

Abstract
Rationale
3,4-Methylenedioxymethamphetamine (MDMA) is a widely abused illicit drug. In animals, high-dose administration of MDMA produces deficits in serotonin (5-HT) neurons (e.g., depletion of forebrain 5-HT) that have been interpreted as neurotoxicity. Whether such 5-HT deficits reflect neuronal damage is a matter of ongoing debate.
Objective
The present paper reviews four specific issues related to the hypothesis of MDMA neurotoxicity in rats: (1) the effects of MDMA on monoamine neurons, (2) the use of “interspecies scaling” to adjust MDMA doses across species, (3) the effects of MDMA on established markers of neuronal damage, and (4) functional impairments associated with MDMA-induced 5-HT depletions.
Results
MDMA is a substrate for monoamine transporters, and stimulated release of 5-HT, NE, and DA mediates effects of the drug. MDMA produces neurochemical, endocrine, and behavioral actions in rats and humans at equivalent doses (e.g., 1–2 mg/kg), suggesting that there is no reason to adjust doses between these species. Typical doses of MDMA causing long-term 5-HT depletions in rats (e.g., 10–20 mg/kg) do not reliably increase markers of neurotoxic damage such as cell death, silver staining, or reactive gliosis. MDMA-induced 5-HT depletions are accompanied by a number of functional consequences including reductions in evoked 5-HT release and changes in hormone secretion. Perhaps more importantly, administration of MDMA to rats induces persistent anxiety-like behaviors in the absence of measurable 5-HT deficits.
Conclusions
MDMA-induced 5-HT depletions are not necessarily synonymous with neurotoxic damage. However, doses of MDMA which do not cause long-term 5-HT depletions can have protracted effects on behavior, suggesting even moderate doses of the drug may pose risks.
Keywords: MDMA, Serotonin, Depletion, Neurotoxicity, Release, Hormones, Behavior
Introduction
3,4-Methylenedioxymethamphetamine (MDMA or Ecstasy) is an illicit drug used by young adults in the US, Europe, and elsewhere. The appeal of MDMA is related to its unique profile of psychotropic actions, which includes amphetamine-like stimulant effects, coupled with feelings of increased emotional sensitivity and closeness to others (Liechti and Vollenweider 2001; Vollenweider et al. 1998). MDMA misuse among children and adolescents is widespread in the US (Landry 2002; Yacoubian 2003); a recent sampling of high school students found 10% of 12th graders reported using MDMA at least once (Banken 2004). MDMA-related medical complications have risen more than 20-fold in recent years, consistent with increasing popularity of the drug. Serious adverse effects of MDMA intoxication include cardiac arrhythmias, hypertension, hyperthermia, serotonin (5-HT) syndrome, hyponatremia, liver problems, seizures, coma, and, in rare cases, death (Schifano 2004). Accumulating evidence indicates that long-term MDMA abuse is associated with cognitive impairments and mood disturbances, which can last for months after cessation of drug intake (Morgan 2000; Parrott 2002). Despite the risks of illicit MDMA use, some clinicians believe the drug may have therapeutic potential in the treatment of psychiatric disorders, such as post-traumatic stress disorder, and clinical studies with MDMA are underway (Doblin 2002). It is worth noting that MDMA has been administered to human subjects in controlled research settings, and few side effects are observed under these circumstances (Harris et al. 2002; Mas et al. 1999).
These considerations provide compelling reasons to evaluate the pharmacology and toxicology of MDMA and related compounds. In this review, we will examine four topics related to the hypothesis of MDMA-induced 5-HT neurotoxicity in rats: (1) the effects of MDMA on monoamine neurons, (2) the use of interspecies scaling to extrapolate doses of MDMA across species, (3) the effects of MDMA on established markers of neurotoxic damage, and (4) the functional impairments associated with MDMA-induced 5-HT depletions. The review will focus on data obtained from rats since most preclinical MDMA research has been carried out in this animal model. Previously published and new data from our laboratory at the National Institute on Drug Abuse (NIDA) will be included to supplement literature reports. Clinical findings will be mentioned in specific instances to note comparisons between rats and humans. All experiments in our laboratory utilized male Sprague–Dawley rats (Wilmington, MA, USA) weighing 300–350 g. Rats were maintained in facilities accredited by the American Association of the Accreditation of Laboratory Animal Care, and procedures were carried out in accordance with the Animal Care and Use Committee of the NIDA Intramural Research Program (IRP). Data from mice will not be considered here because this animal species displays the unusual characteristic of long-term DA depletions (i.e., DA neurotoxicity) in response to MDMA, rather than long-term 5-HT depletions observed in rats, nonhuman primates, and most other animals [reviewed by Colado et al. (2004)]. Finally, the present paper will not address possible molecular mechanisms underlying MDMA-induced 5-HT deficits, as several excellent reviews have covered this subject (Lyles and Cadet 2003; Monks et al. 2004; Sprague et al. 1998).
Effects of MDMA on monoamine neurons
To address the topic of MDMA-induced 5-HT neurotoxicity, the pharmacology of MDMA must be briefly reviewed. Figure 1 shows that MDMA is a ring-substituted analog of methamphetamine. Ecstasy tablets ingested by humans contain a racemic mixture of (+) and (−) isomers of MDMA, and both stereoisomers are known to be bioactive (Johnson et al. 1986; Schmidt et al. 1987). Ecstasy tablets often contain other psychoactive substances such as substituted amphetamines, caffeine, or ketamine, which can contribute to the overall effects of the ingested preparation (Parrott 2004). Upon systemic administration, N-demethylation of MDMA occurs via first-pass metabolism to yield the ring-substituted amphetamine analog 3,4-methylenedioxyamphetamine (MDA) (de la Torre et al. 2004). Initial studies carried out in the 1980s showed that MDMA and MDA stimulate efflux of preloaded [3H]5-HT, and to a lesser extent [3H]DA, in nervous tissue (Johnson et al. 1986; Nichols et al. 1982; Schmidt et al. 1987). Subsequent findings revealed that MDMA interacts with monoamine transporter proteins to stimulate non-exocytotic release of 5-HT, DA and norepinephrine (NE) in rat brain (Berger et al. 1992; Crespi et al. 1997; Fitzgerald and Reid 1993).
Fig. 1.

Chemical structures of MDMA and related compounds
Table 1 summarizes previously published data from our laboratory showing structure–activity relationships for stereoisomers of MDMA, MDA, and related drugs as monoamine releasers in rat brain synaptosomes (Partilla et al. 2000; Rothman et al. 2001; Setola et al. 2003). Like other substrate-type releasers, MDMA and MDA bind to plasma membrane monoamine transporters and are translocated into the cytoplasm. The ensuing transmitter release occurs by a two-pronged mechanism: (1) transmitter molecules exit the cell along their concentration gradients via reversal of normal transporter function, and (2) cytoplasmic concentrations of transmitter are increased due to drug-induced disruption of vesicular storage [reviewed by Rothman and Baumann (2002) and Rudnick and Clark (1993)]. As shown in Table 1, stereoisomers of MDMA and MDA are substrates for 5-HT transporters (SERT), NE transporters (NET) and DA transporters (DAT), with (+) isomers exhibiting greater potency as releasers. In particular, (+) isomers of MDMA and MDA are much more effective DA releasers than their corresponding (−) isomers. It is noteworthy that (+) isomers of MDMA and MDA are rather nonselective in their ability to stimulate monoamine release in vitro. When compared to amphetamine and methamphetamine, the major effect of methylenedioxy ring substitution is enhanced potency for 5-HT release and reduced potency for DA release. For example, (+)-MDMA releases 5-HT (EC50=70.8 nM) about ten times more potently than (+)-methamphetamine (EC50=736 nM), whereas (+)-MDMA releases DA (EC50=142 nM) about six times less potently than (+)-methamphetamine (EC50=24 nM).
Table 1.
Profile of MDMA and related compounds as monoamine transporter substrates in rat brain synaptosomes
| Drug | 5-HT release EC50 (nM±SD) | NE release EC50 (nM±SD) | DA release EC50 (nM±SD) |
|---|---|---|---|
| (+)-Methamphetamine | 736±45 | 12±0.7 | 24±2 |
| (−)-Methamphetamine | 4,640±240 | 29±3 | 416±20 |
| (±)-MDMA | 74.3±5.6 | 136±17 | 278±12 |
| (+)-MDMA | 70.8±5.2 | 110±16 | 142±6 |
| (−)-MDMA | 337±34 | 564±60 | 3,682±178 |
| (+)-Amphetamine | 1,765±94 | 7.1±1.0 | 25±4 |
| (±)-MDA | 159±12 | 108±12 | 290±10 |
| (+)-MDA | 99.6±7.4 | 98.5±6.1 | 50.0±8.0 |
| (−)-MDA | 313±21 | 287±23 | 900±49 |
Consistent with in vitro results, in vivo microdialysis experiments demonstrate that MDMA increases extracellular 5-HT and DA in rat brain, with effects on 5-HT being greater in magnitude (Baumann et al. 2005; Gudelsky and Nash 1996; Kankaanpaa et al. 1998; Yamamoto et al. 1995). Figure 2 depicts new data from our laboratory showing the stimulatory effects of MDMA and MDA on extracellular 5-HT and DA in rat nucleus accumbens. In these experiments, i.v. injections of saline (0 dose) or drug were administered to conscious male rats undergoing in vivo microdialysis. Doses of 0.3 and 1.0 mg/kg were chosen because these doses of MDMA are self-administered by rats (Ratzenboeck et al. 2001; Schenk et al. 2003). Dialysate samples were collected every 20 min beginning 2 h before injections until 2 h thereafter; samples were assayed for 5-HT and DA by high-performance liquid chromatography coupled to electrochemical detection (HPLC–ECD) as described elsewhere (Baumann and Rutter 2003). Neurochemical data were converted to percentage of control values based on three preinjection control samples. The effects of MDMA and MDA are depicted as peak effects, which were observed in the first 20 min after injection. Peak effect data were analyzed using a one-way ANOVA (drug dose) followed by Duncan’s post hoc test. MDMA caused significant increases in dialysate 5-HT [F2,15=19.47, P<0.001] and DA [F2,15=14.46, P<0.01]. MDA caused similar increases in 5-HT [F2,15=17.95; P<0.001] and DA [F2,15=7.69, P<0.01], but appeared to be slightly more potent at releasing DA. Both drugs produced elevations in 5-HT that were greater than the corresponding effects on DA. For example, the 1 mg/kg dose of MDMA produced a tenfold rise in 5-HT and a twofold rise in DA.
Fig. 2.

Effects of (±)-MDMA and (±)-MDA on extracellular levels of 5-HT (top panel) and DA (bottom panel) in rat nucleus accumbens. Male rats undergoing in vivo microdialysis received i.v. injections of saline (0 dose) or drug, and dialysate levels of 5-HT and DA were assayed by HPLC–ECD (Baumann and Rutter 2003). Data are expressed as the percentage of three pre-injection baseline samples; each bar represents the mean±SEM peak effect measured 20 min posttreatment, N=6 rats/group. Baseline levels of 5-HT and DA were 0.17±0.01 and 1.31±0.05 pg/5 μl, respectively. Asterisk denotes significance with respect to zero dose control (P<0.05 Duncan’s)
Acute central nervous system (CNS) effects of MDMA are mediated by the release of monoamine transmitters, with the subsequent activation of presynaptic and postsynaptic receptors [reviewed by Cole and Sumnall (2003) and Green et al. (2003)]. As specific examples in rats, MDMA suppresses 5-HT cell firing, evokes neuroendocrine secretion, and stimulates locomotor activity. MDMA-induced suppression of 5-HT cell firing in the dorsal and median raphe involves activation of presynaptic 5-HT1A autoreceptors by endogenous 5-HT (Gartside et al. 1997; Sprouse et al. 1989). Neuroendocrine effects of MDMA include secretion of prolactin from the anterior pituitary and corticosterone from the adrenal glands (Nash et al. 1988). Evidence indicates that these MDMA-induced hormonal effects are mediated via postsynaptic 5-HT2 receptors in the hypothalamus, which are activated by released 5-HT. MDMA elicits a unique profile of locomotor effects, characterized by forward locomotion and elements of the 5-HT behavioral syndrome such as forepaw treading, flattened body posture, and head weaving (Gold et al. 1988; Slikker et al. 1989; Spanos and Yamamoto 1989). The complex motor effects of MDMA are dependent upon monoamine release followed by activation of multiple 5-HT and DA receptor subtypes in the brain [reviewed by Bankson and Cunningham (2001); Geyer (1996)].
Adverse effects of acute MDMA administration, including cardiovascular stimulation and elevated body temperature, are thought to involve monoamine release from sympathetic nerves in the periphery or nerve terminals in the CNS. MDMA increases heart rate and mean arterial pressure in conscious rats (O’Cain et al. 2000); this cardiovascular stimulation is likely mediated by MDMA-induced release of peripheral NE stores, similar to the effects of amphetamine (Fitzgerald and Reid 1994). MDMA has weak agonist actions at α2-adrenoreceptors and 5-HT2 receptors, which might influence its cardiac and pressor effects (Battaglia and De Souza 1989; Lavelle et al. 1999; Lyon et al. 1986). Moreover, MDA is a potent 5-HT2B agonist and this property could contribute to adverse cardiovascular effects (Setola et al. 2003). The ability of MDMA to elevate body temperature is well-characterized in rats (Dafters 1995; Dafters and Lynch 1998; Nash et al. 1988), and this response has been considered a 5-HT-mediated process. However, a recent study by Mechan et al. (2002) provides convincing evidence that MDMA-induced hyperthermia involves the activation of postsynaptic D1 receptors by released DA.
The long-term adverse effects of MDMA on 5-HT systems have attracted substantial interest because studies in rats and nonhuman primates show that high-dose MDMA administration produces persistent reductions in markers of 5-HT nerve terminal integrity [reviewed by Lyles and Cadet (2003); Sprague et al. (1998)]. Table 2 summarizes findings of investigators who first demonstrated that MDMA causes long-term (>1 week) inactivation of tryptophan hydroxylase activity, depletions of brain tissue 5-HT, and reductions in SERT binding and function (Battaglia et al. 1987; Commins et al. 1987; Schmidt 1987; Stone et al. 1987). These serotonergic deficits are observed in various regions of rat forebrain, including frontal cortex, striatum, hippocampus, and hypothalamus. Immunohistochemical analysis of 5-HT in cortical and subcortical areas reveals an apparent loss of 5-HT axons and terminals in MDMA-treated rats, especially the fine-diameter projections arising from the dorsal raphe nucleus (O’Hearn et al. 1988). Moreover, 5-HT axons and terminals remaining after MDMA treatment appear swollen and fragmented, suggesting structural damage.
Table 2.
Long-term effects of (±)-MDMA on 5-HT neuronal markers in rats
| 5-HT deficit | Dose | Survival interval | Reference |
|---|---|---|---|
| Depletions of 5-HT in cortex, as measured by HPLC–ECD | 10 mg/kg, s.c., single dose | 1 week | Schmidt (1987) |
| Depletions of 5-HT in forebrain regions as measured by HPLC–ECD | 10–40 mg/kg, s.c., twice daily, 4 days | 2 weeks | Commins et al. (1987) |
| Reductions in tryptophan hydroxylase activity in forebrain regions | 10 mg/kg, s.c., single dose | 2 weeks | Stone et al. (1987) |
| Loss of [3H]-paroxetine-labeled SERT binding sites in forebrain regions | 20 mg/kg, s.c., twice daily, 4 days | 2 weeks | Battaglia et al. (1987) |
| Deceased immunoreactive 5-HT in fine axons and terminals in forebrain regions | 20 mg/kg, s.c., twice daily, 4 days | 2 weeks | O’Hearn et al. (1988) |
Time-course studies indicate that MDMA-induced 5-HT depletion occurs in a biphasic manner, with a rapid acute phase followed by a delayed long-term phase (Schmidt 1987; Stone et al. 1987). In the acute phase, which lasts for the first few hours after drug administration, massive depletion of brain tissue 5-HT is accompanied by inactivation of tryptophan hydroxylase. Twenty-four hours later, tissue 5-HT recovers to normal levels but hydroxylase activity remains diminished. In the long-term phase, which begins within 1 week and lasts for months, marked depletion of 5-HT is accompanied by sustained inactivation of tryptophan hydroxylase and loss of SERT binding and function (Battaglia et al. 1988; Scanzello et al. 1993). The findings in Table 2 have been replicated by many investigators, and the spectrum of decrements is typically described as 5-HT neurotoxicity. Most of the studies designed to examine MDMA neurotoxicity in rats have employed i.p. or s.c. injections of 10 mg/kg or higher, either as single or repeated treatments. These MDMA dosing regimens are known to produce significant hyperthermia, which can exacerbate 5-HT depletions caused by the drug (Green et al. 2004; Malberg and Seiden 1998). All of these experiments have involved administration of MDMA that is not contingent on a specific behavior and this factor could significantly influence effects of the drug.
There are caveats to the hypothesis that MDMA produces 5-HT neurotoxicity. O’Hearn et al. (1988) showed that high-dose MDMA administration has no effect on 5-HT cell bodies in the dorsal raphe, despite profound loss of 5-HT in forebrain projection areas. Thus, the effects of MDMA on 5-HT neurons are sometimes referred to as “axotomy,” to account for the fact that perikarya are not damaged (Molliver et al. 1990; O’Hearn et al. 1988). MDMA-induced reductions in 5-HT levels and SERT binding eventually recover (Battaglia et al. 1988; Scanzello et al. 1993), suggesting the possibility that 5-HT terminals are not destroyed. Many drugs used clinically produce effects that are similar to those produced by MDMA. For instance, reserpine causes sustained depletions of brain tissue 5-HT, yet reserpine is not considered a neurotoxin (Carlsson 1976). Chronic administration of 5-HT selective reuptake inhibitors (SSRIs), like paroxetine and sertraline, leads to a marked loss of SERT binding and function comparable to MDMA, but these agents are therapeutic drugs rather than neurotoxins (Benmansour et al. 1999; Frazer and Benmansour 2002). Finally, high-dose administration of SSRIs produces swollen, fragmented, and abnormal 5-HT terminals, which are indistinguishable from the effects of MDMA and other substituted amphetamines (Kalia et al. 2000).
The caveats mentioned above raise a number of questions with respect to MDMA neurotoxicity. Of course, the most important question is whether MDMA abuse causes neurotoxic damage in humans. This complex issue is a matter of ongoing debate, which has been addressed by a number of recent papers (Gouzoulis-Mayfrank et al. 2002; Kish 2002; Reneman 2003). Clinical studies designed to critically evaluate the long-term effects of MDMA are hampered by a number of factors, including comorbid psychopathology and polydrug abuse among MDMA users. Animal models afford the unique opportunity to evaluate the effects of MDMA without many of these complicating factors, and the main focus here will be to review the evidence pertaining to MDMA-induced 5-HT neurotoxicity in rats.
“Interspecies scaling” and MDMA dosing regimens
A major point of controversy relates to the relevance of MDMA doses administered to rats when compared to doses taken by humans [see (Cole and Sumnall (2003)]. As noted above, MDMA regimens that produce 5-HT depletions in rats involve administration of single or multiple injections of 10–20 mg/kg, whereas the typical amount of MDMA abused by humans is one or two tablets of 80–100 mg or 1–3 mg/kg administered orally (Green et al. 2003; Schifano 2004). Based on principles of “interspecies scaling,” some investigators have proposed that neurotoxic doses of MDMA in rats correspond to recreational doses in humans (Ricaurte et al. 2000). To critically evaluate this claim, a brief discussion of interspecies scaling is warranted.
The concept of interspecies scaling is based upon shared biochemical mechanisms among eukaryotic cells (e.g., aerobic respiration), and it was initially developed to describe variations in basal metabolic rate (BMR) between animal species of different sizes [reviewed by White and Seymour (2005)]. In the 1930s, Kleiber (1932) derived what is now called the “allometric equation” to describe the relationship between body mass and BMR. The generic form of the allometric equation is: 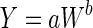 , where Y is the variable of interest, W is body weight, a is the allometric coefficient, and b is the allometric exponent. In the case where Y is BMR, b is accepted to be 0.75. West et al. (2002) have shown that most biological phenomena scale according to a universal quarter–power law, as illustrated by the space-filling fractal networks of branching tubes used by the circulatory system.
, where Y is the variable of interest, W is body weight, a is the allometric coefficient, and b is the allometric exponent. In the case where Y is BMR, b is accepted to be 0.75. West et al. (2002) have shown that most biological phenomena scale according to a universal quarter–power law, as illustrated by the space-filling fractal networks of branching tubes used by the circulatory system.
Given that the allometric equation is grounded in fundamental commonalities across organisms, it is not surprising this equation can describe the relationship between body mass and physiological variables, such as BMR, heart rate, and circulation time [e.g., Noujaim et al. (2004)]. Because circulation time and organ blood flow strongly influence drug pharmacokinetics, the allometric equation has been used in the medication development process to “scale-up” dosages from animal models to man [reviewed by Mahmood (1999)]. In general, smaller animals have faster heart rates and circulation times, leading to faster clearance of exogenous drugs. However, this relationship does not hold true for all classes of drugs, especially those that are extensively metabolized (Lin 1998).
The most important variable to consider when examining therapeutic or adverse effects of any drug is the concentration of bioactive compound reaching target tissues. Tissue drug concentrations are governed by drug absorption, distribution, metabolism, and elimination (ADME) in a complex manner. Many factors such as dose, route, species, strain, age, and gender can affect the ADME profile of a given drug, thereby affecting tissue concentrations. Campbell (1995) has described numerous limitations of interspecies scaling to predict pharmacokinetic parameters in different species. In particular, the allometric equation does not account for species-specific variability in tissue uptake of substituted amphetamines, as illustrated by the fact that brain-to-plasma ratios of the 5-HT releaser fenfluramine are 30–50 for the rat, yet <10 for humans. No studies have compared the brain tissue uptake of MDMA in various animal models. Perhaps the most problematic issue in extrapolating doses across species is the unpredictable nature of drug metabolism, which occurs chiefly in the liver [see Campbell (1996)]. Marked species differences in hepatic drug metabolism are due to variations in expression and activity of cytochrome P450 enzymes, which catalyze biotransformation reactions (Lin 1995).
MDMA is extensively metabolized in humans, as depicted in Fig. 3, and the major pathway of biotransformation involves: (1) O-demethylenation catalyzed by cytochrome P450 2D6 (CYP2D6) and (2) O-methylation catalyzed by catechol-O-methyltransferase (COMT) [reviewed by de la Torre et al. (2004)]. CYP2D6 and COMT are both polymorphic in humans, and differential expression of CYP2D6 isoforms leads to interindividual variations in the metabolism of serotonergic medications (e.g., SSRIs) (Charlier et al. 2003). Interestingly, CYP2D6 is not present in rats, which express a homologous but functionally distinct cytochrome P450 2D1 (Malpass et al. 1999; Maurer et al. 2000). A minor pathway of MDMA biotransformation in humans involves N-demethylation of MDMA to form MDA, which is subsequently O-demethylenated and O-methylated. N-demethylation of MDMA represents a more important pathway for rats when compared to humans (de la Torre and Farre 2004). The metabolism of MDMA and MDA generates a number of metabolites, some of which may be active [e.g., Escobedo et al. (2005); Forsling et al. (2002)]. Determining the potential neurotoxic properties of the various metabolites of MDMA is an important area of research [reviewed by Baumgarten and Lachenmayer (2004); Monks et al. (2004)].
Fig. 3.

Metabolism of MDMA in man. CYP2D6 Cytochrome P450 2D6, CYP3A4 cytochrome P450 3A4, COMT catechol-O-methyltransferase. This is adapted from de la Torre et al. (2004)
To complicate matters further, de la Torre et al. (2000) have shown that MDMA displays nonlinear kinetics in humans such that administration of increasing doses, or multiple doses, leads to unexpectedly high plasma levels of the drug. Enhanced plasma and tissue levels of MDMA are most likely related to auto-inhibition of MDMA metabolism, mediated via formation of a metabolite–enzyme complex that irreversibly inactivates CYP2D6 (Wu et al. 1997). Because MDMA displays nonlinear kinetics, repeated drug dosing could produce serious adverse consequences due to unusually high blood and tissue levels of the drug (Parrott 2002; Schifano 2004). The existing database of MDMA pharmacokinetic studies represents a curious situation where clinical findings are well-documented, while preclinical data are lacking. Specifically, few studies in animals have assessed the relationship between pharmacodynamic effects and pharmacokinetics of MDMA after administration of single or repeated doses [but see Chu et al. (1996)]. No studies have systematically characterized the nonlinear kinetics of MDMA in animal models. Collectively, the available data demonstrate that potential species differences in tissue drug uptake, variations in metabolic enzymes and their activities, and the phenomenon of nonlinear kinetics, preclude the use of interspecies scaling to extrapolate MDMA doses between animals and humans [reviewed by de la Torre and Farre (2004)].
The uncertainties and limitations of allometric scaling led us to investigate the method of “effect scaling” as an alternative strategy for matching equivalent doses of MDMA in rats and humans [see Winneke and Lilienthal (1992)]. In this approach, the lowest dose of drug that produces a specific pharmacological response is determined for rats and humans, and subsequent dosing regimens in rats are calculated with reference to the predetermined threshold dose. In the case of MDMA, this strategy is simplified because CNS drug effects, such as neuroendocrine and behavioral changes, have already been investigated in different species. Theoretically, equivalent drug effects in vivo should reflect similar drug concentrations reaching active sites in tissue, suggesting that the method of effect scaling can account for differences in ADME across species (at least for low drug doses). Table 3 shows the doses of MDMA that produce comparable CNS effects in rats and humans. Remarkably, the findings reveal that doses of MDMA in the range of 1–2 mg/kg produce pharmacological effects that are equivalent in both species. It is noteworthy that MDMA is typically administered to rats via the i.p. or s.c. route, whereas humans take the drug orally. Given the similar effects of MDMA in rats and humans at the same doses, it appears that drug bioavailability is comparable after i.p., s.c., or oral administration [e.g., Finnegan et al. (1988)], but verification of this hypothesis awaits further investigation.
Table 3.
Comparative neurobiological effects of (±)-MDMA administration in rats and humans
| CNS effect | Dose in rats | Dose in humans | |
|---|---|---|---|
| In vivo release of 5-HT and DA | 2.5 mg/kg, i.p. (Gudelsky and Nash 1996); 1 mg/kg, s.c. (Kankaanpaa et al. 1998) | 1.5 mg/kg p.o.a (Liechti et al. 2000; Liechti and Vollenweider 2001) | |
| Secretion of prolactin and glucocorticoids | 1–3 mg/kg, i.p. (Nash et al. 1988) | 1.67 mg/kg, p.o. (Mas et al. 1999); | |
| 1.5 mg/kg, p.o. (Harris et al. 2002) | |||
| Drug discrimination | 1.5 mg/kg, i.p. (Oberlender and Nichols 1988; Schechter 1988). | 1.5 mg/kg, p.o. (Johanson et al. 2006) | |
| Drug reinforcement | 1 mg/kg, i.v. (Wakonigg et al. 2003). | 1–2 mg/kg, p.o.b (Tancer and Johanson 2003) | |
aSubjective effects were attenuated by 5-HT uptake blockers, suggesting the involvement of transporter-mediated 5-HT release
bReinforcing effects were determined based on a multiple choice procedure
Administration of MDMA at i.p. doses of 1–3 mg/kg causes marked elevations in extracellular 5-HT and DA in rat brain, as determined by in vivo microdialysis (Baumann et al. 2005; Gudelsky and Nash 1996; Kankaanpaa et al. 1998). The data from Fig. 2 illustrate that doses of MDMA as low as 0.3 mg/kg i.v. stimulate a significant rise in extracellular 5-HT in rat nucleus accumbens. Although it is impossible to directly measure 5-HT and DA release in living human brain, clinical studies indicate that subjective effects of recreational doses of MDMA (1.5 mg/kg, p.o.) involve transporter-mediated release of 5-HT (Liechti et al. 2000; Liechti and Vollenweider 2001). Nash et al. (1988) showed that i.p. injections of 1–3 mg/kg of MDMA stimulate prolactin and corticosterone secretion in rats, and similar oral doses increase plasma prolactin and cortisol in human drug users (Harris et al. 2002; Mas et al. 1999). The dose of MDMA discriminated by rats and humans is identical: 1.5 mg/kg, i.p. for rats (Glennon and Higgs 1992; Oberlender and Nichols 1988; Schechter 1988) and 1.5 mg/kg, p.o. for humans (Johanson et al. 2006). A few studies have shown that rats will self-administer MDMA at doses ranging from 0.25–1.0 mg/kg i.v., indicating these doses possess reinforcing efficacy (Ratzenboeck et al. 2001; Schenk et al. 2003). Wakonigg et al. (2003) demonstrated that a single i.v. injection of 1 mg/kg MDMA serves a powerful reinforcer in an operant runway procedure, and MDMA displays similar reinforcing potency in Sprague–Dawley and Long–Evans rat strains. Tancer and Johanson (2003) reported that 1 and 2 mg/kg of MDMA have reinforcing properties in humans that resemble those of (+)-amphetamine. The findings summarized in Table 3 suggest that there is no scientific justification for using interspecies scaling to “adjust” MDMA doses between rats and humans.
Based on this analysis, we devised an MDMA dosing regimen in rats, which attempts to mimic binge use of MDMA in humans. Male Sprague–Dawley rats weighing 300–350 g were double-housed in plastic cages, under conditions of constant ambient temperature (22°C) and humidity (70%) in a vivarium. In our initial studies, three i.p. injections of 1.5 or 7.5 mg/kg MDMA were administered, one dose every 2 h, to yield cumulative doses of 4.5 or 22.5 mg/kg. Control rats received saline vehicle according to the same schedule. Rats were removed from their cages to receive i.p. injections but were otherwise confined to their home cages. The 1.5-mg/kg dose was used as a low “behavioral” dose, whereas the 7.5-mg/kg dose was used as a high “noxious” dose (i.e., a dose fivefold greater than threshold). Our repeated dosing regimen was designed to account for the common practice of sequential dosing (i.e., “bumping”) used by human subjects during rave parties (Parrott 2002). During the binge dosing procedure, body temperatures were measured by insertion of a thermometer probe into the rectum, and 5-HT-mediated behaviors were scored every hour. Rats were decapitated 2 weeks after dosing, brain regions were dissected, and tissue levels of 5-HT and DA were determined by HPLC–ECD as described previously (Baumann et al. 2001). Neurochemical data were normalized to percentage of saline control values for each brain region examined. Data were analyzed using a one-way ANOVA (MDMA dose), followed by Duncan’s post hoc test.
Figure 4 illustrates new data showing that our binge MDMA dosing regimen increases core body temperature in rats (F2,48=40.44, P<0.001). Specifically, repeated i.p. doses of 7.5 mg/kg MDMA elicited persistent hyperthermia on the day of treatment, whereas doses of 1.5 mg/kg did not. The 7.5-mg/kg dose caused temperature increases that were about 2°C greater than control treatment. The data in Fig. 5 demonstrate that binge MDMA treatment significantly decreases tissue 5-HT levels in the frontal cortex (F2,12=42.96, P<0.0001), striatum (F2,12=11.46, P<0.001), and olfactory tubercles (F2,12=21.27, P<0.0001) when assessed 2 weeks later. Post hoc tests revealed that high-dose MDMA produced long-term depletions of tissue 5-HT (∼50% reductions) in all three regions examined, but the low-dose group had 5-HT concentrations similar to saline controls. Transmitter depletion was selective for 5-HT neurons since tissue DA levels were unaffected. The magnitude of 5-HT depletions depicted in Fig. 5 is similar to that observed by others (Battaglia et al. 1987; Schmidt 1987; Stone et al. 1987). Our findings demonstrate that repeated treatment with behaviorally relevant doses of MDMA does not cause acute hyperthermia or long-term 5-HT depletions. In contrast, repeated administration of MDMA at a dose that is fivefold higher than the behavioral dose causes both of these adverse effects. The data are consistent with those of O’Shea et al. (1998), who reported that high-dose MDMA (10 or 15 mg/kg, i.p.) but not low-dose MDMA (4 mg/kg, i.p.) causes acute hyperthermia and long-term 5-HT depletion in Dark Agouti rats.
Fig. 4.

Acute effects of (±)-MDMA on core body temperature in rats. Male rats received three i.p. injections of 1.5 or 7.5 mg/kg MDMA, one dose every 2 h (i.e., injections at 0, 2, and 4 h). Saline was administered on the same schedule. Core temperature was recorded via insertion of a rectal thermometer probe every 2 h. Data are mean±SEM for N=5 rats/group. Asterisk denotes significance with respect to saline-injected control at each time point (P<0.05 Duncan’s)
Fig. 5.

Long-term effects of (±)-MDMA on tissue levels of 5-HT (top panel) and DA (bottom panel) in brain regions. Male rats received three i.p. injections of 1.5 or 7.5 mg/kg MDMA, one dose every 2 h. Saline was administered on the same schedule. Rats were killed 2 weeks after injections; brain regions were dissected, and tissue 5-HT and DA were assayed by HPLC–ECD (Baumann et al. 2001). Data are mean±SEM expressed as the percentage of saline-treated control values for each region, N=5 rats/group. Control values of 5-HT and DA were 557±24 and 28±4 pg/mg tissue for frontal cortex (CTX), 429±36 and 10,755±780 pg/mg tissue for striatum (STR), and 1,174±114 and 4,545±426 pg/mg tissue for olfactory tubercle (OT). Asterisk denotes significance compared to saline-injected control for each region (P<0.05 Duncan’s)
Effects of MDMA on established markers of neurotoxicity
Hallmark indicators of neurotoxicity include cell death, silver positive staining (i.e., argyrophilia), and glial cell hypertrophy (Baumgarten and Lachenmayer 2004; O’Callaghan and Sriram 2005; Switzer 2000). Despite the abundance of evidence showing that MDMA causes 5-HT depletions, few studies have examined the effects of MDMA on established markers of neurotoxic damage. Indeed, some investigators have argued that based on 5-HT abnormalities alone, one cannot infer the presence of neurotoxicity (O’Callaghan and Miller 1993; Wang et al. 2004). Support for the hypothesis of MDMA-induced axotomy relies heavily upon immunohistochemical analysis of 5-HT levels, and the use of semiquantitative methods could produce misleading results if not validated by other means. For example, the MDMA-induced disappearance of immunoreactive 5-HT in specific brain regions could reflect depletion of transmitter to a low level (i.e., below the level of detection) in intact axons and nerve terminals. Thus, an alternative hypothesis consistent with available data is that MDMA-induced deficits in 5-HT systems are due to persistent adaptive changes in gene expression or protein function, and these changes reflect a state of metabolic quiescence or exhaustion, rather than neurotoxic damage. Table 4 summarizes the findings from studies that have examined the effects of MDMA on established indicators of neurotoxic damage. All of the studies employed large doses of MDMA, either as single or repeated injections, which cause hyperthermia and its associated complications.
Table 4.
Effects of (±)-MDMA administration on markers of neuronal degeneration in rats
| CNS marker | Dosing regimen | Survival interval | Reference | |
|---|---|---|---|---|
| Increased silver-positive staining in degenerating neurons | 80 mg/kg, s.c., twice daily, 4 days | 15–48 h | Commins et al. (1987) | |
| 25–150 mg/kg, s.c., twice daily, 2 days | 2 days | Jensen et al. (1993) | ||
| Increased Fluoro-Jade B staining of degenerating neurons | 20–40 mg/kg, i.p., single dose | 2 days | Schmued (2003) | |
| Reactive astrogliosis as measured by elevations in GFAP | 75–150 mg/kg, s.c., twice daily, 2 days | 2 days | O’Callaghan and Miller (1993)a | |
| 20 mg/kg, i.p., single dose | 1 week | Aguirre et al. (1999) | ||
| 20 mg/kg, s.c., twice daily, 4 days | 3 days, 1 week | Pubill et al. (2003)a | ||
| 7.5 mg/kg, i.p., three doses | 2 weeks | Wang et al. (2004)a | ||
aThese investigators found no effect of MDMA on GFAP at doses which significantly depleted 5-HT levels in brain tissue
Silver staining methods are commonly utilized to identify degenerating neuronal elements in the CNS [reviewed by Switzer (2000)]. In particular, silver stains are sensitive indicators of damage caused by neurotoxic chemicals. Commins et al. (1987) examined the neurotoxic potential of MDMA by using the Fink–Heimer procedure to detect argyrophilic neuronal structures. In their study, male Sprague–Dawley rats received single or multiple s.c. injections of 80 mg/kg MDMA and were killed 15 to 48 h later. Single doses of MDMA increased silver-positive staining only in the frontoparietal cortex. Multiple doses caused more extensive damage; degenerating nerve terminals were found in the striatum, while degenerating terminals, axons, and cell bodies were observed throughout layers III and IV of the parietal cortex. In a comparable study, Jensen et al. (1993) used the de Olmos cupric–silver procedure to delineate areas of the brain damaged by MDMA. Male Long–Evans rats received twice daily s.c. injections of 25–150 mg/kg MDMA for 2 days and were sacrificed 48 h thereafter. In this case, MDMA produced dose-dependent increases in silver-positive staining in the parietal cortex, with some irregular staining in the striatum and thalamus at higher doses. Most of the staining in the cortex was associated with degenerating axons and terminals, but cell bodies were also stained. The collective results show that administration of MDMA at sufficient doses (i.e., >25 mg/kg) can cause neuronal damage, as indicated by sensitive silver staining methods.
There are several key factors to consider when interpreting the effects of MDMA on silver staining in rat brain. Most importantly, both of the cited studies employed massive cumulative doses of MDMA ranging from 80 to 600 mg/kg. Based on the concept of effect scaling described previously (see Table 3), the single injections of MDMA administered by Commins et al. and Jensen et al. are 16–100 times higher than a behaviorally relevant dose of 1.5 mg/kg. At such extraordinary doses of MDMA, excessive sympathetic activation and hyperthermia could cause physiological dysregulation sufficient to elicit nonspecific neuropathy. The idea that high-dose MDMA causes nonspecific neuronal damage is supported by the observation that increases in silver staining are not confined to 5-HT cells. Both studies noted the presence of argyrophilic cell bodies in the cortex of MDMA-treated rats, yet 5-HT cell bodies are not present in the cortex (Steinbusch 1981). Furthermore, the pattern of MDMA-induced silver staining, which is largely confined to the frontoparietal cortex, does not correspond to the pattern of 5-HT innervation or the pattern of 5-HT depletions. Given these findings, it seems feasible that increases in silver-positive staining produced by high-dose MDMA do not reflect 5-HT neurotoxicity per se.
Schmued (2003) used a novel histological stain, Fluoro-Jade B, to examine neuronal damage produced by MDMA administration in rats. Fluoro-Jade B is an anionic fluorescein derivative that selectively stains degenerating terminals, axons, and cell bodies (Schmued and Hopkins 2000). Male Sprague–Dawley rats received single i.p. injections of 10–40 mg/kg MDMA and were killed 48 h later. After 10 mg/kg MDMA, only one of four rats exhibited Fluoro-Jade staining in the cortex, and this rat experienced significant hyperthermia at the time of treatment. At higher doses of MDMA, most rats displayed Fluoro-Jade staining in a variety of brain areas. For example, degenerating pyramidal and nonpyramidal cells were stained in the parietal cortex, while degenerating multipolar cells were stained in the ventral thalamus. The findings with Fluoro-Jade B indicate that sufficient doses of MDMA (i.e., >20 mg/kg) can produce neuronal degeneration. However, analogous to the findings with silver staining methods, many of the damaged cells identified by Fluoro-Jade B are nonserotonergic, and the pattern of staining does not overlap with the pattern of 5-HT deficits. Additionally, neuronal degeneration was only found in rats that experienced hyperthermia of >41°C, again suggesting an important link between elevated body temperature and adverse effects.
A universal reaction to damage in the CNS is hypertrophy of astrocytes, or “reactive gliosis” [reviewed by O’Callaghan and Sriram (2005)]. This response is accompanied by enhanced expression of glial-specific structural proteins such as, glial fibrillary acidic protein (GFAP). O’Callaghan et al. (1995) have verified that a wide range of neurotoxic chemicals increase the levels of GFAP in rodent CNS, indicating this protein can be used as a sensitive marker of neuronal damage. These investigators (O’Callaghan and Miller 1993) carried out extensive studies to evaluate the effects of MDMA administration on GFAP expression in rat brain. Male Long–Evans rats were treated twice daily with s.c. injections of 10–30 mg/kg MDMA for 7 days and were sacrificed 48 h after the last dose. This high-dose regimen of MDMA did not alter the expression of GFAP, even though 5-HT levels were markedly reduced in the cortex, hippocampus, and striatum. A separate group of rats received twice daily s.c. injections of 75–150 mg/kg MDMA for 2 days, and these rats were killed 48 h later. In response to these massive doses of MDMA, levels of GFAP were significantly elevated in various brain regions but these increases did not correlate with the degree of 5-HT depletions. Finally, the effects of MDMA were compared to the effects of the 5-HT neurotoxin, 5,7-dihydroxytryptamine (5,7-DHT). At doses of MDMA and 5,7-DHT which caused comparable 5-HT depletions, only 5,7-DHT increased GFAP.
In a more recent study, Pubill et al. (2003) compared the effects of MDMA and methamphetamine on SERT binding and a number of glial-specific markers. Male Sprague–Dawley rats received twice daily s.c. injections of 20 mg/kg MDMA for 4 days, and were killed at 3 and 7 days posttreatment. MDMA caused marked decreases in [3H]-paroxetine-labeled SERT binding but had no effect on any measures of reactive gliosis. In particular, MDMA treatment failed to induce astroglial activation as indicated by the lack of changes in immunoreactive GFAP. MDMA had no effect on microglial markers such as peripheral benzodiazepine receptors and OX-6 protein. Interestingly, methamphetamine produced significant increases in astroglial and microglial markers in this study, indicating an important distinction between methamphetamine and MDMA. Consistent with the findings of Pubill and colleagues, Bai et al. (2001) found that 10 mg/kg s.c. MDA failed to increase GFAP expression in Sprague–Dawley rats. In contrast, Aguirre et al. (1999) reported that a single i.p. dose of 20 mg/kg MDMA enhanced histochemical staining for GFAP in the hippocampus of Wistar rats. The reasons for the discrepancies between the findings of Aguirre et al. vs the findings of others are not readily apparent, but could be due to differences in experimental methods across studies (i.e., different rat strains, different brain regions examined, etc).
The majority of data indicate that doses of MDMA causing significant 5-HT depletions (i.e., single or repeated doses of 10–20 mg/kg) do not reliably induce silver staining or glial activation. Recent results from our laboratory support this assertion (Rothman et al. 2003; Wang et al. 2004). We examined the possible neurotoxic effects of MDMA and two other substituted amphetamines, (+)-fenfluramine and p-chloroamphetamine (PCA). Specifically, the effects of drug treatments on the expression of SERT and GFAP were evaluated using Western blot methods. In the first study, male Sprague–Dawley rats received four i.p. injections of 6 mg/kg (+)-fenfluramine or one i.p. injection of 5 mg/kg PCA, and rats were killed 2 days or 2 weeks thereafter. (+)-Fenfluramine and PCA decreased cortical 5-HT levels (∼50–60%) but neither drug caused long-term changes in the expression of SERT or GFAP (Rothman et al. 2003). Similar results were found when three i.p. injections of 7.5 mg/kg MDMA were administered to rats according to the binge dosing regimen described above (see Figs. 4 and 5). MDMA did not alter levels of immunoreactive SERT or GFAP when examined 2 weeks after treatment, even though the same rats displayed substantial depletions of 5-HT in the cortex, striatum, and hippocampus (Wang et al. 2004). Thus, doses of MDMA that deplete tissue 5-HT do not alter expression of SERT or GFAP. The SERT data are especially surprising given that MDMA administration is known to reduce the number of SERT binding sites, thereby decreasing [3H]5-HT uptake capability (Battaglia et al. 1987; Commins et al. 1987; Schmidt 1987).
To explore this phenomenon further, we compared the effects of MDMA and 5,7-DHT on the expression of SERT, GFAP, and other markers of gliosis (Wang et al. 2005). In all cases, proteins were measured by Western blot analysis using sensitive polyclonal antibodies directed against the protein of interest. Sprague–Dawley rats received three i.p. injections of 7.5 mg/kg MDMA or saline vehicle, and were killed at 3 days and 2 weeks later. A separate group of rats received single intracerebroventricular (i.c.v.) infusions of 150 μg 5,7-DHT or 0.1% ascorbic acid vehicle. Both MDMA and 5,7-DHT caused 50–70% depletions of 5-HT in the frontal cortex at 3 days and 2 weeks after treatments. Figure 6 depicts previously published data from our laboratory showing that MDMA does not affect tissue levels of SERT at 2 weeks posttreatment, whereas 5,7-DHT causes significant decreases in this protein (Wang et al. 2005). The data in Fig. 7 reveal that binge MDMA treatment does not affect GFAP, while 5,7-DHT increases levels of this protein. It should be mentioned that treatment with 5,7-DHT, but not MDMA, produces persistent increases in another astrocyte marker, heat shock protein 32. On-going studies in our laboratory indicate that MDMA does not alter the intracellular trafficking of SERT proteins from endosomes to the plasma membrane (Wang et al. 2005). Rather, it seems that MDMA renders SERT proteins nonfunctional, though the mechanism responsible for this effect is not known. Our findings provide evidence that 5,7-DHT decreases SERT and increases GFAP, whereas MDMA does not alter expression of either protein. Accordingly, at the doses tested in these studies, 5,7-DHT displays the profile of a 5-HT neurotoxin, whereas MDMA does not.
Fig. 6.

Comparative effects of (±)-MDMA (top panel) and 5,7-DHT (bottom panel) on SERT protein expression in dissected brain regions. One group of rats received three i.p. injections of saline or 7.5 mg/kg MDMA, one dose every 2 h. Another group received single i.c.v. infusions of 5,7-DHT or vehicle. Rats were killed 2 weeks later and brain regions were dissected. Western blot analysis of SERT immunoreactivity in the frontal cortex (CTX), striatum (STR), and hippocampus (HIPP) was carried out as described (Wang et al. 2005). Blots were digitized and quantified using densitometry (NIH IMAGE software). Changes in immunoreactivity are expressed relative to their corresponding control (defined as 100% value). Each value is the mean±SEM for N=4 rats/group. Asteriskdenotes significance with respect to control in each region (P<0.01 Student’s t test). Data taken from Wang et al. 2005
Fig. 7.

Comparative effects of (±)-MDMA (top panel) and 5,7-DHT (bottom panel) on GFAP expression in dissected brain regions. One group of rats received three i.p. injections of saline or 7.5 mg/kg MDMA, one dose every 2 h. Another group received single i.c.v. infusions of 5,7-DHT or vehicle. Rats were killed 2 weeks later and brain regions were dissected. Western blot analysis of GFAP immunoreactivity in the frontal cortex (CTX), striatum (STR), and hippocampus (HIPP) was carried out as described (Wang et al. 2005). Blots were digitized and quantified using densitometry (NIH IMAGE software). Changes in immunoreactivity are expressed relative to their corresponding control (defined as 100% value). Each value is the mean±SEM for N=4 rats/group. Asterisk denotes significance with respect to control in each region (P<0.01 Student’s t test). Data were taken from Wang et al. 2005
Functional consequences of MDMA-induced 5-HT depletion
Any operational definition of “neurotoxicity” must include the precept that functional impairments accompany neuronal damage (Moser 2000; Winneke and Lilienthal 1992). As noted previously, high-dose MDMA causes persistent inactivation of tryptophan hydroxylase, which leads to inhibition of 5-HT synthesis and loss of 5-HT (O’Hearn et al. 1988; Stone et al. 1987). Moreover, MDMA-induced reduction in the density of SERT binding sites leads to decreased capacity for 5-HT uptake in nervous tissue (Battaglia et al. 1987; Schmidt 1987). Regardless of whether these deficits reflect neurotoxic damage or long-term adaptation, such changes would be expected to have discernible in vivo correlates. Many investigators have examined functional consequences of high-dose MDMA administration, and a comprehensive review of this subject is beyond the scope of the present paper [reviewed by Cole and Sumnall 2003; Green et al. (2003)]. The following discussion will consider long-term effects of MDMA (i.e., >1 week) on in vivo indicators of 5-HT function in rats, as measured by electrophysiological recording, microdialysis sampling, neuroendocrine secretion, and specific aspects of behavior. A number of key findings are summarized in Table 5. In general, few published studies have been able to relate the magnitude of MDMA-induced 5-HT depletion to the degree of specific functional impairment. Furthermore, MDMA administration rarely causes persistent changes in baseline measures of neural function, and deficits are most readily demonstrated by provocation of the 5-HT system by pharmacological (e.g., drug challenge) or physiological means (e.g., environmental stress).
Table 5.
Effects of (±)-MDMA administration on functional indices of 5-HT transmission in rats
| CNS effect | Dosing regimen | Survival interval | Reference |
|---|---|---|---|
| No change in 5-HT cell firing | 20 mg/kg, s.c., twice daily, 4 days | 2 weeks | Gartside et al. (1996) |
| Reductions in evoked 5-HT release in vivo | 20 mg/kg, s.c., twice daily, 4 days | 2 weeks | Series et al. (1994) |
| 10 mg/kg, i.p., twice daily, 4 days | 1 week | Shankaran and Gudelsky (1999) | |
| Changes in corticosterone and prolactin secretion | 20 mg/kg, s.c., single dose | 2 weeks | Poland et al. (1997) |
| 20 mg/kg, s.c. twice daily, 4 days; | 4, 8, and 12 months | ||
| Increased anxiety-like behaviors | 5 mg/kg, s.c., 1 or four injections, 2 days | 3 months | McGregor et al. (2003)a and Morley et al. (2001) |
| 7.5 mg/kg, sc, twice daily, 3 days | 2 weeks | Fone et al. (2002)a |
aThese investigators noted marked increases in anxiogenic behaviors in the absence of significant MDMA-induced 5-HT depletion in brain
5-HT projections innervating the rat forebrain have cell bodies residing in the raphe nuclei (Steinbusch 1981). These neurons exhibit pacemaker-like electrical activity, which can be recorded using electrophysiological techniques (Aghajanian et al. 1978; Sprouse et al. 1989). Gartside et al. (1996) used extracellular recording methods to examine 5-HT cell firing in the dorsal raphe of rats previously treated with MDMA. Rats received two daily injections of 20 mg/kg, s.c. MDMA for 4 days and were tested under chloral hydrate anesthesia 2 weeks later. MDMA pretreatment had no effect on the number of classical or burst-firing 5-HT cells encountered during recording. Additionally, 5-HT cell firing rates and action potential characteristics were not different between MDMA- and saline-pretreated groups. These data show that 5-HT neurons and their firing properties are not altered after MDMA administration, and this agrees with immunohistochemical evidence demonstrating that MDMA does not destroy 5-HT perikarya. The electrophysiological data from MDMA-pretreated rats differ from the findings reported with 5,7-DHT. In rats treated with i.c.v. 5,7-DHT, the number of classical and burst-firing 5-HT neurons is dramatically decreased in the dorsal raphe, in conjunction with a loss of 5-HT fluorescence (Aghajanian et al. 1978; Hajos and Sharp 1996). Thus, 5,7-DHT produces reductions in 5-HT cell firing that are attributable to cell death, but MDMA does not.
In vivo microdialysis allows continuous sampling of extracellular fluid from intact brain, and this method has been used to evaluate persistent neurochemical consequences of MDMA exposure (Gartside et al. 1996; Matuszewich et al. 2002; Series et al. 1994; Shankaran and Gudelsky 1999). Series et al. 1994 carried out microdialysis in rat frontal cortex to examine long-term effects of MDMA administration. Male Sprague–Dawley rats received twice daily s.c. injections of 20 mg/kg MDMA for 4 days, and were tested under chloral hydrate anesthesia 2 weeks later. Prior MDMA exposure did not affect baseline extracellular levels of 5-HT, but levels of the 5-HT metabolite, 5-hydroxyindoleacetic acid (5-HIAA), were decreased to ∼30% of control. Moreover, the ability of (+)-fenfluramine (10 mg/kg, i.p.) to evoke 5-HT release was markedly blunted in MDMA-pretreated rats when compared to saline-pretreated controls. In an analogous investigation, Shankaran and Gudelsky (1999) assessed neurochemical effects of acute MDMA challenge in rats that had previously received four i.p. injections of 10 mg/kg MDMA. In vivo microdialysis was performed in the striatum of conscious rats 1 week after high-dose MDMA treatment. Baseline levels of dialysate 5-HT were not altered by prior MDMA exposure, even though tissue levels of 5-HT in striatum were depleted by 50%. The ability of MDMA to evoke 5-HT release was severely impaired in MDMA-pretreated rats, while the concurrent DA response was normal. In this same study, effects of MDMA on body temperature and 5-HT syndrome were attenuated in MDMA-pretreated rats, suggesting the development of tolerance. Other investigations using in vivo microdialysis methods have shown that 5-HT release in response to physiological or stressful stimuli is impaired in rats pretreated with high-dose MDMA (Gartside et al. 1996; Matuszewich et al. 2002).
Taken together, the microdialysis studies reveal important long-term consequences of MDMA administration: (1) baseline levels of dialysate 5-HT are not altered despite depletions of tissue transmitter, (2) baseline levels of dialysate 5-HIAA are consistently decreased, and (3) stimulated release of 5-HT is blunted in response to pharmacological or physiological provocation. The microdialysis findings with MDMA resemble those obtained from rats treated with 5,7-DHT, in which lesioned rats display normal baseline dialysate 5-HT but decreased 5-HIAA (Hall et al. 1999; Kirby et al. 1995; Romero et al. 1998). In a representative study, Kirby et al. (1995) used microdialysis in rat striatum to evaluate the long-term neurochemical effects of i.c.v. 5,7-DHT; these investigators showed that reductions in dialysate 5-HIAA and impairments in stimulated 5-HT release are highly correlated with loss of tissue 5-HT, whereas baseline dialysate 5-HT is not. In fact, depletions of brain tissue 5-HT up to 90% did not affect baseline levels of dialysate 5-HT (Kirby et al. 1995). It seems that adaptive mechanisms serve to maintain normal concentrations of synaptic 5-HT, even under conditions of severe transmitter depletion. A comparable situation exists after lesions of the nigrostriatal DA system, where baseline levels of extracellular DA are maintained in the physiological range despite substantial loss of tissue DA [see Zigmond et al. (1990)]. In the case of high-dose MDMA treatment, it seems feasible that reductions in 5-HT uptake (e.g., less functional SERT protein) and metabolism (e.g., decreased monoamine oxidase activity) compensate for 5-HT depletions to keep optimal concentrations of 5-HT bathing nerve cells. On the other hand, deficits in 5-HT release are readily demonstrated in MDMA-pretreated rats when 5-HT systems are taxed by drug challenge or environmental stressors.
5-HT neurons projecting to the hypothalamus provide stimulatory input for the secretion of adrenocorticotropin (ACTH) and prolactin from the anterior pituitary (Van de Kar 1991). Accordingly, 5-HT releasers (e.g., fenfluramine) and 5-HT receptor agonists increase plasma levels of these hormones in rats and humans [reviewed by Levy et al. (1994)]. Neuroendocrine challenge experiments have been used to demonstrate changes in serotonergic responsiveness in rats treated with MDMA (Poland 1990; Poland et al. 1997; Series et al. 1995). In the most comprehensive study, Poland et al. (1997) examined effects of high-dose MDMA on hormone responses elicited by acute fenfluramine challenge. Male Sprague–Dawley rats received single s.c. injections of 20 mg/kg MDMA and were tested 2 weeks later. Prior MDMA exposure did not alter baseline levels of circulating ACTH or prolactin. However, in MDMA-pretreated rats, fenfluramine-induced ACTH secretion was reduced, while prolactin secretion was enhanced. The MDMA dosing regimen caused significant depletions of tissue 5-HT in various brain regions, including hypothalamus. In a follow-up time-course study, rats received twice daily s.c. injections of 20 mg/kg MDMA for 4 days, and were challenged with fenfluramine (6 mg/kg, s.c.) at 4, 8, and 12 months thereafter. As observed in the single dose MDMA study, rats exposed to multiple MDMA doses displayed blunted ACTH responses and augmented prolactin responses to fenfluramine. Interestingly, the impaired ACTH response persisted for 12 months in MDMA-pretreated rats, even though tissue levels of 5-HT were not depleted at this time point. The data show that high-dose MDMA can cause functional abnormalities for up to 1 year, and changes in 5-HT responsiveness do not necessarily parallel the extent of recovery from 5-HT depletions in brain.
In our laboratory, we wished to further explore the long-term neuroendocrine consequences of MDMA administration. Utilizing the binge MDMA dosing regimen described previously (see Figs. 4 and 5), male Sprague–Dawley rats received three i.p. injections of 1.5 or 7.5 mg/kg MDMA, one dose every 2 h. Control rats received saline vehicle according to the same schedule. One week after MDMA treatment, rats were fitted with indwelling jugular catheters under pentobarbital anesthesia. After 1 week of recovery from surgery (i.e., 2 weeks after MDMA or saline), rats were brought into the testing room and i.v. catheters were connected to extension tubes. Each rat received an i.v. injection of 1 mg/kg MDMA at time zero, followed by an injection of 3 mg/kg MDMA 60 min later. Blood samples were withdrawn via the catheters immediately before and 30 min after each dose of MDMA. Plasma levels of corticosterone and prolactin were measured by radioimmunoassay methods (Baumann et al. 1998). Hormone data were normalized to percentage of preinjection control and analyzed using a two-way ANOVA (pretreatment × acute treatment), followed by Duncan’s post hoc test. Figure 8 shows new data from our laboratory demonstrating the effects of binge MDMA pretreatment on hormone responses examined 2 weeks later. MDMA pretreatment did not alter baseline levels of either hormone. Acute administration of MDMA elicited dose-related elevations in circulating corticosterone (F2,63=62.03, P<0.0001) and prolactin (F2,63=45.41, P<0.0001) as shown by others (Nash et al. 1988). Importantly, MDMA pretreatment significantly attenuated MDMA-induced corticosterone (F2,63=6.89, P<0.01) and prolactin (F2,63=8.30, P<0.001) responses. Post hoc tests revealed that rats exposed to high-dose MDMA pretreatment displayed reductions in corticosterone and prolactin secretion in response to acute MDMA challenge, whereas hormone responses in the low-dose MDMA rats were indistinguishable from controls.
Fig. 8.

Effects of (±)-MDMA pretreatment on secretion of corticosterone (top panel) and prolactin (bottom panel) evoked by acute (±)-MDMA challenge. Male rats received three i.p. injections of 1.5 or 7.5 mg/kg MDMA, one dose every 2 h. Saline was administered on the same schedule. Two weeks later, each rat received a single i.v. injection of 1 mg/kg MDMA at time zero, followed by a single injection of 3 mg/kg MDMA 60 min later. Blood samples were drawn via indwelling catheters immediately before injections and 30 min after each injection; plasma corticosterone and prolactin were measured by RIA (Baumann et al. 1998). Data are mean±SEM expressed as the percentage of baseline hormone levels for N=8 rats/group. Baseline corticosterone and prolactin levels were 73±18 and 2.4±0.6 ng/ml of plasma, respectively. Asterisk denotes significance compared to saline-pretreated control group (P<0.05 Duncan’s)
Our neuroendocrine results are consistent with the development of tolerance to hormonal effects of MDMA. These findings do not agree completely with the data of Poland et al. (1997) discussed above; however, our findings do agree with previous data showing blunted hormonal responses to fenfluramine in rats with fenfluramine-induced 5-HT depletions (Baumann et al. 1998). Perhaps more importantly, the data shown in Fig. 8 are strikingly similar to clinical findings in which cortisol and prolactin responses to acute (+)-fenfluramine administration are reduced in human MDMA users (Gerra et al. 1998, 2000; Gouzoulis-Mayfrank et al. 2002). Indeed, Gerra et al. (2000) reported that (+)-fenfluramine-induced prolactin secretion is blunted in abstinent MDMA users for up to 1 year after cessation of drug use. The mechanism(s) underlying reduced sensitivity to (+)-fenfluramine and MDMA are not known, but it is tempting to speculate that MDMA-induced impairments in evoked 5-HT release are involved, as shown by in vivo microdialysis studies. While some investigators have cited neuroendocrine changes in human MDMA users as evidence for 5-HT neurotoxicity, Gouzoulis-Mayfrank et al. (2002) provide a compelling argument that endocrine abnormalities in MDMA users could be related to cannabis use rather than MDMA. Further experiments will be required to resolve the precise nature of neuroendocrine changes in MDMA users.
One of the more serious and disturbing clinical findings is that MDMA causes persistent cognitive deficits in some users (Morgan 2000; Reneman 2003). Numerous research groups have examined the effects of MDMA treatment on learning and memory processes in rats, yet most studies failed to identify persistent impairments, even when extensive 5-HT depletions were present (Byrne et al. 2000; McNamara et al. 1995; Ricaurte et al. 1993; Robinson et al. 1993; Seiden et al. 1993; Slikker et al. 1989). While an exhaustive review of this literature is not possible here, representative findings will be mentioned. In an extensive series of experiments, Seiden et al. (1993) evaluated the effects of high-dose MDMA on a battery of tests including open-field behavior, schedule-controlled behavior, one-way avoidance, discriminated two-way avoidance, forced swim, and radial maze performance. Male Sprague–Dawley rats received twice daily s.c. injections of 10–40 mg/kg MDMA for 4 days and were tested beginning 2 weeks after treatment. Despite large depletions of brain tissue 5-HT, MDMA-pretreated rats exhibited normal behaviors in all paradigms. Likewise, Robinson et al. (1993) found that MDMA-induced depletion of cortical 5-HT up to 70% did not alter spatial navigation, skilled forelimb use, or foraging behavior in rats. On the other hand, Marston et al. (1999) reported that MDMA administration produces persistent deficits in a delayed nonmatch to performance (DNMTP) procedure when long delay intervals are employed (i.e., 30 s). Specifically, saline-pretreated rats exhibited progressive improvement in task performance over successive days of testing, whereas MDMA-pretreated rats did not. The authors theorized that delay-dependent impairments in the DNMTP procedure reflect MDMA-induced deficits in short-term memory, possibly attributable to 5-HT depletion.
With the exception of the findings of Marston et al., the collective behavioral data in rats indicate that MDMA-induced depletions of brain 5-HT have little effect on cognitive processes. There are several potential explanations for this apparent paradox. First, high-dose MDMA administration produces only partial depletion of 5-HT in the range of 40–60% in most brain areas. This level of 5-HT loss may not be sufficient to elicit behavioral alterations, as compensatory adaptations in 5-HT neurons could maintain normal physiological function. Second, MDMA appears to selectively affect fine diameter fibers arising from the dorsal raphe, and it is possible that these 5-HT circuits may not subserve the behaviors being monitored. Third, the behavioral tests utilized in rat studies might not be sensitive enough to detect subtle changes in learning and memory processes. Finally, the functional reserve capacity in the CNS might be sufficient to compensate for even large depletions of a single transmitter.
While MDMA appears to have few long-term effects on cognition in rats, a growing body of evidence demonstrates that MDMA administration can cause persistent anxiety-like behaviors in this species (Fone et al. 2002; Gurtman et al. 2002; Morley et al. 2001). Morley et al. (2001) first reported that MDMA exposure induces long-term anxiety in rats. These investigators gave male Wistar rats one or four i.p. injections of 5 mg/kg MDMA for two consecutive days. Subjects were tested 3 months later in a battery of anxiety-related paradigms, including elevated plus maze, emergence, and social interaction tests. Rats receiving either single or multiple MDMA injections displayed marked increases in anxiogenic behaviors in all three tests when compared to control rats. Because no 5-HT endpoints were examined, it was impossible to relate changes in behavior to changes in 5-HT transmission. In a follow-up study, Gurtman et al. (2002) replicated the original findings of Morley et al. using rats pretreated with four i.p. injections of 5 mg/kg MDMA for 2 days; persistent anxiogenic effects of MDMA were associated with depletions of 5-HT in the amygdala, hippocampus, and striatum. Interestingly, Fone et al. (2002) showed that twice daily injections of 7.5 mg/kg MDMA for 3 days caused impairments in social interaction in adolescent Lister rats, even in the absence of 5-HT depletions or reductions in [3H]-paroxetine-labeled SERT binding sites. These data suggested the possibility that MDMA-induced anxiety does not require 5-HT deficits.
In an attempt to determine potential mechanisms underlying MDMA-induced anxiety, McGregor et al. (2003) evaluated effects of the drug on anxiety-related behaviors and a number of postmortem parameters including autoradiography for SERT and 5-HT receptor subtypes. Rats received moderate (5 mg/kg, i.p., 2 days) or high (5 mg/kg, i.p., four injections, 2 days) doses of MDMA, and tests were conducted 10 weeks later. This study confirmed that moderate doses of MDMA could cause protracted increases in anxiety-like behaviors without significant 5-HT depletions. Furthermore, the autoradiographic analysis revealed that anxiogenic effects of MDMA may involve long-term reductions in 5-HT2A/2C receptors, rather than reductions in SERT binding. Additional work by Bull et al. (2003, 2004) suggests that decreases in the sensitivity of 5-HT2A receptors, but not 5-HT2C receptors, could underlie MDMA-associated anxiety. Clearly, more investigation into this important area of research is warranted.
Summary
The findings reviewed in this paper allow a number of conclusions to be drawn with regard to MDMA-induced neurotoxicity. First, there is little doubt that MDMA targets monoamine transporters, and transporter-mediated release of 5-HT, DA, and NE underlies pharmacological effects of the drug. While MDMA has been considered a predominately serotonergic agent, certain adverse effects including cardiovascular stimulation and hyperthermia likely involve NE and DA mechanisms, respectively. There seems to be no scientific rationale for using allometric scaling to adjust doses of MDMA between rats and humans because the pharmacologically relevant doses are similar in both species (e.g., 1–2 mg/kg). Nonetheless, the complex metabolism of MDMA needs to be examined in various animal species to permit comparison with clinical literature and to validate appropriate preclinical models. With regard to MDMA-induced neurotoxicity, it seems that 5-HT deficits are not always synonymous with axonal death because doses of MDMA which cause marked depletions of brain tissue 5-HT in rats (e.g., 10–20 mg/kg) are not associated with silver-positive staining, reactive gliosis, or loss of SERT protein. Like many other psychotropic drugs, MDMA is capable of producing bona fide neurotoxicity at sufficient doses (e.g., >25 mg/kg), and damage is not confined to 5-HT neurons. Many aspects of 5-HT function appear to be normal in MDMA-pretreated rats despite significant loss of brain 5-HT, perhaps illustrating the profound adaptive capability of the CNS. On the other hand, MDMA-induced 5-HT depletions are accompanied by impairments in evoked 5-HT release and neuroendocrine secretion that suggest tolerance development. The clinical relevance of preclinical findings is often uncertain, but the fact that MDMA can produce persistent increases in anxiety-like behaviors without measurable 5-HT deficits suggests that even moderate doses may pose risks.
Acknowledgements
This research was generously supported by the NIDA IRP. The authors are indebted to John Partilla, Chris Dersch, Mario Ayestas, Robert Clark, Fred Franken, and John Rutter for their expert technical assistance during these studies.
References
- Aghajanian GK, Wang RY, Baraban J. Serotonergic and non-serotonergic neurons of the dorsal raphe: reciprocal changes in firing induced by peripheral nerve stimulation. Brain Res. 1978;153:169–175. doi: 10.1016/0006-8993(78)91140-x. [DOI] [PubMed] [Google Scholar]
- Aguirre N, Barrionuevo M, Ramirez MJ, Del Rio J, Lasheras B. Alpha-lipoic acid prevents 3,4-methylenedioxy-methamphetamine (MDMA)-induced neurotoxicity. Neuroreport. 1999;10:3675–3680. doi: 10.1097/00001756-199911260-00039. [DOI] [PubMed] [Google Scholar]
- Bai F, Jones DC, Lau SS, Monks TJ. Serotonergic neurotoxicity of 3,4-(+/−)-methylenedioxyamphetamine and 3,4-(+/−)-methylendioxymethamphetamine (ecstasy) is potentiated by inhibition of gamma-glutamyl transpeptidase. Chem Res Toxicol. 2001;14:863–870. doi: 10.1021/tx010011l. [DOI] [PubMed] [Google Scholar]
- Banken JA. Drug abuse trends among youth in the United States. Ann N Y Acad Sci. 2004;1025:465–471. doi: 10.1196/annals.1316.057. [DOI] [PubMed] [Google Scholar]
- Bankson MG, Cunningham KA. 3,4-Methylenedioxymethamphetamine (MDMA) as a unique model of serotonin receptor function and serotonin–dopamine interactions. J Pharmacol Exp Ther. 2001;297:846–852. [PubMed] [Google Scholar]
- Battaglia G, De Souza EB. Pharmacologic profile of amphetamine derivatives at various brain recognition sites: selective effects on serotonergic systems. NIDA Res Monogr. 1989;94:240–258. [PubMed] [Google Scholar]
- Battaglia G, Yeh SY, O’Hearn E, Molliver ME, Kuhar MJ, De Souza EB. 3,4-Methylenedioxymethamphetamine and 3,4-methylenedioxyamphetamine destroy serotonin terminals in rat brain: quantification of neurodegeneration by measurement of [3H]paroxetine-labeled serotonin uptake sites. J Pharmacol Exp Ther. 1987;242:911–916. [PubMed] [Google Scholar]
- Battaglia G, Yeh SY, De Souza EB. MDMA-induced neurotoxicity: parameters of degeneration and recovery of brain serotonin neurons. Pharmacol Biochem Behav. 1988;29:269–274. doi: 10.1016/0091-3057(88)90155-4. [DOI] [PubMed] [Google Scholar]
- Baumann MH, Rutter JJ. Application of in vivo microdialysis methods to the study of psychomotor stimulant drugs. In: Waterhouse BD, editor. Methods in drug abuse research, cellular and circuit Analysis. Boca Raton, FL: CRC Press; 2003. pp. 51–86. [Google Scholar]
- Baumann MH, Ayestas MA, Rothman RB. Functional consequences of central serotonin depletion produced by repeated fenfluramine administration in rats. J Neurosci. 1998;18:9069–9077. doi: 10.1523/JNEUROSCI.18-21-09069.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann MH, Ayestas MA, Dersch CM, Rothman RB. 1-(m-chlorophenyl)piperazine (mCPP) dissociates in vivo serotonin release from long-term serotonin depletion in rat brain. Neuropsychopharmacology. 2001;24:492–501. doi: 10.1016/S0893-133X(00)00221-9. [DOI] [PubMed] [Google Scholar]
- Baumann MH, Clark RD, Budzynski AG, Partilla JS, Blough BE, Rothman RB. N-substituted piperazines abused by humans mimic the molecular mechanism of 3,4-methylenedioxymethamphetamine (MDMA, or ‘Ecstasy’) Neuropsychopharmacology. 2005;30:550–560. doi: 10.1038/sj.npp.1300585. [DOI] [PubMed] [Google Scholar]
- Baumgarten HG, Lachenmayer L. Serotonin neurotoxins—past and present. Neurotox Res. 2004;6:589–614. doi: 10.1007/BF03033455. [DOI] [PubMed] [Google Scholar]
- Benmansour S, Cecchi M, Morilak DA, Gerhardt GA, Javors MA, Gould GG, Frazer A. Effects of chronic antidepressant treatments on serotonin transporter function, density, and mRNA level. J Neurosci. 1999;19:10494–10501. doi: 10.1523/JNEUROSCI.19-23-10494.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger UV, Gu XF, Azmitia EC. The substituted amphetamines 3,4-methylenedioxymethamphetamine, methamphetamine, p-chloroamphetamine and fenfluramine induce 5-hydroxytryptamine release via a common mechanism blocked by fluoxetine and cocaine. Eur J Pharmacol. 1992;215:153–160. doi: 10.1016/0014-2999(92)90023-w. [DOI] [PubMed] [Google Scholar]
- Bull EJ, Hutson PH, Fone KC. Reduced social interaction following 3,4-methylenedioxymethamphetamine is not associated with enhanced 5-HT 2C receptor responsivity. Neuropharmacology. 2003;44:439–448. doi: 10.1016/s0028-3908(02)00407-0. [DOI] [PubMed] [Google Scholar]
- Bull EJ, Hutson PH, Fone KC. Decreased social behaviour following 3,4-methylenedioxymethamphetamine (MDMA) is accompanied by changes in 5-HT2A receptor responsivity. Neuropharmacology. 2004;46:202–210. doi: 10.1016/j.neuropharm.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Byrne T, Baker LE, Poling A. MDMA and learning: effects of acute and neurotoxic exposure in the rat. Pharmacol Biochem Behav. 2000;66:501–508. doi: 10.1016/s0091-3057(00)00227-6. [DOI] [PubMed] [Google Scholar]
- Campbell DB. Are interspecies comparisons in the toxicity of centrally acting drugs valid without brain concentrations? A commentary. Neurochem Int. 1995;26:103–110. doi: 10.1016/0197-0186(94)00104-3. [DOI] [PubMed] [Google Scholar]
- Campbell DB. Extrapolation from animals to man. The integration of pharmacokinetics and pharmacodynamics. Ann N Y Acad Sci. 1996;801:116–135. doi: 10.1111/j.1749-6632.1996.tb17436.x. [DOI] [PubMed] [Google Scholar]
- Carlsson A. The contribution of drug research to investigating the nature of endogenous depression. Pharmakopsychiatr Neuropsychopharmakol. 1976;9:2–10. [PubMed] [Google Scholar]
- Charlier C, Broly F, Lhermitte M, Pinto E, Ansseau M, Plomteux G. Polymorphisms in the CYP 2D6 gene: association with plasma concentrations of fluoxetine and paroxetine. Ther Drug Monit. 2003;25:738–742. doi: 10.1097/00007691-200312000-00014. [DOI] [PubMed] [Google Scholar]
- Chu T, Kumagai Y, DiStefano EW, Cho AK. Disposition of methylenedioxymethamphetamine and three metabolites in the brains of different rat strains and their possible roles in acute serotonin depletion. Biochem Pharmacol. 1996;51:789–796. doi: 10.1016/0006-2952(95)02397-6. [DOI] [PubMed] [Google Scholar]
- Colado MI, O’Shea E, Green AR. Acute and long-term effects of MDMA on cerebral dopamine biochemistry and function. Psychopharmacology (Berl) 2004;173:249–263. doi: 10.1007/s00213-004-1788-8. [DOI] [PubMed] [Google Scholar]
- Cole JC, Sumnall HR. The pre-clinical behavioural pharmacology of 3,4-methylenedioxymethamphetamine (MDMA) Neurosci Biobehav Rev. 2003;27:199–217. doi: 10.1016/s0149-7634(03)00031-9. [DOI] [PubMed] [Google Scholar]
- Commins DL, Vosmer G, Virus RM, Woolverton WL, Schuster CR, Seiden LS. Biochemical and histological evidence that methylenedioxymethylamphetamine (MDMA) is toxic to neurons in the rat brain. J Pharmacol Exp Ther. 1987;241:338–345. [PubMed] [Google Scholar]
- Crespi D, Mennini T, Gobbi M. Carrier-dependent and Ca(2+)-dependent 5-HT and dopamine release induced by (+)-amphetamine, 3,4-methylendioxymethamphetamine, p-chloroamphetamine and (+)-fenfluramine. Br J Pharmacol. 1997;121:1735–1743. doi: 10.1038/sj.bjp.0701325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dafters RI. Hyperthermia following MDMA administration in rats: effects of ambient temperature, water consumption, and chronic dosing. Physiol Behav. 1995;58:877–882. doi: 10.1016/0031-9384(95)00136-7. [DOI] [PubMed] [Google Scholar]
- Dafters RI, Lynch E. Persistent loss of thermoregulation in the rat induced by 3,4-methylenedioxymethamphetamine (MDMA or “Ecstasy”) but not by fenfluramine. Psychopharmacology (Berl) 1998;138:207–212. doi: 10.1007/s002130050664. [DOI] [PubMed] [Google Scholar]
- de la Torre R, Farre M. Neurotoxicity of MDMA (ecstasy): the limitations of scaling from animals to humans. Trends Pharmacol Sci. 2004;25:505–508. doi: 10.1016/j.tips.2004.08.001. [DOI] [PubMed] [Google Scholar]
- de la Torre R, Farre M, Ortuno J, Mas M, Brenneisen R, Roset PN, Segura J, Cami J. Non-linear pharmacokinetics of MDMA (‘ecstasy’) in humans. Br J Clin Pharmacol. 2000;49:104–109. doi: 10.1046/j.1365-2125.2000.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre R, Farre M, Roset PN, Pizarro N, Abanades S, Segura M, Segura J, Cami J. Human pharmacology of MDMA: pharmacokinetics, metabolism, and disposition. Ther Drug Monit. 2004;26:137–144. doi: 10.1097/00007691-200404000-00009. [DOI] [PubMed] [Google Scholar]
- Doblin R. A clinical plan for MDMA (Ecstasy) in the treatment of posttraumatic stress disorder (PTSD): partnering with the FDA. J Psychoactive Drugs. 2002;34:185–194. doi: 10.1080/02791072.2002.10399952. [DOI] [PubMed] [Google Scholar]
- Escobedo I, O’Shea E, Orio L, Sanchez V, Segura M, de la Torre R, Farre M, Green AR, Colado MI. A comparative study on the acute and long-term effects of MDMA and 3,4-dihydroxymethamphetamine (HHMA) on brain monoamine levels after i.p. or striatal administration in mice. Br J Pharmacol. 2005;144:231–241. doi: 10.1038/sj.bjp.0706071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan KT, Ricaurte GA, Ritchie LD, Irwin I, Peroutka SJ, Langston JW. Orally administered MDMA causes a long-term depletion of serotonin in rat brain. Brain Res. 1988;447:141–144. doi: 10.1016/0006-8993(88)90974-2. [DOI] [PubMed] [Google Scholar]
- Fitzgerald JL, Reid JJ. Interactions of methylenedioxymethamphetamine with monoamine transmitter release mechanisms in rat brain slices. Naunyn Schmiedebergs Arch Pharmacol. 1993;347:313–323. doi: 10.1007/BF00167451. [DOI] [PubMed] [Google Scholar]
- Fitzgerald JL, Reid JJ. Sympathomimetic actions of methylenedioxymethamphetamine in rat and rabbit isolated cardiovascular tissues. J Pharm Pharmacol. 1994;46:826–832. doi: 10.1111/j.2042-7158.1994.tb03738.x. [DOI] [PubMed] [Google Scholar]
- Fone KC, Beckett SR, Topham IA, Swettenham J, Ball M, Maddocks L. Long-term changes in social interaction and reward following repeated MDMA administration to adolescent rats without accompanying serotonergic neurotoxicity. Psychopharmacology (Berl) 2002;159:437–444. doi: 10.1007/s00213-001-0931-z. [DOI] [PubMed] [Google Scholar]
- Forsling ML, Fallon JK, Shah D, Tilbrook GS, Cowan DA, Kicman AT, Hutt AJ. The effect of 3,4-methylenedioxymethamphetamine (MDMA, ‘ecstasy’) and its metabolites on neurohypophysial hormone release from the isolated rat hypothalamus. Br J Pharmacol. 2002;135:649–656. doi: 10.1038/sj.bjp.0704502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer A, Benmansour S. Delayed pharmacological effects of antidepressants. Mol Psychiatry. 2002;7(Suppl 1):S23–S28. doi: 10.1038/sj.mp.4001015. [DOI] [PubMed] [Google Scholar]
- Gartside SE, McQuade R, Sharp T. Effects of repeated administration of 3,4-methylenedioxymethamphetamine on 5-hydroxytryptamine neuronal activity and release in the rat brain in vivo. J Pharmacol Exp Ther. 1996;279:277–283. doi: 10.1163/2211730x96x00153. [DOI] [PubMed] [Google Scholar]
- Gartside SE, McQuade R, Sharp T. Acute effects of 3,4-methylenedioxymethamphetamine (MDMA) on 5-HT cell firing and release: comparison between dorsal and median raphe 5-HT systems. Neuropharmacology. 1997;36:1697–1703. doi: 10.1016/s0028-3908(97)00171-8. [DOI] [PubMed] [Google Scholar]
- Gerra G, Zaimovic A, Giucastro G, Maestri D, Monica C, Sartori R, Caccavari R, Delsignore R. Serotonergic function after (+/−)3,4-methylene-dioxymethamphetamine (‘Ecstasy’) in humans. Int Clin Psychopharmacol. 1998;13:1–9. doi: 10.1097/00004850-199801000-00001. [DOI] [PubMed] [Google Scholar]
- Gerra G, Zaimovic A, Ferri M, Zambelli U, Timpano M, Neri E, Marzocchi GF, Delsignore R, Brambilla F. Long-lasting effects of (+/−)3,4-methylenedioxymethamphetamine (ecstasy) on serotonin system function in humans. Biol Psychiatry. 2000;47:127–136. doi: 10.1016/s0006-3223(99)00180-8. [DOI] [PubMed] [Google Scholar]
- Geyer MA. Serotonergic functions in arousal and motor activity. Behav Brain Res. 1996;73:31–35. doi: 10.1016/0166-4328(96)00065-4. [DOI] [PubMed] [Google Scholar]
- Glennon RA, Higgs R. Investigation of MDMA-related agents in rats trained to discriminate MDMA from saline. Pharmacol Biochem Behav. 1992;43:759–763. doi: 10.1016/0091-3057(92)90405-5. [DOI] [PubMed] [Google Scholar]
- Gold LH, Koob GF, Geyer MA. Stimulant and hallucinogenic behavioral profiles of 3,4-methylenedioxymethamphetamine and N-ethyl-3,4-methylenedioxyamphetamine in rats. J Pharmacol Exp Ther. 1988;247:547–555. [PubMed] [Google Scholar]
- Gouzoulis-Mayfrank E, Becker S, Pelz S, Tuchtenhagen F, Daumann J. Neuroendocrine abnormalities in recreational ecstasy (MDMA) users: is it ecstasy or cannabis? Biol Psychiatry. 2002;51:766–769. doi: 10.1016/s0006-3223(01)01306-3. [DOI] [PubMed] [Google Scholar]
- Green AR, Mechan AO, Elliott JM, O’Shea E, Colado MI. The pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”) Pharmacol Rev. 2003;55:463–508. doi: 10.1124/pr.55.3.3. [DOI] [PubMed] [Google Scholar]
- Green AR, O’Shea E, Colado MI. A review of the mechanisms involved in the acute MDMA (ecstasy)-induced hyperthermic response. Eur J Pharmacol. 2004;500:3–13. doi: 10.1016/j.ejphar.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Gudelsky GA, Nash JF. Carrier-mediated release of serotonin by 3,4-methylenedioxymethamphetamine: implications for serotonin–dopamine interactions. J Neurochem. 1996;66:243–249. doi: 10.1046/j.1471-4159.1996.66010243.x. [DOI] [PubMed] [Google Scholar]
- Gurtman CG, Morley KC, Li KM, Hunt GE, McGregor IS. Increased anxiety in rats after 3,4-methylenedioxymethamphetamine: association with serotonin depletion. Eur J Pharmacol. 2002;446:89–96. doi: 10.1016/s0014-2999(02)01820-4. [DOI] [PubMed] [Google Scholar]
- Hajos M, Sharp T. A 5-hydroxytryptamine lesion markedly reduces the incidence of burst-firing dorsal raphe neurones in the rat. Neurosci Lett. 1996;204:161–164. doi: 10.1016/0304-3940(96)12333-8. [DOI] [PubMed] [Google Scholar]
- Hall FS, Devries AC, Fong GW, Huang S, Pert A. Effects of 5,7-dihydroxytryptamine depletion of tissue serotonin levels on extracellular serotonin in the striatum assessed with in vivo microdialysis: relationship to behavior. Synapse. 1999;33:16–25. doi: 10.1002/(SICI)1098-2396(199907)33:1<16::AID-SYN2>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Harris DS, Baggott M, Mendelson JH, Mendelson JE, Jones RT. Subjective and hormonal effects of 3,4-methylenedioxymethamphetamine (MDMA) in humans. Psychopharmacology (Berl) 2002;162:396–405. doi: 10.1007/s00213-002-1131-1. [DOI] [PubMed] [Google Scholar]
- Jensen KF, Olin J, Haykal-Coates N, O’Callaghan J, Miller DB, de Olmos JS. Mapping toxicant-induced nervous system damage with a cupric silver stain: a quantitative analysis of neural degeneration induced by 3,4-methylenedioxymethamphetamine. NIDA Res Monogr. 1993;136:133–149. doi: 10.1037/e495922006-008. [DOI] [PubMed] [Google Scholar]
- Johanson CE, Kilbey M, Gatchalian K, Tancer M. Discriminative stimulus effects of 3,4-methylenedioxymethamphetamine (MDMA) in humans trained to discriminate among d-amphetamine, meta-chlorophenylpiperazine and placebo. Drug Alcohol Depend. 2006;81(1):27–36. doi: 10.1016/j.drugalcdep.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Johnson MP, Hoffman AJ, Nichols DE. Effects of the enantiomers of MDA, MDMA and related analogues on [3H]serotonin and [3H]dopamine release from superfused rat brain slices. Eur J Pharmacol. 1986;132:269–276. doi: 10.1016/0014-2999(86)90615-1. [DOI] [PubMed] [Google Scholar]
- Kalia M, O’Callaghan JP, Miller DB, Kramer M. Comparative study of fluoxetine, sibutramine, sertraline and dexfenfluramine on the morphology of serotonergic nerve terminals using serotonin immunohistochemistry. Brain Res. 2000;858:92–105. doi: 10.1016/s0006-8993(99)02430-0. [DOI] [PubMed] [Google Scholar]
- Kankaanpaa A, Meririnne E, Lillsunde P, Seppala T. The acute effects of amphetamine derivatives on extracellular serotonin and dopamine levels in rat nucleus accumbens. Pharmacol Biochem Behav. 1998;59:1003–1009. doi: 10.1016/s0091-3057(97)00527-3. [DOI] [PubMed] [Google Scholar]
- Kirby LG, Kreiss DS, Singh A, Lucki I. Effect of destruction of serotonin neurons on basal and fenfluramine-induced serotonin release in striatum. Synapse. 1995;20:99–105. doi: 10.1002/syn.890200202. [DOI] [PubMed] [Google Scholar]
- Kish SJ. How strong is the evidence that brain serotonin neurons are damaged in human users of ecstasy? Pharmacol Biochem Behav. 2002;71:845–855. doi: 10.1016/s0091-3057(01)00708-0. [DOI] [PubMed] [Google Scholar]
- Kleiber M. Body size and metabolism. Hilgardia. 1932;6:315–353. [Google Scholar]
- Landry MJ. MDMA: a review of epidemiologic data. J Psychoactive Drugs. 2002;34:163–169. doi: 10.1080/02791072.2002.10399950. [DOI] [PubMed] [Google Scholar]
- Lavelle A, Honner V, Docherty JR. Investigation of the prejunctional alpha2-adrenoceptor mediated actions of MDMA in rat atrium and vas deferens. Br J Pharmacol. 1999;128:975–980. doi: 10.1038/sj.bjp.0702875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy AD, Baumann MH, Van de Kar LD. Monoaminergic regulation of neuroendocrine function and its modification by cocaine. Front Neuroendocrinol. 1994;15:85–156. doi: 10.1006/frne.1994.1006. [DOI] [PubMed] [Google Scholar]
- Liechti ME, Baumann C, Gamma A, Vollenweider FX. Acute psychological effects of 3,4-methylenedioxymethamphetamine (MDMA, “Ecstasy”) are attenuated by the serotonin uptake inhibitor citalopram. Neuropsychopharmacology. 2000;22:513–521. doi: 10.1016/S0893-133X(99)00148-7. [DOI] [PubMed] [Google Scholar]
- Liechti ME, Vollenweider FX. Which neuroreceptors mediate the subjective effects of MDMA in humans? A summary of mechanistic studies. Hum Psychopharmacol. 2001;16:589–598. doi: 10.1002/hup.348. [DOI] [PubMed] [Google Scholar]
- Lin JH. Species similarities and differences in pharmacokinetics. Drug Metab Dispos. 1995;23:1008–1021. [PubMed] [Google Scholar]
- Lin JH. Applications and limitations of interspecies scaling and in vitro extrapolation in pharmacokinetics. Drug Metab Dispos. 1998;26:1202–1212. [PubMed] [Google Scholar]
- Lyles J, Cadet JL. Methylenedioxymethamphetamine (MDMA, Ecstasy) neurotoxicity: cellular and molecular mechanisms. Brain Res Brain Res Rev. 2003;42:155–168. doi: 10.1016/s0165-0173(03)00173-5. [DOI] [PubMed] [Google Scholar]
- Lyon RA, Glennon RA, Titeler M. 3,4-Methylenedioxymethamphetamine (MDMA): stereoselective interactions at brain 5-HT1 and 5-HT2 receptors. Psychopharmacology (Berl) 1986;88:525–526. doi: 10.1007/BF00178519. [DOI] [PubMed] [Google Scholar]
- Mahmood I. Allometric issues in drug development. J Pharm Sci. 1999;88:1101–1106. doi: 10.1021/js9902163. [DOI] [PubMed] [Google Scholar]
- Malberg JE, Seiden LS. Small changes in ambient temperature cause large changes in 3,4-methylenedioxymethamphetamine (MDMA)-induced serotonin neurotoxicity and core body temperature in the rat. J Neurosci. 1998;18:5086–5094. doi: 10.1523/JNEUROSCI.18-13-05086.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malpass A, White JM, Irvine RJ, Somogyi AA, Bochner F. Acute toxicity of 3,4-methylenedioxymethamphetamine (MDMA) in Sprague–Dawley and Dark Agouti rats. Pharmacol Biochem Behav. 1999;64:29–34. doi: 10.1016/s0091-3057(99)00116-1. [DOI] [PubMed] [Google Scholar]
- Marston HM, Reid ME, Lawrence JA, Olverman HJ, Butcher SP. Behavioural analysis of the acute and chronic effects of MDMA treatment in the rat. Psychopharmacology (Berl) 1999;144:67–76. doi: 10.1007/s002130050978. [DOI] [PubMed] [Google Scholar]
- Mas M, Farre M, de la Torre R, Roset PN, Ortuno J, Segura J, Cami J. Cardiovascular and neuroendocrine effects and pharmacokinetics of 3, 4-methylenedioxymethamphetamine in humans. J Pharmacol Exp Ther. 1999;290:136–145. [PubMed] [Google Scholar]
- Matuszewich L, Filon ME, Finn DA, Yamamoto BK. Altered forebrain neurotransmitter responses to immobilization stress following 3,4-methylenedioxymethamphetamine. Neuroscience. 2002;110:41–48. doi: 10.1016/s0306-4522(01)00539-5. [DOI] [PubMed] [Google Scholar]
- Maurer HH, Bickeboeller-Friedrich J, Kraemer T, Peters FT. Toxicokinetics and analytical toxicology of amphetamine-derived designer drugs (‘Ecstasy’) Toxicol Lett. 2000;112–113:133–142. doi: 10.1016/s0378-4274(99)00207-6. [DOI] [PubMed] [Google Scholar]
- McGregor IS, Clemens KJ, Van der Plasse G, Li KM, Hunt GE, Chen F, Lawrence AJ. Increased anxiety 3 months after brief exposure to MDMA (“Ecstasy”) in rats: association with altered 5-HT transporter and receptor density. Neuropsychopharmacology. 2003;28:1472–1484. doi: 10.1038/sj.npp.1300185. [DOI] [PubMed] [Google Scholar]
- McNamara MG, Kelly JP, Leonard BE. Some behavioural and neurochemical aspects of subacute (+/−)3,4-methylenedioxymethamphetamine administration in rats. Pharmacol Biochem Behav. 1995;52:479–484. doi: 10.1016/0091-3057(95)00206-c. [DOI] [PubMed] [Google Scholar]
- Mechan AO, Esteban B, O’Shea E, Elliott JM, Colado MI, Green AR. The pharmacology of the acute hyperthermic response that follows administration of 3,4-methylenedioxymethamphetamine (MDMA, ‘ecstasy’) to rats. Br J Pharmacol. 2002;135:170–180. doi: 10.1038/sj.bjp.0704442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliver ME, Berger UV, Mamounas LA, Molliver DC, O’Hearn E, Wilson MA. Neurotoxicity of MDMA and related compounds: anatomic studies. Ann N Y Acad Sci. 1990;600:649–661. doi: 10.1111/j.1749-6632.1990.tb16916.x. [DOI] [PubMed] [Google Scholar]
- Monks TJ, Jones DC, Bai F, Lau SS. The role of metabolism in 3,4-(+)-methylenedioxyamphetamine and 3,4-(+)-methylenedioxymethamphetamine (ecstasy) toxicity. Ther Drug Monit. 2004;26:132–136. doi: 10.1097/00007691-200404000-00008. [DOI] [PubMed] [Google Scholar]
- Morgan MJ. Ecstasy (MDMA): a review of its possible persistent psychological effects. Psychopharmacology (Berl) 2000;152:230–248. doi: 10.1007/s002130000545. [DOI] [PubMed] [Google Scholar]
- Morley KC, Gallate JE, Hunt GE, Mallet PE, McGregor IS. Increased anxiety and impaired memory in rats 3 months after administration of 3,4-methylenedioxymethamphetamine (“ecstasy”) Eur J Pharmacol. 2001;433:91–99. doi: 10.1016/s0014-2999(01)01512-6. [DOI] [PubMed] [Google Scholar]
- Moser VC. The functional observational battery in adult and developing rats. Neurotoxicology. 2000;21:989–996. [PubMed] [Google Scholar]
- Nash JF, Jr, Meltzer HY, Gudelsky GA. Elevation of serum prolactin and corticosterone concentrations in the rat after the administration of 3,4-methylenedioxymethamphetamine. J Pharmacol Exp Ther. 1988;245:873–879. [PubMed] [Google Scholar]
- Nichols DE, Lloyd DH, Hoffman AJ, Nichols MB, Yim GK. Effects of certain hallucinogenic amphetamine analogues on the release of [3H]serotonin from rat brain synaptosomes. J Med Chem. 1982;25:530–535. doi: 10.1021/jm00347a010. [DOI] [PubMed] [Google Scholar]
- Noujaim SF, Lucca E, Munoz V, Persaud D, Berenfeld O, Meijler FL, Jalife J. From mouse to whale: a universal scaling relation for the PR Interval of the electrocardiogram of mammals. Circulation. 2004;110:2802–2808. doi: 10.1161/01.CIR.0000146785.15995.67. [DOI] [PubMed] [Google Scholar]
- O’Cain PA, Hletko SB, Ogden BA, Varner KJ. Cardiovascular and sympathetic responses and reflex changes elicited by MDMA. Physiol Behav. 2000;70:141–148. doi: 10.1016/s0031-9384(00)00235-3. [DOI] [PubMed] [Google Scholar]
- O’Callaghan JP, Miller DB. Quantification of reactive gliosis as an approach to neurotoxicity assessment. NIDA Res Monogr. 1993;136:188–212. [PubMed] [Google Scholar]
- O’Callaghan JP, Jensen KF, Miller DB. Quantitative aspects of drug and toxicant-induced astrogliosis. Neurochem Int. 1995;26:115–124. doi: 10.1016/0197-0186(94)00106-5. [DOI] [PubMed] [Google Scholar]
- O’Callaghan JP, Sriram K. Glial fibrillary acidic protein and related glial proteins as biomarkers of neurotoxicity. Expert Opin Drug Saf. 2005;4:433–442. doi: 10.1517/14740338.4.3.433. [DOI] [PubMed] [Google Scholar]
- O’Hearn E, Battaglia G, De Souza EB, Kuhar MJ, Molliver ME. Methylenedioxyamphetamine (MDA) and methylenedioxymethamphetamine (MDMA) cause selective ablation of serotonergic axon terminals in forebrain: immunocytochemical evidence for neurotoxicity. J Neurosci. 1988;8:2788–2803. doi: 10.1523/JNEUROSCI.08-08-02788.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea E, Granados R, Esteban B, Colado MI, Green AR. The relationship between the degree of neurodegeneration of rat brain 5-HT nerve terminals and the dose and frequency of administration of MDMA (‘ecstasy’) Neuropharmacology. 1998;37:919–926. doi: 10.1016/s0028-3908(98)00029-x. [DOI] [PubMed] [Google Scholar]
- Oberlender R, Nichols DE. Drug discrimination studies with MDMA and amphetamine. Psychopharmacology (Berl) 1988;95:71–76. doi: 10.1007/BF00212770. [DOI] [PubMed] [Google Scholar]
- Parrott AC. Recreational ecstasy/MDMA, the serotonin syndrome, and serotonergic neurotoxicity. Pharmacol Biochem Behav. 2002;71:837–844. doi: 10.1016/s0091-3057(01)00711-0. [DOI] [PubMed] [Google Scholar]
- Parrott AC. Is ecstasy MDMA? A review of the proportion of ecstasy tablets containing MDMA, their dosage levels, and the changing perceptions of purity. Psychopharmacology (Berl) 2004;173:234–241. doi: 10.1007/s00213-003-1712-7. [DOI] [PubMed] [Google Scholar]
- Partilla JS, Dersch CM, Yu H, Rice KC, Rothman RB. Neurochemical neutralization of amphetamine-type stimulants in rat brain by the indatraline analog (−)-HY038. Brain Res Bull. 2000;53:821–826. doi: 10.1016/s0361-9230(00)00419-6. [DOI] [PubMed] [Google Scholar]
- Poland RE. Diminished corticotropin and enhanced prolactin responses to 8-hydroxy-2(di-n-propylamino)tetralin in methylenedioxymethamphetamine pretreated rats. Neuropharmacology. 1990;29:1099–1101. doi: 10.1016/0028-3908(90)90120-g. [DOI] [PubMed] [Google Scholar]
- Poland RE, Lutchmansingh P, McCracken JT, Zhao JP, Brammer GL, Grob CS, Boone KB, Pechnick RN. Abnormal ACTH and prolactin responses to fenfluramine in rats exposed to single and multiple doses of MDMA. Psychopharmacology (Berl) 1997;131:411–419. doi: 10.1007/s002130050311. [DOI] [PubMed] [Google Scholar]
- Pubill D, Canudas AM, Pallas M, Camins A, Camarasa J, Escubedo E. Different glial response to methamphetamine- and methylenedioxymethamphetamine-induced neurotoxicity. Naunyn Schmiedebergs Arch Pharmacol. 2003;367:490–499. doi: 10.1007/s00210-003-0747-y. [DOI] [PubMed] [Google Scholar]
- Ratzenboeck E, Saria A, Kriechbaum N, Zernig G. Reinforcing effects of MDMA (“ecstasy”) in drug-naive and cocaine-trained rats. Pharmacology. 2001;62:138–144. doi: 10.1159/000056086. [DOI] [PubMed] [Google Scholar]
- Reneman L. Designer drugs: how dangerous are they? J Neural Transm Suppl. 2003;66:61–83. doi: 10.1007/978-3-7091-0541-2_4. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Markowska AL, Wenk GL, Hatzidimitriou G, Wlos J, Olton DS. 3,4-Methylenedioxymethamphetamine, serotonin and memory. J Pharmacol Exp Ther. 1993;266:1097–1105. [PubMed] [Google Scholar]
- Ricaurte GA, Yuan J, McCann UD. (+/−)3,4-Methylenedioxymethamphetamine (‘Ecstasy’)-induced serotonin neurotoxicity: studies in animals. Neuropsychobiology. 2000;42:5–10. doi: 10.1159/000026664. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Castaneda E, Whishaw IQ. Effects of cortical serotonin depletion induced by 3,4-methylenedioxymethamphetamine (MDMA) on behavior, before and after additional cholinergic blockade. Neuropsychopharmacology. 1993;8:77–85. doi: 10.1038/npp.1993.9. [DOI] [PubMed] [Google Scholar]
- Romero L, Jernej B, Bel N, Cicin-Sain L, Cortes R, Artigas F. Basal and stimulated extracellular serotonin concentration in the brain of rats with altered serotonin uptake. Synapse. 1998;28:313–321. doi: 10.1002/(SICI)1098-2396(199804)28:4<313::AID-SYN7>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH. Therapeutic and adverse actions of serotonin transporter substrates. Pharmacol Ther. 2002;95:73–88. doi: 10.1016/s0163-7258(02)00234-6. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse. 2001;39:32–41. doi: 10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Jayanthi S, Wang X, Dersch CM, Cadet JL, Prisinzano T, Rice KC, Baumann MH. High-dose fenfluramine administration decreases serotonin transporter binding, but not serotonin transporter protein levels, in rat forebrain. Synapse. 2003;50:233–239. doi: 10.1002/syn.10266. [DOI] [PubMed] [Google Scholar]
- Rudnick G, Clark J. From synapse to vesicle: the reuptake and storage of biogenic amine neurotransmitters. Biochim Biophys Acta. 1993;1144:249–263. doi: 10.1016/0005-2728(93)90109-s. [DOI] [PubMed] [Google Scholar]
- Scanzello CR, Hatzidimitriou G, Martello AL, Katz JL, Ricaurte GA. Serotonergic recovery after (+/−)3,4-(methylenedioxy) methamphetamine injury: observations in rats. J Pharmacol Exp Ther. 1993;264:1484–1491. [PubMed] [Google Scholar]
- Schechter MD. Serotonergic–dopaminergic mediation of 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”) Pharmacol Biochem Behav. 1988;31:817–824. doi: 10.1016/0091-3057(88)90390-5. [DOI] [PubMed] [Google Scholar]
- Schenk S, Gittings D, Johnstone M, Daniela E. Development, maintenance and temporal pattern of self-administration maintained by ecstasy (MDMA) in rats. Psychopharmacology (Berl) 2003;169:21–27. doi: 10.1007/s00213-003-1407-0. [DOI] [PubMed] [Google Scholar]
- Schifano F. A bitter pill. Overview of ecstasy (MDMA, MDA) related fatalities. Psychopharmacology (Berl) 2004;173:242–248. doi: 10.1007/s00213-003-1730-5. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ. Neurotoxicity of the psychedelic amphetamine, methylenedioxymethamphetamine. J Pharmacol Exp Ther. 1987;240:1–7. [PubMed] [Google Scholar]
- Schmidt CJ, Levin JA, Lovenberg W. In vitro and in vivo neurochemical effects of methylenedioxymethamphetamine on striatal monoaminergic systems in the rat brain. Biochem Pharmacol. 1987;36:747–755. doi: 10.1016/0006-2952(87)90729-5. [DOI] [PubMed] [Google Scholar]
- Schmued LC. Demonstration and localization of neuronal degeneration in the rat forebrain following a single exposure to MDMA. Brain Res. 2003;974:127–133. doi: 10.1016/s0006-8993(03)02563-0. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Seiden LS, Woolverton WL, Lorens SA, Williams JE, Corwin RL, Hata N, Olimski M. Behavioral consequences of partial monoamine depletion in the CNS after methamphetamine-like drugs: the conflict between pharmacology and toxicology. NIDA Res Monogr. 1993;136:34–46. doi: 10.1037/e495922006-004. [DOI] [PubMed] [Google Scholar]
- Series HG, Cowen PJ, Sharp T. p-Chloroamphetamine (PCA), 3,4-methylenedioxy-methamphetamine (MDMA) and d-fenfluramine pretreatment attenuates d-fenfluramine-evoked release of 5-HT in vivo. Psychopharmacology (Berl) 1994;116:508–514. doi: 10.1007/BF02247485. [DOI] [PubMed] [Google Scholar]
- Series HG, le Masurier M, Gartside SE, Franklin M, Sharp T. Behavioural and neuroendocrine responses to d-fenfluramine in rats treated with neurotoxic amphetamines. J Psychopharmacology. 1995;9:214–222. doi: 10.1177/026988119500900303. [DOI] [PubMed] [Google Scholar]
- Setola V, Hufeisen SJ, Grande-Allen KJ, Vesely I, Glennon RA, Blough B, Rothman RB, Roth BL. 3,4-methylenedioxymethamphetamine (MDMA, “Ecstasy”) induces fenfluramine-like proliferative actions on human cardiac valvular interstitial cells in vitro. Mol Pharmacol. 2003;63:1223–1229. doi: 10.1124/mol.63.6.1223. [DOI] [PubMed] [Google Scholar]
- Shankaran M, Gudelsky GA. A neurotoxic regimen of MDMA suppresses behavioral, thermal and neurochemical responses to subsequent MDMA administration. Psychopharmacology (Berl) 1999;147:66–72. doi: 10.1007/s002130051143. [DOI] [PubMed] [Google Scholar]
- Slikker W, Jr., Holson RR, Ali SF, Kolta MG, Paule MG, Scallet AC, McMillan DE, Bailey JR, Hong JS, Scalzo FM. Behavioral and neurochemical effects of orally administered MDMA in the rodent and nonhuman primate. Neurotoxicology. 1989;10:529–542. [PubMed] [Google Scholar]
- Spanos LJ, Yamamoto BK. Acute and subchronic effects of methylenedioxymethamphetamine [(+/−)MDMA] on locomotion and serotonin syndrome behavior in the rat. Pharmacol Biochem Behav. 1989;32:835–840. doi: 10.1016/0091-3057(89)90044-0. [DOI] [PubMed] [Google Scholar]
- Sprague JE, Everman SL, Nichols DE. An integrated hypothesis for the serotonergic axonal loss induced by 3,4-methylenedioxymethamphetamine. Neurotoxicology. 1998;19:427–441. [PubMed] [Google Scholar]
- Sprouse JS, Bradberry CW, Roth RH, Aghajanian GK. MDMA (3,4-methylenedioxymethamphetamine) inhibits the firing of dorsal raphe neurons in brain slices via release of serotonin. Eur J Pharmacol. 1989;167:375–383. doi: 10.1016/0014-2999(89)90446-9. [DOI] [PubMed] [Google Scholar]
- Steinbusch HW. Distribution of serotonin-immunoreactivity in the central nervous system of the rat-cell bodies and terminals. Neuroscience. 1981;6:557–618. doi: 10.1016/0306-4522(81)90146-9. [DOI] [PubMed] [Google Scholar]
- Stone DM, Merchant KM, Hanson GR, Gibb JW. Immediate and long-term effects of 3,4-methylenedioxymethamphetamine on serotonin pathways in brain of rat. Neuropharmacology. 1987;26:1677–1683. doi: 10.1016/0028-3908(87)90117-1. [DOI] [PubMed] [Google Scholar]
- Switzer RC., 3rd Application of silver degeneration stains for neurotoxicity testing. Toxicol Pathol. 2000;28:70–83. doi: 10.1177/019262330002800109. [DOI] [PubMed] [Google Scholar]
- Tancer M, Johanson CE. Reinforcing, subjective, and physiological effects of MDMA in humans: a comparison with d-amphetamine and mCPP. Drug Alcohol Depend. 2003;72:33–44. doi: 10.1016/s0376-8716(03)00172-8. [DOI] [PubMed] [Google Scholar]
- Van de Kar LD. Neuroendocrine pharmacology of serotonergic (5-HT) neurons. Annu Rev Pharmacol Toxicol. 1991;31:289–320. doi: 10.1146/annurev.pa.31.040191.001445. [DOI] [PubMed] [Google Scholar]
- Vollenweider FX, Gamma A, Liechti M, Huber T. Psychological and cardiovascular effects and short-term sequelae of MDMA (“ecstasy”) in MDMA-naive healthy volunteers. Neuropsychopharmacology. 1998;19:241–251. doi: 10.1016/S0893-133X(98)00013-X. [DOI] [PubMed] [Google Scholar]
- Wakonigg G, Sturm K, Saria A, Zernig G. Methylenedioxymethamphetamine (MDMA, ‘ecstasy’) serves as a robust positive reinforcer in a rat runway procedure. Pharmacology. 2003;69:180–182. doi: 10.1159/000073661. [DOI] [PubMed] [Google Scholar]
- Wang X, Baumann MH, Xu H, Rothman RB. 3,4-methylenedioxymethamphetamine (MDMA) administration to rats decreases brain tissue serotonin but not serotonin transporter protein and glial fibrillary acidic protein. Synapse. 2004;53:240–248. doi: 10.1002/syn.20058. [DOI] [PubMed] [Google Scholar]
- Wang X, Baumann MH, Xu H, Morales M, Rothman RB. ({+/−})-3,4-Methylenedioxymethamphetamine (MDMA) administration to rats does not decrease levels of the serotonin transporter protein or alter its distribution between endosomes and the plasma membrane. J Pharmacol Exp Ther. 2005;314(3):1002–1012. doi: 10.1124/jpet.105.088476. [DOI] [PubMed] [Google Scholar]
- West GB, Woodruff WH, Brown JH. Allometric scaling of metabolic rate from molecules and mitochondria to cells and mammals. Proc Natl Acad Sci USA. 2002;99(Suppl 1):2473–2478. doi: 10.1073/pnas.012579799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CR, Seymour RS. Allometric scaling of mammalian metabolism. J Exp Biol. 2005;208:1611–1619. doi: 10.1242/jeb.01501. [DOI] [PubMed] [Google Scholar]
- Winneke G, Lilienthal H. Extrapolation from animals to humans: scientific and regulatory aspects. Toxicol Lett. 1992;64–65 Spec No:239–246. doi: 10.1016/0378-4274(92)90195-p. [DOI] [PubMed] [Google Scholar]
- Wu D, Otton SV, Inaba T, Kalow W, Sellers EM. Interactions of amphetamine analogs with human liver CYP2D6. Biochem Pharmacol. 1997;53:1605–1612. doi: 10.1016/s0006-2952(97)00014-2. [DOI] [PubMed] [Google Scholar]
- Yacoubian GS., Jr. Tracking ecstasy trends in the United States with data from three national drug surveillance systems. J Drug Educ. 2003;33:245–258. doi: 10.2190/BDQB-2NPV-PAW3-VUXV. [DOI] [PubMed] [Google Scholar]
- Yamamoto BK, Nash JF, Gudelsky GA. Modulation of methylenedioxymethamphetamine-induced striatal dopamine release by the interaction between serotonin and gamma-aminobutyric acid in the substantia nigra. J Pharmacol Exp Ther. 1995;273:1063–1070. [PubMed] [Google Scholar]
- Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM. Compensations after lesions of central dopaminergic neurons: some clinical and basic implications. Trends Neurosci. 1990;13:290–296. doi: 10.1016/0166-2236(90)90112-n. [DOI] [PubMed] [Google Scholar]