analytical manual volume I (PAM) (1994, updated October 1999), 3rd edn. US Food and Drug Administration, Washington, DC
- 22.International Council for Exploration of the Sea (ICES) (2003) Guidelines for monitoring programmes. ICES, Copenhagen, Denmark
- 23.Oslo-Paris Commission (OSPAR) (2000) JAMP guidelines for monitoring contaminants in sediments. OSPAR Secretariat, London, UK
- 24.Keith LH (1996) Compilation of EPA’s sampling and analysis methods, 2nd edn. CRC Press, Boca Raton, FL
- 25.Wells DE, Hess P (2000) Separation, clean-up and recoveries of persistant trace organic contaminants from soils, sediment and biological matrices. In: Barceló E (ed) Sample handling and trace analysis of pollutants, techniques, applications and quality assurance. Elsevier, Amsterdam, pp 73–116
- 26.Wells DE, Hess P (2000) Determination and evaluation of chlorinated biphenyls. In: Barceló E (ed) Sample handling and trace analysis of pollutants, techniques, applications and quality assurance. Elsevier, Amsterdam, pp 239–285
- 27.de Boer J, Law RJ (2003) J Chromatogr A 1000:223–251 [DOI] [PubMed]
- 28.US EPA (1997) Performance-based measurement system. US Environmental Protection Agency, Washington, DC, pp 52098–52100
- 29.Ferrario J, Byrne C, Dupuy AEJ (1997) Chemosphere 34:2451–2465 [DOI] [PubMed]
- 30.Kohler M, Tremp J, Zennegg M, Seiler C, Minder-Kohler S, Beck M, Lienemann P, Wegmann L, Schmid P (2005) Environ Sci Technol 39:1967–1973 [DOI] [PubMed]
- 31.Azcue JM, Mudroch A (1995) Manual of aquatic sediment sampling. CRC Press, Boca Raton, FL
- 32.Fellin P, Barrie LA, Dougherty D, Toom D, Muir D, Grift N, Lockhart L, Billeck B (1996) Environ Toxicol Chem 15:253–261 [DOI]
- 33.Wells DE (1994) Trends Anal Chem 13:339–343 [DOI]
- 34.US Environmental Protection Agency (1995) Method 508.1, Rev 2.0. methods for the determination of organic compounds in drinking water-supplement III (EPA/600/R-95-131). US EPA, Washington, DC
- 35.US Environmental Protection Agency (1999) Method 1668, revision A. Chlorinated biphenyl congeners in water, soil, sediment and tissue by HRGC/HRMS. US EPA, Office of Water, Washington, DC
- 36.Zuagg SD, Sandstrom MW, Smith SG, Fehlberg KM (1995) Methods of analysis by the U.S. Geological Survey national water quality laboratory—determination of pesticides in water by C-18 solid-phase extraction and capillary-column gas chromatography/mass spectrometry with selected-ion monitoring. US Geological Survey, Denver, CO
- 37.Achman DR, Hornbuckle KC, Eisenreich SJ (1993) Environ Sci Technol 27:75–87 [DOI]
- 38.Leister DL, Baker JE (1994) Atmos Environ 28:1499–1520 [DOI]
- 39.Sun P, Backus S, Blanchard P, Hites RA (2006) Environ Sci Technol 40:2135–2141 [DOI] [PubMed]
- 40.Neilson MA, Stevens RJJ, Biberhofer J, Goulden PD, Anthony DHJ (1988) A Large-sample extractor for determining organic contaminants in the great lakes. Technical Bulletin No. 157. Environment Canada, Burlington, ON, Canada
- 41.Petrick G, Schultz-Bull DE, Martens V, Scholz K, Duinker JC (1996) Mar Chem 54:97–105
- 42.Sobek A, Gustafsson Ö (2004) Environ Sci Technol 38:2751–2846 [DOI] [PubMed]
- 43.Lu YB, Wang ZJ, Huckins J (2002) Aquat Toxicol 60:139–153 [DOI] [PubMed]
- 44.Shen L, Wania F, Lei YD, Teixeira C, Muir DC, Bidleman TF (2004) Environ Sci Technol 38:965–975 [DOI] [PubMed]
- 45.Huckins JN, Petty JD, Lebo JA, Almeida FV, Booij K, Alvarez DA, Cranor LW, Clark RC, Mogensen BB (2002) Environ Sci Technol 36:85–91 [DOI] [PubMed]
- 46.Ockenden WA, Corrigan BP, Howsam M, Jones KC (2001) Environ Sci Technol 35:4536–4543 [DOI] [PubMed]
- 47.Shoeib M, Harner T (2002) Environ Sci Technol 36:4142–4151 [DOI] [PubMed]
- 48.Wania F, Shen L, Lei YD, Teixeira C, Muir DCG (2003) Environ Sci Technol 37:1352–1359 [DOI]
- 49.Meijer SN, Ockenden WA, Steinnes E, Corrigan BP, Jones KC (2003) Environ Sci Technol 37:454–461 [DOI] [PubMed]
- 50.Kiriluk RM, Hyatt WH, Keir MJ, Whittle DM (1996) Fluctuations in levels of total PCB, organochlorine residues, lipid, and moisture in whole lake trout homogenate samples during four years of frozen storage. Fisheries and Oceans Canada, Ottawa, ON, p 32
- 51.Norstrom RJ, Won HT (1985) J Assoc Offic Anal Chem 68:129–135 [PubMed]
- 52.de Boer J, Smedes F (1997) Mar Poll Bull 35:93–108 [DOI]
- 53.Wells DE, Chiffoleau J-F, Klungsøyr J (1997) Mar Poll Bull 35:109–124 [DOI]
- 54.Söderström M, Nylund K, Jarnberg U, Alsberg T, Asplund L (2005) Chemosphere 58:355–366 [DOI] [PubMed]
- 55.Gevao B (2000) PhD thesis. Lancaster University, Lancaster, UK
- 56.Karl H (2000) Inf Fischwirtsch Fischereiforsch 1:52–57
- 57.Wallace JC, Brzuzy LP, Simonich SL, Visscher SM, Hites RA (1996) Environ Sci Technol 30:2730–2734
- 58.Koester CJ, Simonich SL, Esser BK (2003) Anal Chem 75:2813–2829 [DOI]
- 59.Björklund E, Nilsson T, Bøwadt S (2000) Trends Anal Chem 19:434–445 [DOI]
- 60.Schantz MM (2006) Anal Bioanal Chem DOI 10.1007/s00216-006-0648-2 [DOI] [PubMed]
- 61.US Environmental Protection Agency (1995) EPA Method 3545, pressurised fluid extraction, test methods for evaluating solid waste, 3rd edn. Update. III, EPA SW-846. US EPA, Washington, DC
- 62.Lopez-Avila V, Young R, Beckert WF (1994) Anal Chem 66:1097–1106 [DOI]
- 63.de Boer J (1988) Chemosphere 17:1803–1810 [DOI]
- 64.Lopez-Avila V (1999) Crit Rev Anal Chem 29:195–230
- 65.US Environmental Protection Agency (2000) SW-846, test methods for evaluating solid waste, 3rd edn. Update IVB. Chapter 4, organic analytes. US EPA, Washington, DC
- 66.Specht W, Pelz S, Gilsbach W (1995) Fresen J Anal Chem 353:183–190 [DOI] [PubMed]
- 67.Ribick MA, Dubay GR, Petty JD, Stalling DL, Schmitt CJ (1982) Environ Sci Technol 16:310–318 [DOI] [PubMed]
- 68.Guerin TF (1999) J Environ Monitor 1:63–67 [DOI] [PubMed]
- 69.Lou X, Janssen H-G, Cramers CA (1997) Anal Chem 69:1598–1603 [DOI]
- 70.Ramos L, Kristenson EM, Brinkman UAT (2002) J Chromatogr A 975:3–29 [DOI] [PubMed]
- 71.Bjerregaard P, Dewailly E, Ayotte P, Pars T, Ferron L, Mulvad G (2001) J Toxicol Environ Health A 62:69–81 [DOI] [PubMed]
- 72.Ayotte P, Dewailly É, Ryan JJ, Bruneau S, Lebel G (1997) Chemosphere 34:1459–1468 [DOI] [PubMed]
- 73.Sandau CD, Sjodin A, Davis MD, Barr JR, Maggio VL, Waterman AL, Preston KE, Preau JLJ, Barr DB, Needham LL, Patterson DGJ (2003) Anal Chem 75:71–77 [DOI] [PubMed]
- 74.Needham LL, Ryan JJ, Fürst P (2002) J Toxicol Environ Health A65:1893–1908 [DOI] [PubMed]
- 75.Burke ER, Holden AJ, Shaw IC (2003) Chemosphere 50:529–535 [DOI] [PubMed]
- 76.AOAC International (2003) Official methods of analysis of AOAC International, 17th edition current through 2nd revision, vol 1. Agricultural chemicals, contaminants, drugs. AOAC International, Gaithersburg, MD
- 77.Focant J-F, Pirard C, Massart A-C, De Pauw E (2003) Chemosphere 52:725–733 [DOI] [PubMed]
- 78.Usenko S, Hageman KJ, Schmedding DW, Wilson GR, Simonich SL (2005) Environ Sci Technol 39:6006–6015 [DOI] [PubMed]
- 79.Sauret-Szczepanski N, Mirabel P, Wortham H (2006) Environ Poll 139:133–142 [DOI] [PubMed]
- 80.Zhang Z, Yang MJ, Pawliszyn J (1994) Anal Chem 66:844–853
- 81.Baily SK, Wells DE, de Boer J, Delbeke K (1994) The measurement of lipids as a co-factor for organic contaminants in biota. FRS Marine Laboratory, Aberdeen, UK
- 82.Bligh EG, Dyer WJ (1959) Can J Biochem Physiol 37:911–917 [DOI] [PubMed]
- 83.Lee CM, Trevino B, Chaiyawat M (1996) J Assoc Offic Anal Chem Int 79:487–492 [PubMed]
- 84.Smedes F (1999) Analyst 124:1711–1718 [DOI]
- 85.Frings CS, Fendley TW, Dunn RT, Queen CA (1972) Clin Chem 18:673–674 [PubMed]
- 86.International Organization for Standardization (2002) Soil quality, determination of organochlorine pesticides and polychlorinated biphenyls. Gas-chromatographic method with electron capture detection. ISO, Geneva, Switzerland, p 22
- 87.Tanabe S, Kannan N, Wakimoto T, Tatsukawa R (1987) Int J Environ Anal Chem 29:199–213 [DOI] [PubMed]
- 88.US Environmental Protection Agency (1995) Method 525.2, Rev 2.0. Methods for the determination of organic compounds in drinking water-supplement iii (EPA/600/R-95-131). US EPA, Washington, DC
- 89.van der Valk F, Wester PG (1991) Chemosphere 22:57–66 [DOI]
- 90.Chan HM, Zhu J, Yeoboa F (1998) Chemosphere 36:2135–2148 [DOI]
- 91.Rimkus GG, Rummler M, Nausch I (1996) J Chromatogr 737:9–14 [DOI] [PubMed]
- 92.Jansson B, Wideqvist U (1983) Int J Environ Anal Chem 13:309–321 [DOI] [PubMed]
- 93.Jansson B, Andersson R, Asplund L, Litzen K, Nylund K, Sellstrom U, Uvemo U-B, Wahlberg C, Wideqvist U (1993) Environ Toxicol Chem 12:1163–1174
- 94.Giesy JP, Hilscherova K, Jones PD, Kannan K, Machala M (2002) Mar Pollut Bull 45:3–16 [DOI] [PubMed]
- 95.Jensen S, Renberg L, Reutergardh L (1977) Anal Chem 49:316–318 [DOI] [PubMed]
- 96.Environment Canada (1997) Manual for analytical methods, vol 3. National Laboratory for Environmental Testing, Burlington, ON, Canada
- 97.Cochran JW, Frame GM (1999) J Chromatogr A 843:323–368 [DOI] [PubMed]
- 98.Tuinstra LGMT, Roos AH, Griepink B, Wells DE (1985) J High Res Chromatogr 8:475–480
- 99.Lang V (1992) J Chromatogr 595:1–43 [DOI] [PubMed]
- 100.Mullins MD, Pochini CM, McCrindle S, Romkes M, Safe SH, Safe LM (1984) Environ Sci Technol 18:468–476 [DOI] [PubMed]
- 101.Frame GM (1997) Anal Chem 69:468A–475A
- 102.Alaee M, Backus S, Cannon C (2001) J Sep Sci 24:465–469 [DOI]
- 103.Vetter W, Schurig V (1997) J Chromatogr A 774:143–175 [DOI] [PubMed]
- 104.Schurig V (2002) Trends Anal Chem 21:647–661 [DOI]
- 105.Jantunen LMM, Bidleman TF, Harner T, Parkhurst WJ (2000) Environ Sci Technol 34:5097–5105 [DOI]
- 106.Leone AD, Amato S, Falconer RL (2001) Environ Sci Technol 35:4592–4596 [DOI] [PubMed]
- 107.Wong CS, Mabury SA, Whittle DM, Backus SM, Teixeira C, DeVault DS, Bronte CR, Muir DCG (2004) Environ Sci Technol 38:84–92 [DOI] [PubMed]
- 108.Klee MS, Williams MB, Chang I (1999) J High Res Chromatogr 22:24–28 [DOI]
- 109.Richardson SD (2001) Chem Rev 101:211–254 [DOI] [PubMed]
- 110.Leonards PEG, Brinkman UAT, Cofino WP (1996) Chemosphere 32:2381–2387 [DOI]
- 111.Carlson DL ,Hites RA (2005) Environ Sci Technol 39:7389–7395 [DOI] [PubMed]
- 112.Chu S, Hong CS, Rattner BA, McGowan PC (2003) Anal Chem 75:1058–1066 [DOI] [PubMed]
- 113.Behnisch PA, Hosoea K, Sakai S (2001) Environ Int 27:413–439 [DOI] [PubMed]
- 114.Sherry J (1997) Chemosphere 34:1011–1025 [DOI] [PubMed]
- 115.Meulenberg EP, Mulder WH, Stoks PG (1995) Environ Sci Technol 29:554–560 [DOI] [PubMed]
- 116.Beasley HL, Pasha A, Guihot SL, Skerritt JH (2000) Food Agric Immunol 12:203–215 [DOI]
- 117.Beasley HL, Phongkham T, Daunt MH, Guihot SL, Skerritt JH (1998) J Agric Food Chem 46:3339–3352 [DOI]
- 118.Shivaramaiah HM, Odeh IOA, Kennedy IR, Skerritt JH (2002) J Agric Food Chem 50:5360–5367 [DOI] [PubMed]
- 119.Galloway TS, Sanger RC, Smith KL, Fillmann G, Readman JW, Ford TE, Depledge MH (2002) Environ Sci Technol 36:2219–2226 [DOI] [PubMed]
- 120.Skerritt JH, Hill AS, Sashidhar Rao RB, Beasley HL, Rani BEA, Udaya Kumari CG, Vijayashankar YN, Venugopal NBRK, Karanth NGK (2003) Food Agric Immunol 15:17–34 [DOI]
- 121.de Boer J (2003) Quality assurance/quality control and data treatment. Discussion paper No.4 for UNEP POP Workshop. In: Proceedings of the UNEP workshop to develop a global POPs monitoring programme to support the effectiveness evaluation of the Stockholm convention, UNEP Chemicals, Geneva, Switzerland
- 122.Gomez-Taylor M, Kahn HD, Telliard WA, Ditthavong K, Kopylev L, McCarty H, Riddick L, Miller K, Cuddeback J, Rushneck D, Dedah S, Stralka K (2003) Technical support document for the assessment of detection and quantitation approaches. US Environmental Protection Agency, Washington, DC, p 124
- 123.Currie LA (1995) Pure Appl Chem 67:1699–1722
- 124.MacDougall D, Crummett WB et al. (1980) Anal Chem 52:2242–2249
- 125.Keith LH, Crummett W, Deegan J, Libby RA, Taylor JK, Wentler G (1983) Anal Chem 55:2210–2218 [DOI]
- 126.Keith LH (1991) Environmental sampling and analysis: a practical guide. Lewis, Boca Raton, FL
- 127.Liem AKD (1999) Trends Anal Chem 18:429–439
- 128.de Boer J, McGovern E (2001) Trends Anal Chem 20:140–159 [DOI]
- 129.Poster DL, Schantz MM, Kucklick JR, de Alda MJL, Porter BJ, Pugh R, Wise SA (2004) Anal Bioanal Chem 378:1213–1231 [DOI] [PubMed]
- 130.Wise SA, Poster DL, Schantz MM, Kucklick JR, Sander LC, de Alda ML, Schubert P, Parris RM, Porter BJ (2004) Anal Bioanal Chem 378:1251–1264 [DOI] [PubMed]
- 131.Kucklick JR, Tuerk KJS, Vander Pol SS, Schantz MM, Wise SA (2004) Anal Bioanal Chem 378:1147–1151 [DOI] [PubMed]
- 132.Thompson M, Ellison SLR, Wood R (2002) Pure Appl Chem 74:835–856
- 133.Weigert P, Gilbert J, Patey AL, Key PE, Wood R, Barylko-Pikielna N (1997) Food Addit Contam 14:399–410 [DOI] [PubMed]
- 134.Weber J-P (2001) Report on the AMAP ringtest, round 2. Institut National de Santé Publique du Québec, Sainte-Foy, Canada
- 135.Nicholson MD (1989) Mar Pollut Bull 20:33–40
- 136.EC (2002) Commission decision implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Doc. 2002/657/EC. European Commission, Brussels, Belgium
- 137.Koester CJ, Moulik A (2005) Anal Chem 77:3737–3754 [DOI] [PubMed]
- 138.Clement RE, Yang PW, Koester CJ (2001) Anal Chem 73:2761–2790 [DOI] [PubMed]
- 139.Björklund E, Müller A, von Holst C (2001) Anal Chem 73:4050–4053 [DOI] [PubMed]
- 140.Frysinger GS, Gaines RB, Reddy CM (2002) Environ Forens 3:27–34
- 141.Korytar P, Leonards PEG, de Boer J, Brinkman UAT (2002) J Chromatogr A 958:203–218 [DOI] [PubMed]
- 142.Covaci A, Schepens P (2001) J Chromatogr A 923:287–293 [DOI] [PubMed]
- 143.Mackay D, Shui WY, Ma KC, Lee SC (2006) Physical-chemical properties and environmental handbook, 2nd edn (CD ROM version). CRC Press, Boca Raton, FL, p 5664
- 144.Fisk AT, Cymbalisty CD, Tomy GT, Stern GA, Muir DCG (1999) Chemosphere 39:2549–2562 [DOI]
- 145.de Wit CA, Fisk AT, Hobbs KE, Muir DCG, Gabrielsen GW, Kallenborn R, Krahn MM, Norstrom RJ, Skaare JU (2004) AMAP assessment 2002: persistent organic pollutants in the Arctic. AMAP, Oslo, Norway
- 146.US Food and Drug Administration (2001) Fish and fisheries products hazards and controls guidance, 3rd edn. US FDA, Center for Food Safety & Applied Nutrition, Washington, DC
- 147.IJC (1986) Great lakes water quality agreement. Annex 1—objectives for protection of aquatic life and wildlife. International Joint Commission, Windsor, ON Canada
- 148.Environment Canada (2002) Canadian tissue residue guidelines for the protection of wildlife consumers of aquatic biota. Environment Canada, Ottawa, ON, Canada
- 149.US Environmental Protection Agency (1995) Great lakes water quality initiative criteria documents for the protection of wildlife, EPA-820-B-95-008. US EPA, Office of Science and Technology, Washington, DC
- 150.OME (1993) Guidelines for the protection and management of aquatic sediment quality in Ontario. Ontario Ministry of Environment, Toronto, ON, Canada, p 27
- 151.Long ER, Macdonald DD, Smith SL, Calder FD (1995) Environ Manag 19:81–97 [DOI]
- 152.Van den Berg M, Birnbaum L, Bosveld BTC, Brunström B, Cook P, Feeley M, Giesy J, Hanberg A, Hasegawa R, Kennedy SW, Kubiak T, Larsen JC, van Leeuwen FXR, Liem AKD, Nolt C, Peterson RE, Poellinger L, Safe S, Schrenk D, Tillitt D, Tysklind M, Younes M, Wærn F, Zacharewski R (1998) Environ Health Perspect 106:775–792 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below is the link to the electronic supplementary material.
(DOC 4 kb)
(DOC 13 kb)
(DOC 6 kb)