

Abstract
Cells must regulate both biosynthesis and degradation to ensure proper homeostasis of cellular organelles and proteins. This balance is demonstrated in a unique way in the yeast Saccharomyces cerevisiae, which possesses two distinct, yet mechanistically related trafficking routes mediating the delivery of proteins from the cytoplasm to the vacuole: the biosynthetic cytoplasm to vacuole targeting (Cvt) and the degradative autophagy pathways. Several components employed by these two transport routes have been identified, but their mechanistic interactions remain largely unknown. Here we report a novel gene involved in these pathways, which we have named ATG23. Atg23 localizes to the pre-autophagosomal structure but also to other cytosolic punctate compartments. Our characterization of the Atg23 protein indicates that it is required for the Cvt pathway and efficient autophagy but not pexophagy. In the absence of Atg23, cargo molecules such as prApe1 are correctly recruited to a pre-autophagosomal structure that is unable to give rise to Cvt vesicles. We also demonstrate that Atg23 is a peripheral membrane protein that requires the presence of Atg9/Apg9 to be specifically targeted to lipid bilayers. Atg9 transiently interacts with Atg23 suggesting that it participates in the recruitment of this protein.
Cells must carefully maintain homeostasis by balancing biosynthetic and catabolic pathways as well as by being able to sense changes in their environments and respond accordingly. This balance can be seen quite dramatically when considering the delivery of proteins and substrates to the lysosome-like vacuole of the yeast Saccharomyces cerevisiae. The vacuole is the primary degradative organelle within the yeast cell. In accordance with its catabolic role, the vacuole contains a large number of resident hydrolases that must be targeted to this compartment. Multiple pathways are used for the proper localization of these vacuolar hydrolases (reviewed in Ref. 1). Similarly, substrates destined for degradation, including both proteins and entire organelles, must be delivered to the vacuole where they become accessible to the lumenal hydrolases. S. cerevisiae possesses two distinct, yet mechanistically overlapping trafficking pathways to allow for the delivery of proteins directly from the cytoplasm to the vacuole, termed cytoplasm to vacuole targeting (Cvt)1 and autophagy (Atg). Under rich growth conditions, these mechanistic components are almost exclusively dedicated to the biosynthetic trafficking of specific resident vacuolar proteins via the Cvt pathway. However, during periods of starvation, these components switch to mediate autophagy, in which bulk cellular material is transported non-specifically to the vacuole for degradation (reviewed in Refs. 2-5). The generated pool of nutrients can then be used by the cells to survive such starvation periods for several weeks. Autophagy occurs not only in starving yeast but is also important in mammalian development, cellular differentiation, and non-apoptotic programmed cell death (6). Moreover, defects in autophagy have been implicated in a growing list of mammalian disease states, including cancer (7), neurodegenerative disorders such as Parkinson's and Alzheimer's diseases (8, 9), and cardiomyopathy associated with Danon's disease (10, 11). Therefore, understanding the molecular basis underlying autophagy is a subject of intense interest. Because many of the mechanisms mediating the Cvt and autophagy pathways are held in common, advances in the understanding of one pathway often yield insight into the other.
Genetic screens identifying autophagy- or Cvt-deficient mutants (termed atg) have identified many identical genetic requirements between the two pathways (12-18). In this study we report the characterization of ATG23, a gene identified in a screen of a systematic ORF deletion library to isolate genes that, when deleted, render cells incompetent for Cvt trafficking during nitrogen-rich growth conditions. We demonstrate that atg23Δ mutants are defective for Cvt trafficking as well as exhibiting decreased levels of autophagy upon challenge by nitrogen starvation. The Atg23 protein is a peripheral membrane protein distributed in subcellular punctate organelles. One of the multiple Atg23-positive structures co-localizes with the pre-autophagosomal structure (PAS), the potential site of organization for Cvt vesicle/autophagosome formation (19, 20). Atg23 interacts with Atg9, a transmembrane protein essential for Cvt vesicle and autophagosome formation with an identical localization pattern (21). The association between those two proteins is critical to recruit Atg23 to membranes.
EXPERIMENTAL PROCEDURES
Strains, Plasmids, and Media
The S. cerevisiae knockout library in strain BY4742 was purchased from ResGen™ (Invitrogen). The strains used in this study are listed in Table I. For ATG23 gene disruptions, the entire coding region was replaced with either the Escherichia coli kanr or the S. cerevisiae TRP1 gene using PCR primers containing ∼40 bases of identity to the regions flanking the open reading frame. PCR-based integrations of the triple HA tag, the 13×Myc tag, protein A (PA), GFP, and YFP at the 3′ end of ATG23 and ATG9 were used to generate strains expressing fusion proteins under the control of their native promoters. The templates for integration were pFA6a-3HA-TRP1, pFA6a-GFP-TRP1, pFA6a-GFP-KanMX, pFA6a-13Myc-TRP1, pDH3, and pHAB102 (22-24). PCR verification and prApe1 processing were used to confirm the functionality of all genomic fusions.
Table I.
Yeast strains used in this study
| Strain | Genotype | Source or Ref. |
|---|---|---|
| BY4742 | MATα his3Δ leu2Δ lys2Δ ura3Δ | ResGen™ |
| SEY6210 | MATα ura3–52 leu2–3,112 his3-Δ200 trp1-Δ901 lys2–801 suc2-Δ9 mel GAL | 46 |
| TN124 | MATα leu2–3,112 ura3–52 trp1 pho8Δ::PHO8Δ60 pho13Δ::LEU2 | 39 |
| atg9Δ | BY4742 atg9Δ::KAN | ResGen™ |
| atg23Δ | BY4742 atg23Δ::KAN | ResGen™ |
| D3Y102 | SEY6210 vac8Δ::TRP1 | 38 |
| D3Y103 | TN124 atg13Δ | 38 |
| FRY134 | SEY6210 ATG9-YFP::HIS5 S.p. promCFPATG8::URA3 | This study |
| JKY007 | SEY6210 atg9Δ::HIS3 | 21 |
| KTY7 | SEY6210 ATG23-13xMyc::TRP1 | This study |
| KTY8 | SEY6210 ATG23-HA::TRP1 | This study |
| KTY9 | TN124 atg23Δ::TRP1 | This study |
| KTY14 | SEY6210 atg23Δ::KAN | This study |
| KTY22 | BY4742 atg23Δ::KAN pep4Δ::LEU2 | This study |
| KTY26 | SEY6210 ATG23-YFP::HIS5 S.p. | This study |
| KTY32 | SEY6210 ATG23-GFP::TRP1 atg9Δ::HIS3 S.k. | This study |
| KTY51 | SEY6210 ATG9-YFP::HIS5 S.p. atg23Δ::KAN | This study |
| KTY74 | SEY6210 ATG9-PA::HIS5 S.p. ATG23-HA::TRP1 | This study |
| TVY1 | SEY6210 pep4Δ::LEU2 | 47 |
Plasmids expressing PA (pRS416-CuProtA), CFP-Ape1 (pTS470), YFP-Atg11 (pPS97), CFP-Atg8 (pRS416ECFP-Aut7), YFP-Atg8 (pRS414EYFP-Aut7), and Atg19-CFP (pCVT19CFP(414)) have been described elsewhere (19-21, 25, 26).
Cells were grown in rich (YPD: 1% yeast extract, 2% peptone, 2% glucose) or synthetic minimal media (SMD: 0.67% yeast nitrogen base, 2% glucose, amino acids and vitamins as needed). Starvation experiments were conducted in synthetic media lacking nitrogen (SD-N: 0.17% yeast nitrogen base without amino acids, 2% glucose).
Cell Labeling and Immunoprecipitation
Cells were grown to A600 = 1.0 in SMD. Cells (10 ml) were collected and labeled in 100 μl of SMD with 20 μCi of Tran-35S-label (ICN, Costa Mesa, CA) for 10 min and then subjected to a nonradioactive chase in 1 ml of SMD supplemented with 0.2% yeast extract, 2 mm methionine, and 2 mm cysteine. Samples were removed at the time points indicated, precipitated with 10% trichloroacetic acid, and washed with acetone. Extracts were generated using glass bead lysis and subjected to immunoprecipitation as described elsewhere (17).
Filtration Lysis and Membrane Flotation
Indicated cells were grown in SMD + casamino acids to early log phase (A600 = 0.5) and converted to spheroplasts by treatment with 5 μg/A600 oxalyticase (Enzogenetics, Eugene, OR). Spheroplasts were resuspended in PS600 lysis buffer (20 mm K-PIPES, pH 6.8, 600 mm sorbitol) containing 5 mm MgCl2 and filtered through 3.0-μm nucleopore track-etch membranes (Whatman) in a 13-mm syringe filter holder using a 3-cc syringe. The filtrate was pre-cleared with a low speed spin at 800 rpm for 5 min at 4 °C. Cleared filtrate was subjected to centrifugation at 16,000 × g for 10 min at 4 °C, resulting in low speed supernatant (S13) and pellet (P13) fractions.
Subcellular Fractionation
Cells were subjected to filtration lysis as described above, and S13/P13 fractions were collected. The S13 fraction was subjected to further centrifugation at 100,000 × g for 20 min in a TLA100.4 fixed angle rotor to generate high speed supernatant (S100) and pellet (P100) fractions. All fractions were precipitated with trichloroacetic acid, washed with acetone, and analyzed by immunoblot using anti-Myc antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-Pgk1 antiserum (a generous gift from Dr. Jeremy Thorner, University of California, Berkeley).
Miscellaneous Procedures
The analyses of protease sensitivity, cell viability under nitrogen starvation conditions, pexophagy, Pho8Δ60 activity, sucrose step gradient fractionation, PA affinity isolation, and fluorescent and electron microscopy were conducted as described previously (27-33).
RESULTS
atg23Δ Cells Are Defective in the Cvt Pathway and Impaired in Autophagy
The resident vacuolar hydrolase aminopeptidase I (Ape1) is synthesized in the cytosol as an inactive precursor protein (prApe1) and then delivered directly across the limiting membrane of the vacuole via the Cvt pathway during growth conditions, where it is processed to the mature, active protein (14, 18, 34, 35). During periods of nitrogen starvation, both Cvt cargo and bulk cytoplasm are transported across the vacuolar membrane by the autophagy pathway (18, 34, 36). We recently performed a systematic screen of the yeast ORF deletion library by Western blot looking for the accumulation of prApe1 in nitrogen-rich conditions to identify novel genes required for the Cvt pathway. One such gene identified was CVT23 (ORF YLR431c). This gene was subsequently renamed ATG23 in accordance with a new, unified genetic nomenclature for AuTophaGy-related genes in yeast (15). To verify that Atg23 is required for the Cvt pathway, we performed a pulse-chase analysis to determine the kinetics of prApe1 import in atg23Δ cells.
Yeast cells were pulse-labeled with [35S]methionine/cysteine and subjected to a non-radioactive chase. In wild type cells, the 61-kDa prApe1 was processed to the 50-kDa mature form with a half-time of ∼30 min (Fig. 1). In contrast, the atg23Δ mutant specifically accumulated prApe1 in rich medium (Fig. 1), demonstrating that atg23Δ cells are indeed defective for the Cvt pathway. To determine whether this block was specific to the Cvt pathway, we examined the delivery of proteins to the vacuole through the carboxypeptidase Y pathway. Many resident vacuolar hydrolases, including Prc1 (carboxypeptidase Y), are delivered to the vacuole through a portion of the secretory pathway (1). We found that the processing of Prc1 was unaffected in atg23Δ cells (data not shown). This result indicates that the atg23Δ mutant did not display a general block in vacuolar protein delivery, implying a specific function for Atg23 in the Cvt pathway.
Fig. 1.
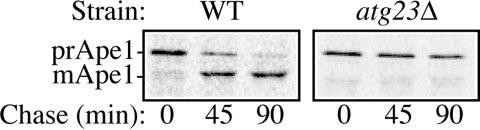
atg23Δ cells are defective in the Cvt pathway. Wild type (WT) (SEY6210) and atg23Δ (KTY14) cells were pulse-labeled for 10 min and subjected to a non-radioactive chase for 90 min. Ape1 was immunoprecipitated from cell lysates and resolved by SDS-PAGE at the indicated time points. The positions of prApe1 and mApe1 are indicated.
We wished to determine whether Atg23 had a function in autophagic trafficking to the vacuole, because many of the proteins utilized by the Cvt pathway are also needed for autophagy. Although most currently characterized atg mutants are defective in delivery through both pathways, there is an increasing list of genes required primarily for the Cvt pathway, including VAC8, ATG11/CVT9, ATG20/CVT20, ATG24/CVT13, and the VFT tethering complex (28, 29, 37, 38). As prApe1 can be specifically transported to the vacuole through both the Cvt pathway and autophagy (34), mutants in Cvt-specific genes are able to “reverse” the prApe1 accumulation defect when challenged with nitrogen starvation (28, 29, 37, 38). As shown in Fig. 2A, atg23Δ cells, like vac8Δ cells, accumulated prApe1 under growth conditions but rapidly processed prApe1 when deprived of nitrogen. As a control, we examined the atg9Δ mutant. Atg9 is a membrane protein essential for both the Cvt and autophagy pathways (21). As expected, atg9Δ cells accumulated prApe1 under both growth and starvation conditions. In contrast, wild type cells were able to mature this hydrolase under either growth or starvation conditions.
Fig.2.
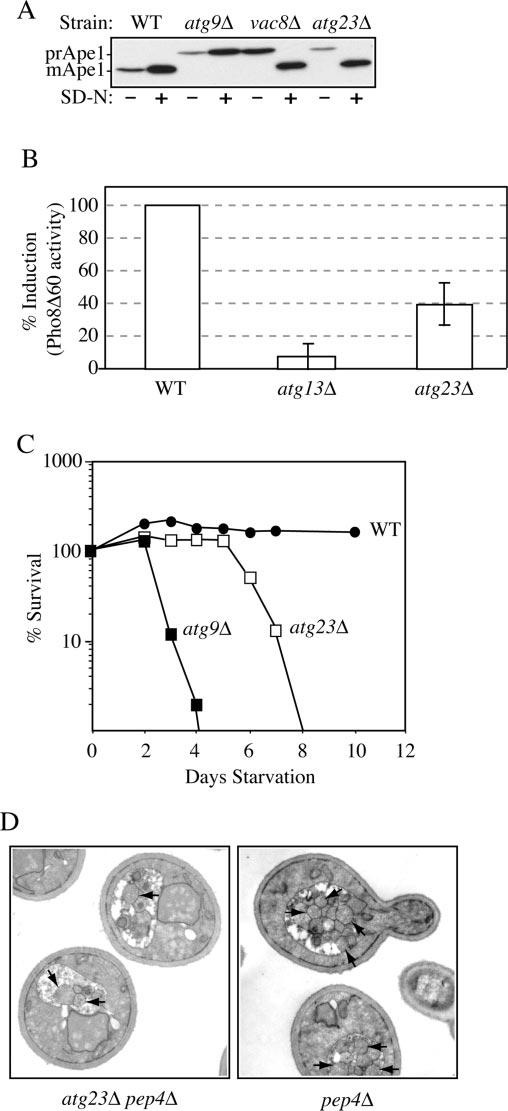
Atg23 is required for autophagy. A, precursor Ape1 matures in atg23Δ cells during starvation conditions. Wild type (WT) (SEY6210), atg23Δ (KTY14), atg9Δ (JKY007), and vac8Δ (D3Y102) cells were grown to A600 = 1.0 in SMD and then kept in SMD or starved in SD-N medium for 2 h. Protein extracts were prepared and subjected to immunoblot analysis with anti-Ape1 antiserum. B, autophagy is partially induced in atg23Δ cells. Wild type (TN124), atg23Δ (KTY9), and atg13Δ (D3Y103) cells were shifted from SMD to SD-N medium for 4 h. Autophagy was measured by the levels of Pho8Δ60 activity in whole cell protein extracts. Activity in the wild type strain was set to 100% and activity in the other strains normalized relative to wild type. Error bars represent the S.D. from three separate experiments. C, the atg23Δ cells exhibit intermediate starvation resistance. Wild type (SEY6210), atg23Δ (KTY14), and atg9Δ (JKY007) cells were grown in SMD to A600 = 1.0 and then shifted to SD-N. At the indicated day, viability was determined by removing aliquots, plating in triplicate, and counting the number of colonies per plate after 2–3 days growth. D, fewer autophagic bodies accumulate in atg23Δ pep4Δ cells during starvation conditions. Cells from the pep4Δ (TVY1) and pep4Δ atg23Δ (KTY22) strains were grown in SMD, then shifted to SD-N for 4 h, fixed in potassium permanganate, and processed for electron microscopy as described under “Experimental Procedures.” Arrows indicate autophagic bodies.
Although the ability of atg23Δ cells to mature prApe1 during starvation conditions indicated that this mutant was at least partly competent for autophagy, it did not necessarily mean that the process functioned normally. For example, Atg17/Apg17 is required for autophagy; however, the corresponding deletion mutant is able to process prApe1 during starvation conditions, presumably through the Cvt pathway (25, 29, 30). Accordingly, we performed additional experiments to analyze autophagy in the atg23Δ mutant. For a quantitative measure of autophagy, we utilized a truncated derivative of the vacuolar alkaline phosphatase (Pho8), Pho8Δ60, which lacks the trans-membrane and vacuolar targeting domains. In the absence of these sequences, Pho8Δ60 accumulates in the cytosol and is only transported to the vacuole by inclusion into autophagosomes. Once delivered to the vacuole, it is processed to the mature, active form (39). Thus, measuring Pho8Δ60 activity under starvation conditions provides a quantitative analysis of autophagy. The activity of Pho8Δ60 was measured in wild type, atg23Δ, and atg13Δ/apg13Δ cells expressing pho8Δ60 during starvation conditions (Fig. 2B). As expected, atg13Δ cells that are defective for autophagy exhibited no induction of Pho8Δ60 activity (40). In contrast, atg23Δ cells induced ∼40% of the amount of Pho8Δ60 activity compared with wild type. To extend our analysis of autophagy in the atg23Δ mutant, we analyzed the ability of the mutant to survive extended periods of starvation (Fig. 2C). Wild type cells survived nitrogen deprivation for at least 2 weeks because of their capacity for autophagy. In contrast, autophagy-defective mutants, such as atg9Δ, began to die rapidly upon nitrogen removal, becoming inviable after 3–4 days. In contrast to either of these extremes, atg23Δ mutants remained viable during starvation conditions for several days and began dying after ∼6–8 days. The intermediate starvation-resistance phenotype and partial induction of Pho8Δ60 activity both indicate that autophagy occurred in atg23Δ cells, unlike the case with mutants such as atg9Δ. However, in the atg23Δ mutant, it did not occur as efficiently as in wild type cells.
Autophagy allows for the nonspecific uptake of bulk cytoplasm into large vesicles called autophagosomes, which are delivered to the vacuole to generate an internal pool of nutrients that can be exploited for survival during prolonged periods of starvation (41, 42). Two possibilities exist for the phenotype of atg23Δ cells during starvation conditions, either a reduction in the size of the forming autophagosomes or reduced numbers of autophagosomes. To address these possibilities, we used electron microscopy to examine the ultrastructure of the autophagic bodies that accumulated in atg23Δ pep4Δ cells during starvation. Autophagic bodies are single membrane intravacuolar vesicles that result from the fusion of autophagosomes with the vacuole; once delivered to the lumen, they are degraded in a Pep4-dependent manner (42). As seen in Fig. 2D, pep4Δ atg23Δ mutants accumulated autophagic bodies of normal size within the vacuolar lumen under starvation conditions. Quantification of the numbers of autophagic bodies, however, revealed approximately a 70% reduction in the number of autophagic bodies accumulating in atg23Δ cells compared with wild type (10.5 ± 3.5 for pep4Δ cells compared with 3.18 ± 2.1 for pep4Δ atg23Δ cells, n = 50 vacuoles). This reduction correlated very well with the biochemical data (Fig. 2B) and indicated that atg23Δ mutants exhibit decreased efficiency of autophagy rather than a structural defect in autophagosome formation.
Finally, we wished to determine a role, if any, for Atg23 in pexophagy. Excess peroxisomes in S. cerevisiae can be delivered to the vacuole for degradation by using machinery that largely overlaps with the Cvt pathway and autophagy; most currently characterized atg mutants, including those specific for the Cvt pathway, are defective in this process (27). Cells were grown in oleic acid to induce peroxisome proliferation and then shifted to glucose-containing medium lacking nitrogen to induce maximum degradation of the superfluous organelles. We measured pexophagy in wild type, atg23Δ, and atg9Δ cells by following the degradation of the peroxisomal thiolase Fox3. As shown in Fig. 3, elimination of Fox3 in atg23Δ cells was comparable with wild type, whereas atg9Δ cells exhibited no such degradation. This indicates that Atg23 is not required for pexophagy. These results place the atg23Δ strain into a unique category of mutants that are defective in the Cvt and autophagy pathways but not in pexophagy.
Fig.3.
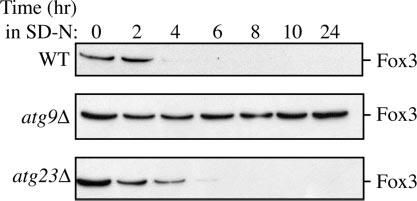
Pexophagy is normal in atg23Δ cells. Cells from wild type (WT), atg23Δ, and atg9Δ mutants in the BY4742 background were grown under peroxisome-inducing conditions and shifted to SD-N for 24 h. At the indicated times, protein extracts were prepared and subjected to immunoblot analysis using anti-Fox3 antiserum.
Atg23 Is Required for Cvt Vesicle Formation
To gain insight into Atg23 function, we decided to identify the step at which Atg23 acts in the Cvt pathway, utilizing the current model of Cvt cargo transport: Cargo selection, sequestration into intact Cvt vesicles, vesicle fusion with the limiting membrane of the vacuole, and maturation of the prApe1 cargo in the vacuolar lumen (reviewed in Ref. 43). Following oligomerization of prApe1 in the cytosol, the Ape1 complex binds to the Atg19/Cvt19 receptor to form the Cvt complex, which is subsequently recruited to the PAS in an Atg11-dependent step (26, 36). We first assayed whether atg23Δ cells were competent to recruit the Cvt complex to lipid bilayers by performing a membrane flotation experiment (Fig. 4A). Following spheroplast lysis, the majority of prApe1 in atg23Δ cells was found in the pellet fraction (P13). In contrast, almost all the cytosolic marker Pgk1 was found in the S13 supernatant fraction, indicating efficient lysis of spheroplasts. The prApe1-containing fraction was then subjected to centrifugation on a Ficoll step gradient as described under “Experimental Procedures.” Float fractions were recovered in the absence and presence of detergent and subjected to a Western blot analysis for prApe1. Due to their buoyancy, all lipids, membranes, and associated proteins will float to the top of the gradient; moreover, the addition of detergent disrupts the membranes and abolishes the flotation of membrane-bound proteins. All prApe1 recovered from the atg23Δ cells was found in the float fraction in the absence of detergent and not in the presence of detergent. This indicates that prApe1 is membrane- or lipid-associated in the atg23Δ strain.
Fig.4.
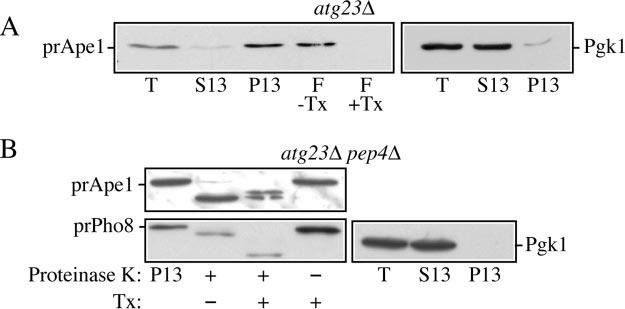
Atg23 functions at the stage of Cvt vesicle formation. A, precursor Ape1 is membrane-associated in atg23Δ cells. The P13 fraction from atg23Δ (KTY14) spheroplasts was collected and loaded on the bottom of a Ficoll step gradient in the presence or absence of the detergent Triton X-100 (Tx). After centrifugation, the float (F) fractions were collected and analyzed by immunoblot using anti-Ape1 antiserum as described under “Experimental Procedures.” Lysis conditions were verified by immunoblot analysis using anti-Pgk1. B, precursor Ape1 is protease-sensitive in atg23Δ pep4Δ cells. The 13,000 × g pellet (P13) fraction was collected from atg23Δ pep4Δ (KTY22) lysed spheroplasts, subjected to treatment with proteinase K in the presence or absence of Triton X-100 (Tx), and analyzed by immunoblot using anti-Ape1 antiserum as described under “Experimental Procedures.” Lysis conditions were verified by immunoblot analysis using anti-Pgk1 and anti-Pho8 antisera. T, total; S13, 13,000 × g supernatant fraction.
The next step in the Cvt pathway is the sequestration of prApe1 into intact Cvt vesicles. Accordingly, we next analyzed the ability of atg23Δ cells to complete the sequestration process through a protease-protection assay (Fig. 4B). If a mutant is blocked at the step of sequestration, Cvt vesicles will not close and prApe1 will accumulate in a form sensitive to exogenous protease following osmotic lysis of the spheroplast plasma membrane. However, if a mutant is competent for sequestration, this hydrolase will be protected from exogenous protease, becoming sensitive only upon membrane disruption by the addition of detergent (17). Spheroplasts were prepared and subjected to osmotic lysis. As before, efficient separation of Pgk1 into the supernatant fraction indicated efficient lysis of spheroplasts. The pelletable cellular fraction from atg23Δ pep4Δ mutants was subjected to treatment by proteinase K in the absence or presence of detergent. As shown in Fig. 4B, the accumulated prApe1 in atg23Δ pep4Δ cells was accessible to the exogenous proteinase K in the presence or absence of detergent. To verify that this sensitivity is not the result of nonspecific disruption of intracellular membranes, we simultaneously examined the protease sensitivity of endogenous Pho8. Precursor Pho8 is accumulated in the limiting membrane of the vacuole in pep4Δ mutants. The precursor form of Pho8 contains a small cytosolically oriented tail, whereas the major part of the protein, including the C-terminal propeptide, resides within the vacuolar lumen. Treatment with proteinase K in the absence of detergent allowed the cleavage of only the cytosolic tail (Fig. 4B). Coupled with the fractionation of Pgk1, this result indicated that the experimental conditions selectively disrupted the plasma membrane but maintained the integrity of the vacuole and presumably other intracellular compartments, i.e. protease sensitivity of prApe1 in atg23Δ cells was not due to osmotic lysis of completed Cvt vesicles. Upon the addition of detergent, all intracellular membranes were disrupted, and the entirety of Pho8 became accessible to the exogenous proteinase K. This can be seen as a second decrease in the molecular mass of Pho8 following SDS-PAGE and Western blot analysis resulting from removal of the C-terminal propeptide. These results indicate a sequestration defect in atg23Δ pep4Δ mutants.
Atg23 Is Not Necessary for Protein Recruitment to the PAS
To continue our analysis of the role of Atg23 in the Cvt and autophagy pathways, we decided to determine whether Atg23 is required for the recruitment of PAS-associated proteins. Although the exact nature and function of the PAS is not yet clear, it appears to play a physiological role in Cvt vesicle and autophagosome formation; many proteins involved in the Cvt pathway and autophagy have been shown to localize at least transiently to the PAS (19, 20). As shown in Fig. 5, wild type cells showed the typical co-localization of YFP-Atg8, YFPAtg11, Atg19-CFP, and prApe1 (CFP-Ape1) to the single punctate, perivacuolar dot that corresponds to the PAS. Similarly, atg23Δ mutants exhibited normal co-localization of these proteins at the PAS. This indicates that the function of Atg23 is not to organize protein recruitment to the PAS.
Fig.5.
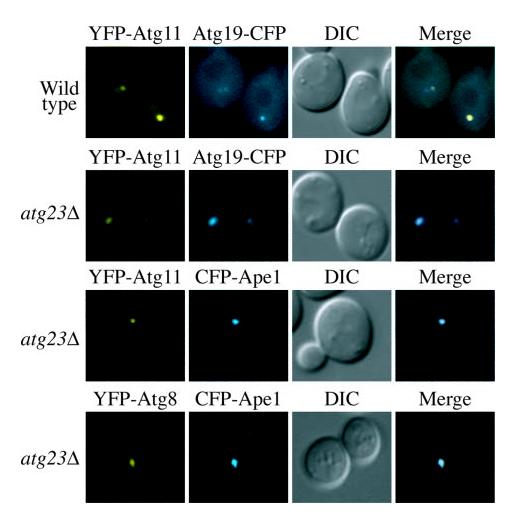
Atg23 is not required for the organization of proteins at the PAS. Wild type and atg23Δ (KTY14) cells were co-transformed with plasmids expressing YFP-Atg11 or YFP-Atg8 and Atg19-CFP or CFPApe1, grown in selective SMD medium to mid-log phase and visualized by fluorescence microscopy. DIC, differential interference contrast.
Atg23 Is a Peripheral Membrane Protein That Localizes to the PAS
In order to gain more information about Atg23, we decided to examine the biosynthesis of the protein. Many of the components involved in the Cvt and autophagy pathways are peripheral membrane proteins that specifically associate with the PAS; as the amino acid sequence of Atg23 yielded no obvious membrane-spanning domains, we hypothesized that this protein may also have similar characteristics. To monitor the localization of Atg23, we generated a functional C-terminal Myc fusion at the chromosomal ATG23 locus. Immunoblot analysis with anti-Myc antiserum detected a protein with the predicted molecular mass. The Myc tag did not interfere with Atg23 function because the strain with the integrated tag displayed normal maturation of prApe1 (data not shown). The strain expressing Atg23-Myc was subjected to subcellular fractionation into soluble and pelletable fractions. As shown in Fig. 6A, the majority of cellular Atg23-Myc was found in the soluble fraction (S100); however, a significant portion of the protein was also found in both the P13 and P100 fractions. In contrast, cytosolic Pgk1 was completely absent from the P13 pellet, again indicating efficient lysis of spheroplasts and separation of the pelletable and soluble fractions. This distribution of Atg23 is similar to other Atg proteins that are peripherally associated with membranes such as Atg2, Atg11, and Atg18 (28, 33, 44).
Fig.6.
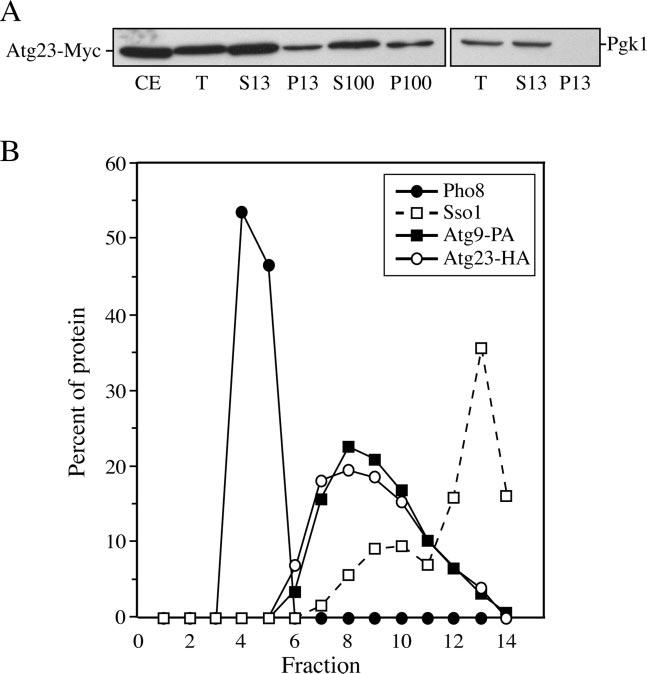
Atg23 exists in both soluble and membrane-associated pools. A, Atg23-Myc is present in both soluble and pelletable fractions by velocity gradient sedimentation. Atg23-Myc (KTY7) cells were converted to spheroplasts, lysed, and subjected to differential centrifugations. All fractions were separated by SDS-PAGE and membranes probed with monoclonal anti-Myc antibody. Lysis conditions were monitored by analysis of Pgk1. CE, whole cell protein extract; T, total spheroplast protein extract; S13, low speed supernatant; P13, low speed pellet; S100, high speed supernatant; P100, high speed pellet. B, Atg23-HA and Atg9 cofractionate at the PAS. Spheroplasts from cells expressing both Atg23-HA and Atg9-PA (KTY74) were osmotically lysed, resulting in total protein extracts, and the membranes were fractionated on a sucrose density gradient as described under “Experimental Procedures.” A total of 14 fractions was collected from the top of the gradient and resolved by SDS-PAGE and membranes probed with antisera to HA, PA, Pho8, and Sso1. Percent of protein per fraction was determined relative to the total protein extracts used as the starting material.
We then decided to separate intracellular membranes on a sucrose step density gradient in order to establish Atg23 localization. We generated a strain simultaneously expressing functional Atg9-protein A (Atg9-PA) and Atg23-hemagglutinin (Atg23-HA) fusions under the control of their authentic promoters. Total cell extracts were prepared and resolved on sucrose density gradients as described under “Experimental Procedures.” Atg9 has been shown to co-localize with other Atg proteins but not with other subcellular membranes by gradient analysis (19); therefore, it was used as a marker for the PAS. Both Atg9-PA and Atg23-HA were primarily concentrated in fractions 7–10, suggesting that these two proteins co-localize (Fig. 6B). The fractions containing Atg9 and Atg23 were well separated from the vacuole (Pho8) and the main plasma membrane peak (Sso1). This result suggests that, like Atg9, Atg23 localizes to the PAS.
Atg9 Is Essential to Recruit Atg23 to Membranes and Both Proteins Localize to Multiple Distinct Punctate Structures in the Cytoplasm
To follow the localization of Atg23 in vivo, we visualized the localization of this protein in living cells by fluorescence microscopy (Fig. 7A). The gene encoding YFP was integrated in-frame at the 3′ end of ATG23 at the chromosomal locus, and the resulting Atg23-YFP fusion protein was functional based on the ability of this strain to import prApe1 (data not shown). Cells expressing both Atg23-YFP and the PAS marker Atg19-CFP were grown to early log phase and imaged with a fluorescent microscope. Atg23-YFP localized to multiple, distinct structures dispersed throughout the cytoplasm and also displayed some diffuse cytoplasmic staining. This distribution was identical under both growth and starvation conditions (data not shown). Co-localization with Atg19-CFP indicated that one of those punctate structures was the PAS (Fig. 7A), consistent with the results from the gradient analysis. The localization pattern of Atg23 was unusual in that it resembled the pattern displayed by the transmembrane protein Atg9 (21), a pattern distinct from other Atg proteins.
Fig .7.
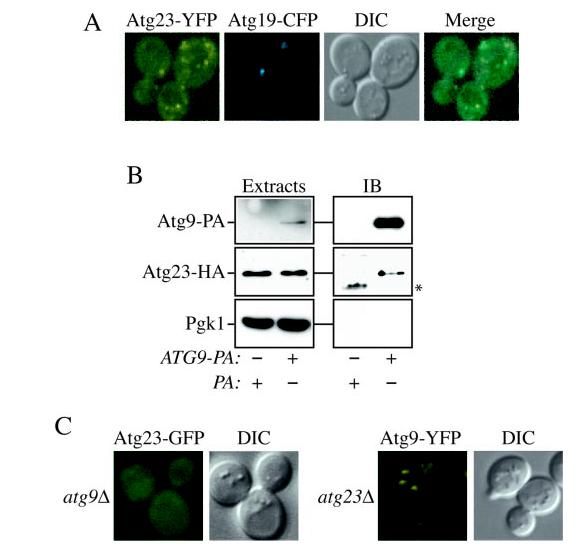
Atg23-GFP and Atg9-YFP exhibit similar localization as well as physical and functional interactions. A, Atg9 and Atg23 localize to several punctate structures dispersed in the cytoplasm. Cells containing Atg9-YFP and CFP-Atg8 integrated at their chromosomal loci (FRY134) or those containing both Atg23-YFP integrated at the chromosomal ATG23 locus (KTY26) and the plasmid bearing Atg19-CFP were grown in selective medium to A600 = 0.8–1.2 and imaged with a fluorescent microscope. B, Atg23 interacts with Atg9. Atg23-HA (KTY8) cells transformed with Atg23-HA and either Atg9-PA (KTY74) or pRS416-CuProtA as a control were used to prepare detergent-solubilized extracts as described under “Experimental Procedures.” IgG-Sepharose beads were used to affinity-purify the PA fusions together with the associated proteins. Eluted polypeptides were separated by SDS-PAGE and then visualized by immunoblotting (IB) with antiserum to PA, HA, and Pgk1. For each experiment, 0.013% of the total lysate or 4.45% of the total eluate was loaded per gel lane. The asterisk marks a contaminating band. C, Atg9 is required to recruit Atg23 to membranes. The atg9Δ mutant bearing an integrated Atg23-GFP (KTY32) or Atg9-YFP cells deleted for the ATG23 ORF (KTY51) were grown and imaged as in A. DIC, differential interference contrast.
Because of this similarity, we decided to examine if Atg23 and Atg9 associated with each other. Accordingly, we utilized the strain expressing the Atg9-PA and Atg23-HA fusions. These cells were converted to spheroplasts, lysed, and the PA chimera isolated using IgG-Sepharose beads. Bound complexes were released from the Sepharose by eluting with a low pH buffer, and the presence of Atg9-PA, Atg23-HA, and Pgk1 was tested by immunoblotting. As shown in Fig. 7B, Atg23 was selectively affinity-isolated by the Atg9-PA fusion, demonstrating an interaction between these two proteins. The presence of Atg23-HA in the eluates was not caused by nonspecific binding to the IgG-Sepharose or PA because the abundant cytosolic protein Pgk1 was not detected in the same samples, and PA alone did not pull down the Atg23-HA fusion.
We then examined if the association between Atg23 and Atg9 was important for their proper localization. Atg9-YFP was expressed in atg23Δ cells, and conversely, Atg23-GFP was examined in the atg9Δ strain. As shown in Fig. 7C, the absence of Atg23 did not interfere with normal Atg9-YFP distribution. In contrast, the punctate localization of Atg23-GFP was completely lost in the atg9Δ mutant and was instead found solely in a diffuse cytosolic pool. Reintroducing plasmid-based Atg9 restored the wild type localization of Atg23-GFP, indicating that its altered cellular distribution was due to the ATG9 deletion (data not shown). We concluded that Atg9 is essential to recruit Atg23 to membranes.
DISCUSSION
Atg23 Is Required for the Cvt Pathway and Efficient Autophagy but Not Pexophagy
The Cvt pathway and autophagy are two vacuolar trafficking pathways that serve drastically different purposes. The Cvt pathway is a biosynthetic trafficking pathway for a distinct subset of resident vacuolar hydrolases, whereas autophagy is a nonspecific degradative mechanism to allow cell survival during starvation. However, the two pathways are morphologically very similar and share a number of identical molecular components. To identify currently unknown genes required for these two pathways, we undertook a screen to identify deletion mutants defective for prApe1 import during growth conditions, and we identified a strain that was disrupted for the ATG23 gene. This gene codes for a 453-amino acid protein that contains three putative coiled-coil domains with clear homologues in the Saccharomyces fungus family but not in other eukaryotic organisms.
The characterization of the atg23Δ strain revealed a pheno-type distinct from all other reported Atg deletion mutants. We demonstrated that Atg23 is used by both the Cvt pathway and autophagy; however, unlike most previously characterized ATG genes, it is not absolutely required for autophagy (Fig. 2, A and B). Mutants lacking Atg23 can form morphologically normal autophagosomes and deliver them to the vacuole, but at too slow a rate to sustain viability during extended periods of starvation (Fig. 2, C and D). Excess peroxisomes in S. cerevisiae can be sequestered into double membrane-bound structures and delivered to the vacuole in a manner similar to autophagy. This process is in unaffected in the atg23Δ mutant (Fig. 3). Thus, atg23 falls into a relatively unique category of mutants that is not defective in pexophagy. This group includes atg19Δ, for example; ATG19 encodes the receptor for prApe1 import and also displays normal autophagy (36).
The gene products specific for the Cvt pathway can be divided in two classes. The first class, consisting of factors required to target cargo molecules to the PAS (26), are proteins with homologues only in other fungi (43). The second class includes components with a role in vesicular traffic; these proteins may be involved in mediating membrane delivery to the PAS (29, 37, 45). Precursor Ape1 is recruited to the PAS in atg23Δ cells (Fig. 5); thus ATG23 may not belong in the first class of genes. However, it cannot be excluded that Atg23 may modulate the interaction of the prApe1-Atg19-Atg11 complex with the rest of the Atg proteins once at the PAS. Interestingly, cells lacking subunits of the VFT tethering complex are defective in the Cvt pathway and have small autophagosomes and normal pexophagy (37). The strong similarity between these phenotypes and those of the atg23Δ mutant suggests that Atg23 may play an important role in mediating membrane delivery to the PAS. A final possibility is that Atg23 has two different functions in the Cvt pathway and autophagy, a concept not without precedent. Increasing evidence suggests that numerous proteins play similarly distinct roles in these two pathways. For example, Atg1 kinase activity may be required for the Cvt pathway, but Atg1 may carry out a non-kinase role in autophagy (24). Similarly, Atg8 is required for the formation of Cvt vesicles but for the expansion rather than the nucleation of autophagosomes (30). Like Atg8, Atg23 may be essential for the formation of vesicles in the Cvt pathway but not autophagy.
Atg23 Recruitment to Membranes Depends on Atg9
Many Atg proteins localize to a single punctate structure (19, 20). The analysis of a fluorescent Atg23 fusion revealed that this protein is concentrated to several punctate structures dispersed in the cytosol (Fig. 7A). Among all the Atg components, Atg9 is the only factor that displays this unique localization pattern (21). Only one of these structures corresponds to the PAS, indicating that these proteins are divided in two qualitatively different populations. Surprisingly, membrane separation by sucrose step gradients showed that Atg23 and Atg9 exclusively cofractionated in a single peak (Fig. 6B). It has been demonstrated previously that those Atg9-containing fractions also include several other Atg components, confirming our fluorescence microscopy data (19). We attempted to separate the two Atg9 and Atg23 populations by differential subcellular centrifugation and OptiPrep™ linear and sucrose step gradient fractionations, but we were unsuccessful (data not shown). This suggests that the punctate structures have a very similar, if not identical, density and size.
Because of the similarity between Atg23 and Atg9 localization, we investigated if those two molecules were associated. Affinity isolation experiments demonstrated that Atg23 can indeed bind Atg9 (Fig. 7B). Cells contain both a soluble and a membrane-bound pool of Atg23 suggesting that it is a peripheral membrane protein (Fig. 6A). Interestingly, in the absence of Atg9, Atg23 lost its punctate localization, becoming completely dispersed (Fig. 7C). The simplest hypothesis would be that Atg23 is associated with membranes via its binding to the integral membrane protein Atg9. However, three separate results indicate that Atg9 catalyzes Atg23 recruitment to membranes in a process temporally and locally restricted to the PAS. First, Atg9 is accumulated at the PAS in the atg2Δ, atg18Δ, and atg14Δ mutants, whereas Atg23 has normal distribution.2 That result indicates that the Atg23 pool not localizing to the PAS does not require the physical presence of Atg9 to remain membrane-associated. Second, Atg23 was affinity-isolated by Atg9 in atg1Δ cells when both proteins were restricted to the PAS.2 Third, Atg23 is completely cytosolic in the atg1Δ atg9Δ double deletion mutant showing that Atg9 mediates the targeting of Atg23 to the PAS (data not shown). The conclusion that Atg23 and Atg9 do not form a functional unit is also supported by the difference in phenotypes between the corresponding deletion mutants. For example, the Cvt pathway, autophagy, and pexophagy are completely blocked in atg9Δ cells (21) (Fig. 2A and Fig. 3). In addition, Atg8 is not concentrated at the PAS in the atg9Δ mutant, whereas it normally localizes to this structure in atg23Δ cells (20) (Fig. 5). It remains to be established if the pools of Atg9 and Atg23 not localizing to the PAS are in the same location. Our co-localization attempts with YFP and CFP fusions of these two proteins failed because of the weak signal.3
Future work will try to identify other Atg23-binding partners in order to unveil the role played by this protein in the Cvt pathway and autophagy. In addition, the study of the relationship between the Atg23 pools at the PAS and at the other punctate structures will provide insights about both the origin and the delivery mechanism of the membranes composing the double-membrane vesicles that are the hallmark of the Cvt and autophagy pathways.
Acknowledgments
We thank Drs. Wei-Pang Huang and Per Stromhaug for valuable discussions.
Footnotes
This work was supported in part by United States Public Health Service Grant GM53396 from the National Institutes of Health (to D. J. K.) and National Science Foundation Grant MCB-9817002 (to W. A. D.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: Cvt, cytoplasm to vacuole targeting; PAS, pre-autophagosomal structure; ORF, open reading frame; HA, hemagglutinin; PA, protein A; GFP, green fluorescent protein; YFP, yellow fluorescent protein; PIPES, 1,4-piperazinediethanesulfonic acid.
F. Reggiori, K. A. Tucker, and D. J. Klionsky, submitted for publication.
Katherine A. Tucker, F. Reggiori, W. A. Dunn, Jr., and D. J. Klionsky, unpublished observations.
REFERENCES
- 1.Bryant NJ, Stevens TH. Microbiol. Mol. Biol. Rev. 1998;62:230–247. doi: 10.1128/mmbr.62.1.230-247.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khalfan WA, Klionsky DJ. Curr. Opin. Cell Biol. 2002;14:468–475. doi: 10.1016/s0955-0674(02)00343-5. [DOI] [PubMed] [Google Scholar]
- 3.Klionsky DJ, Ohsumi Y. Annu. Rev. Cell Dev. Biol. 1999;15:1–32. doi: 10.1146/annurev.cellbio.15.1.1. [DOI] [PubMed] [Google Scholar]
- 4.Stromhaug PE, Klionsky DJ. Traffic. 2001;2:524–531. doi: 10.1034/j.1600-0854.2001.20802.x. [DOI] [PubMed] [Google Scholar]
- 5.Klionsky DJ, editor. Autophagy. Landes Bioscience; Georgetown: 2004. pp. 1–327. TX. [Google Scholar]
- 6.Klionsky DJ, Emr SD. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ogier-Denis E, Codogno P. Biochim. Biophys. Acta. 2003;1603:113–128. doi: 10.1016/s0304-419x(03)00004-0. [DOI] [PubMed] [Google Scholar]
- 8.Nixon RA, Cataldo AM, Mathews PM. Neurochem. Res. 2000;25:1161–1172. doi: 10.1023/a:1007675508413. [DOI] [PubMed] [Google Scholar]
- 9.Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Histol. Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 10.Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, Sue CM, Yamamoto A, Murakami N, Shanske S, Byrne E, Bonilla E, Nonaka I, DiMauro S, Hirano M. Nature. 2000;406:906–910. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka Y, Guhde G, Suter A, Eskelinen E-L, Hartmann D, Lullmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P. Nature. 2000;406:902–906. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- 12.Tsukada M, Ohsumi Y. FEBS Lett. 1993;333:169–174. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- 13.Thumm M, Egner R, Koch B, Schlumpberger M, Straub M, Veenhuis M, Wolf DH. FEBS Lett. 1994;349:275–280. doi: 10.1016/0014-5793(94)00672-5. [DOI] [PubMed] [Google Scholar]
- 14.Scott SV, Baba M, Ohsumi Y, Klionsky DJ. J. Cell Biol. 1997;138:37–44. doi: 10.1083/jcb.138.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klionsky DJ, Cregg JM, Dunn WA, Jr., Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, Ohsumi Y. Dev. Cell. 2003;5:539–545. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 16.Harding TM, Hefner-Gravink A, Thumm M, Klionsky DJ. J. Biol. Chem. 1996;271:17621–17624. doi: 10.1074/jbc.271.30.17621. [DOI] [PubMed] [Google Scholar]
- 17.Harding TM, Morano KA, Scott SV, Klionsky DJ. J. Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baba M, Osumi M, Scott SV, Klionsky DJ, Ohsumi Y. J. Cell Biol. 1997;139:1687–1695. doi: 10.1083/jcb.139.7.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim J, Huang W-P, Stromhaug PE, Klionsky DJ. J. Biol. Chem. 2002;277:763–773. doi: 10.1074/jbc.M109134200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. EMBO J. 2001;20:5971–5981. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noda T, Kim J, Huang W-P, Baba M, Tokunaga C, Ohsumi Y, Klionsky DJ. J. Cell Biol. 2000;148:465–480. doi: 10.1083/jcb.148.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Longtine MS, McKenzie A, III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 23.Drees BL, Sundin B, Brazeau E, Caviston JP, Chen GC, Guo W, Kozminski KG, Lau MW, Moskow JJ, Tong A, Schenkman LR, McKenzie A, III, Brennwald P, Longtine M, Bi E, Chan C, Novick P, Boone C, Pringle JR, Davis TN, Fields S, Drubin DG. J. Cell Biol. 2001;154:549–571. doi: 10.1083/jcb.200104057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abeliovich H, Zhang C, Dunn WA, Jr., Shokat KM, Klionsky DJ. Mol. Biol. Cell. 2003;14:477–490. doi: 10.1091/mbc.E02-07-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. J. Cell Biol. 2000;150:1507–1513. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shintani T, Huang W-P, Stromhaug PE, Klionsky DJ. Dev. Cell. 2002;3:825–837. doi: 10.1016/s1534-5807(02)00373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hutchins MU, Veenhuis M, Klionsky DJ. J. Cell Sci. 1999;112:4079–4087. doi: 10.1242/jcs.112.22.4079. [DOI] [PubMed] [Google Scholar]
- 28.Kim J, Kamada Y, Stromhaug PE, Guan J, Hefner-Gravink A, Baba M, Scott SV, Ohsumi Y, Dunn WA, Jr., Klionsky DJ. J. Cell Biol. 2001;153:381–396. doi: 10.1083/jcb.153.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nice DC, Sato TK, Stromhaug PE, Emr SD, Klionsky DJ. J. Biol. Chem. 2002;277:30198–30207. doi: 10.1074/jbc.M204736200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abeliovich H, Dunn WA, Jr., Kim J, Klionsky DJ. J. Cell Biol. 2000;151:1025–1034. doi: 10.1083/jcb.151.5.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Black MW, Pelham HRB. J. Cell Biol. 2000;151:587–600. doi: 10.1083/jcb.151.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siniossoglou S, Wimmer C, Rieger M, Doye V, Tekotte H, Weise C, Emig S, Segref A, Hurt EC. Cell. 1996;84:265–275. doi: 10.1016/s0092-8674(00)80981-2. [DOI] [PubMed] [Google Scholar]
- 33.Wang C-W, Kim J, Huang W-P, Abeliovich H, Stromhaug PE, Dunn WA, Jr., Klionsky DJ. J. Biol. Chem. 2001;276:30442–30451. doi: 10.1074/jbc.M102342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scott SV, Hefner-Gravink A, Morano KA, Noda T, Ohsumi Y, Klionsky DJ. Proc. Natl. Acad. Sci. U. S. A. 1996;93:12304–12308. doi: 10.1073/pnas.93.22.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim J, Scott SV, Oda MN, Klionsky DJ. J. Cell Biol. 1997;137:609–618. doi: 10.1083/jcb.137.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scott SV, Guan J, Hutchins MU, Kim J, Klionsky DJ. Mol. Cell. 2001;7:1131–1141. doi: 10.1016/s1097-2765(01)00263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reggiori F, Wang C-W, Stromhaug PE, Shintani T, Klionsky DJ. J. Biol. Chem. 2003;278:5009–5020. doi: 10.1074/jbc.M210436200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott SV, Nice DC, III, Nau JJ, Weisman LS, Kamada Y, Keizer-Gunnink I, Funakoshi T, Veenhuis M, Ohsumi Y, Klionsky DJ. J. Biol. Chem. 2000;275:25840–25849. doi: 10.1074/jbc.M002813200. [DOI] [PubMed] [Google Scholar]
- 39.Noda T, Matsuura A, Wada Y, Ohsumi Y. Biochem. Biophys. Res. Commun. 1995;210:126–132. doi: 10.1006/bbrc.1995.1636. [DOI] [PubMed] [Google Scholar]
- 40.Funakoshi T, Matsuura A, Noda T, Ohsumi Y. Gene (Amst.) 1997;192:207–213. doi: 10.1016/s0378-1119(97)00031-0. [DOI] [PubMed] [Google Scholar]
- 41.Baba M, Takeshige K, Baba N, Ohsumi Y. J. Cell Biol. 1994;124:903–913. doi: 10.1083/jcb.124.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. J. Cell Biol. 1992;119:301–311. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reggiori F, Klionsky DJ. Eukaryot. Cell. 2002;1:11–21. doi: 10.1128/EC.01.1.11-21.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guan J, Stromhaug PE, George MD, Habibzadegah-Tari P, Bevan A, Dunn WA, Jr., Klionsky DJ. Mol. Biol. Cell. 2001;12:3821–3838. doi: 10.1091/mbc.12.12.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abeliovich H, Darsow T, Emr SD. EMBO J. 1999;18:6005–6016. doi: 10.1093/emboj/18.21.6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson JS, Klionsky DJ, Banta LM, Emr SD. Mol. Cell. Biol. 1988;8:4936–4948. doi: 10.1128/mcb.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gerhardt B, Kordas TJ, Thompson CM, Patel P, Vida T. J. Biol. Chem. 1998;273:15818–15829. doi: 10.1074/jbc.273.25.15818. [DOI] [PubMed] [Google Scholar]