

Abstract
Autophagy, pexophagy, and the Cvt pathway are processes that deliver hydrolytic enzymes and substrates to the yeast vacuole/lysosome via double-membrane cytosolic vesicles. Whereas these pathways operate under different nutritional conditions, they all employ common machinery with only a few specific factors assisting in the choice of the delivery program and the membrane source for the sequestering vesicle. We found that the YKR020w gene product is essential for Cvt vesicle formation but not for pexophagy or induction of autophagy. Autophagosomes in the ykr020wΔ mutant, however, have a reduced size. We demonstrate that Ykr020 is a subunit of the Vps fifty-three tethering complex, composed of Vps52, Vps53, and Vps54, which is required for retrograde traffic from the early endosome back to the late Golgi, and for this reason we named it Vps51. This complex participates in a fusion event together with Tlg1 and Tlg2, two SNAREs also shown to be necessary for Cvt vesicle assembly. In particular, those factors are essential to correctly target the prApe1-Cvt19-Cvt9 complex to the preautophagosomal structure, the site of Cvt vesicle formation.
Autophagy is a catabolic process conserved among yeast, plants, and animal cells that permits the cell to eliminate unwanted or unnecessary proteins and organelles and to recycle the components for reuse (1, 2). The organellar turnover is exclusively accomplished in the lysosome/vacuole lumen by a wide range of hydrolases capable of breaking down all cellular constituents (1, 2). Autophagy plays an essential role during normal physiological processes such as starvation, cellular differentiation, cell death, and aging, but also in preventing some types of cellular dysfunction including cancer (2).
Studies in the yeast Saccharomyces cerevisiae have led to the identification of a large number of molecular components that form the autophagic machinery (1). Interestingly, most of these proteins are also utilized for the cytoplasm to vacuole targeting (Cvt)1 pathway (3, 4), which assures the delivery of the resident vacuolar hydrolase aminopeptidase I (Ape1) (5, 6). The same components are also required for peroxisome degradation, or pexophagy (7). These various processes operate under different nutritional conditions, but biochemical and morphological analyses have shown that in all cases the cargo material (pre-cursor Ape1 (prApe1), bulk cytoplasm or a specific organelle) is sequestered by a cytosolic double-membrane vesicle (7-11). The basic mechanism that leads to the formation of this structure, called an autophagosome, Cvt vesicle, or pexophagosome, is identical in all three pathways and it can be divided into five discrete steps: vesicle induction/nucleation, cargo selection/packaging, vesicle formation/completion, docking/fusion with the vacuole, and subvacuolar vesicle breakdown (1, 2). In the case of pexophagy and the Cvt pathway, the cargo may be specifically targeted to the sequestering membrane where it starts to be enwrapped by a double lipid bilayer. This process leads to the creation of the cytosolic double membrane vesicle. The completed vesicle docks with the lysosome/vacuole and successively fuses with it. In this way the inner vesicle is liberated into the lysosome/vacuole lumen where it is finally consumed by hydrolases.
Cellular signals dictate the selection of the cargo material but also the size of the forming vesicle (9, 12, 13). The serine/threonine protein kinase Apg1 and its interacting partner Apg13 are two components that play a part in all three pathways. These proteins seem to have a central role in determining the specific cellular response to nutrient conditions (4, 7, 13-15). Phosphorylation and dephosphorylation reactions mediate the association of Apg1 and Apg13 (13) creating a modular core complex able to interact with factors such as Apg17, Cvt9, and Vac8 that are specific only for one or two pathways (13, 16-18) (Table II).
Table II.
Specific factors required for autophagy, pexophagy, and Cvt pathway A plus or a minus mark indicates whether the protein is required for a pathway.
| Protein | Autophagy | Cvt Pathway | Pexophagy |
|---|---|---|---|
| Apg17 | + | − | NDa |
| Cvt9 | − | + | + |
| Vac8 | − | + | +b |
| Tlg1/Tlg2/Vps45 | − | + | ND |
| Cvt13/Cvt20 | − | + | + |
| Etf1 | − | + | ND |
| VFT complex | − | + | − |
| Sec12/Sec16/Sec23/Sec24 | + | − | ND |
ND, not determined.
P. Stromhaug and D. J. Klionsky, unpublished observation.
The rest of the components involved in the biogenesis of autophagosomes and Cvt vesicles include two conjugation systems that lead to the covalent linkage of the ubiquitin-like protein Aut7 to a molecule of phosphatidylethanolamine and the formation of a multimeric complex composed of Apg12-Apg5 and Apg16 (19). In addition, an autophagy-specific phosphatidylinositol (PtdIns) 3-kinase complex is involved in the synthesis of PtdIns(3)P that may serve to recruit downstream effectors that function in autophagy and the Cvt pathway (1, 20-23).
These shared factors and all the regulatory elements localize to a punctate perivacuolar organelle, also called the preautophagosomal structure (PAS), that is believed to be the formation site of autophagosomes and Cvt vesicles (24-26). Most of the autophagy (Apg)/Cvt proteins are cytosolic and achieve their correct localization by interaction with other factors or by specific binding to lipids such as phosphatidylethanolamine or PtdIns(3)P (20, 23, 24, 27). Several lines of evidence suggest that the source of the sequestering vesicles for autophagy and the Cvt pathway differ at least in part. For example, Aut7 is needed for nucleation of Cvt vesicles but not autophagosomes; Aut7 is needed for expansion of the autophagosomal membrane (12). Similarly, Apg1 appears to have different roles in these pathways; a catalytic function is needed for the Cvt pathway, but Apg1 may have a nonkinase role in inducing autophagy (28). With the exception of the proteins interacting with the Apg1-Apg13 complex, the rest of the pathway-specific factors are components of vesicular traffic machineries (Table II). Autophagy, but not the Cvt pathway, requires the GTP exchange factors, Sec12 and Sec16, and the two COPII coat subunits, Sec23 and Sec24 (29). That seems to correlate with studies in mammalian cells indicating that autophagosomes are derived from the endoplasmic reticulum (30). However, the tSNARE Tlg2, the vSNARE Tlg1, and the Sec1 homologue, Vps45, are essential for the formation of Cvt vesicles but not for autophagosome biogenesis (31). That is also true for three PtdIns(3)P-binding proteins, the two sorting nexins Cvt13 and Cvt20 plus the transmembrane protein Etf1 (23, 32).
SNARE-mediated fusion events employ additional proteins called tethering factors that play a role essential in the specificity and coordination of those reactions (33, 34). In the present study, we have identified the fourth subunit of the VFT tethering complex that, together with Tlg2 and Tlg1, is required for a retrieval transport step back to the late Golgi that is essential for the recycling of the vSNARE Snc1, but also for the completion of Cvt vesicles. We also show that in the absence of those fusion factors, prApe1 and its receptor, Cvt19, together with Cvt9 are not correctly targeted to the PAS.
EXPERIMENTAL PROCEDURES
Strains and Growth Media
The S. cerevisiae knockout strains in the BY4742 background used in this study (ykr020wΔ, vps52Δ, vps54Δ, cvt9Δ, apg9Δ, vac8Δ, apg1Δ, vps4Δ, vps5Δ, vps27Δ, vps29Δ, tlg2Δ and ccz1Δ) were from ResGen™ (Invitrogen, Carlsbad, CA). A vps53Δ homozygous diploid strain obtained from the same company was sporulated and dissected to obtain the VPS53 deletion in a similar background. The rest of the employed strains are listed in Table I. For YKR020w and VPS52 gene disruptions, the entire coding regions were replaced with either the URA3 gene from Kluyveromyces lactis flanked by coliphage loxP sites or the HIS5 gene of Schizosaccharomyces pombe, using PCR primers containing ∼40 bases of identity to the regions flanking the open reading frame. The vac8Δ::URA3 disruption cassette, generously provided by Dr. Lois Weisman (University of Iowa), was digested with AflII and EcoRI and the reaction mixture was then used to transform the vps52Δ strain. Double deletants were selected by verifying the absence of Vac8 in the cell extracts with anti-Vac8 immunoblots (18).
Table I.
Saccharomyces cerevisiae strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| BY4742 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | ResGen™ |
| SEY6210 | MATα his3-Δ200 leu2–3,112 lys2–801 ura3–52 trp1-Δ901 suc2-Δ9 | Ref. 49 |
| TN124 | MATa leu2–3,112 ura3–52 trp1 pho8::pho8Δ60 pho13Δ::LEU2 | Ref. 56 |
| D3Y103 | TN124 apg13Δ::URA3 | Ref. 17 |
| FRY107 | MATa his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 vps53Δ::kanMX4 | This study |
| FRY116 | SEY6210 VPS53–3xHA::TRP1 vps52Δ::URA3 K.l. LoxP | This study |
| FRY117 | SEY6210 VPS53–3xHA::TRP1 ykr020wΔ::URA3 K.l. LoxP | This study |
| PSY116 | SEY6210 YKR020w-13xMyc::TRP1 | This study |
| FRY119 | BY4742 vps52Δ::kanMX4 vac8Δ::URA3 | This study |
| FRY122 | TN124 ykr020wΔ::URA3 K.l. LoxP | This study |
| FRY123 | TN124 vps52Δ::URA3 K.l. LoxP | This study |
| FRY124 | SEY6210 vam3Δ::LEU2 ykr020wΔ::HIS5 S.p. LoxP + pVAM3–6.414 | This study |
| FRY125 | SEY6210 vam3Δ::LEU2 vps52Δ::URA3 K.l. LoxP + pVAM3–6.414 | This study |
| FRY126 | SEY6210 ykr020wΔ::HIS5 S.p. LoxP | This study |
| PSY113 | SEY6210 vps52Δ::HIS5 S.p. | This study |
| PSY118 | SEY6210 VPS52–3xHA::TRP1 | This study |
| PSY119 | SEY6210 VPS53–3xHA::TRP1 | This study |
| PSY120 | SEY6210 VPS54–3xHA::HIS5 S.p | This study |
| FRY118 | SEY6210 YKR020w-13xMyc::TRP1 vps52Δ::URA3 K.l. LoxP | This study |
| TDY2 | SEY6210 vam3Δ::LEU2 + pVAM3–6.414 | Ref. 57 |
PCR-based integrations of the triple HA tag and the 13 × Myc tag at the 3′ end of YKR020w, VPS52, VPS53, and VPS54 were used to generate strains expressing fusion proteins under the control of their native promoters. The templates for integration were pFA6a-3HATRP1, pFA6a-13Myc-His3MX6, and pFA6a-13Myc-TRP1 (35). Normal prApe1 processing and vacuolar morphology were used to confirm the functionality of all genomic fusions.
Strains were grown in YPD (1% yeast extract, 2% peptone and 2% glucose) or synthetic minimal medium (SMD; 0.67% yeast nitrogen base without amino acids, 2% glucose, and auxotrophic amino acids as needed). Nitrogen starvation was carried out in SD-N medium (0.17% yeast nitrogen base without amino acids and ammonium sulfate and 2% glucose).
Plasmids
YKR020w flanked by MfeI sites was generated by PCR and cloned into the EcoRI site of the pRS416-CuProtA vector (25) behind sequences expressing two IgG binding domains of protein A (PA) and the CUP1 promoter, and before the CYC1 terminator. The new plasmid was called pCuPAYKR020(416). This construction was also transferred as a KpnI-SacI fragment into a pRS414 plasmid (36) creating pCuPAYKR020(414). All enzymes for manipulation of DNA were from New England Biolabs (Beverly, MA). Plasmids expressing PA (pRS416-CuProtA), GFP-Snc1 (pGS416), GFP-Ape1 (pTS466), CFPApe1 (pTS470), Cvt19-CFP (pCVT19CFP(414)), YFP-Aut7 (pRS414EYFP-Aut7), CFP-Aut7 (pRS316ECFP-Aut7), GFP-Cvt9 (pTS495 and pCuGFPCVT9(416)), YFP-Cvt9 (pPS97), and CFP-Cvt9 (pPS98) have been described elsewhere (24, 25, 37, 38).
Protein Extraction and Western Blot
Cells were inoculated and grown in YPD overnight to early log phase (A600 = 0.6). Cells from this preculture were then either grown again in YPD or nitrogen starved in SD-N medium for 3 h. 1 A600 unit of cells was collected by centrifugation and proteins were precipitated with 500 μl of ice-cold 10% trichloroacetic acid for 30 min. After spinning the samples for 5 min, pellets were washed once with acetone. Pellets were air dried, resuspended in 100 μl of MURB buffer (50 mm Na2HPO4, 25 mm MES, pH 7.0, 1% SDS, 3m urea, 0.5% 2-mercaptoethanol, 1 mm NaN3, and 0.05% bromphenol blue), and heated at 75 °C for 10 min. Aliquots of 10 μl were loaded on 8% SDS-PAGE gels and after Western blotting, membranes were probed with anti-Ape1 polyclonal antiserum (6).
Fluorescence Microscopy
Cells grown to early logarithmic (log) phase in SMD medium or shifted to SD-N for 3 h, were prepared for fluorescence and stained with FM 4-64 (Molecular Probes, Eugene, OR) as described previously (39). Fluorescence signals were visualized with the use of a Nikon E-800 fluorescent microscope (Mager Scientific, Dexter, MI). The images were captured with an ORCA II CCD camera (Hamamatsu, Bridgewater, NJ) with the use of Openlab software (Im-provision, Lexington, MA).
Protein A Affinity Isolation
Cells were first grown overnight in SMD medium, then diluted with YPD and grown for an 3 additional hours. 50 A600 units of cells were harvested, converted to spheroplasts, and kept frozen. Spheroplasts were resuspended and Dounce homogenized in 2 ml of lysis buffer (40) containing 2 mm phenylmethylsulfonyl fluoride. Cell lysates were then centrifuged at 13,000 × g for 15 min and 1.6 ml of supernatant was incubated for 2 h at 4 °C with 20 μl of prewashed IgG Sepharose beads (Amersham Biosciences). Beads were then washed twice with lysis buffer (40), once with lysis buffer containing 300 mm KCl, once with lysis buffer containing 500 mm KCl, then once again with lysis buffer containing 300 mm KCl, and finally 3 times with the initial buffer. Finally, beads were heated at 75 °C for 10 min in 50 μl of MURB buffer. After SDS-PAGE and Western blot, membranes were probed with anti-HA monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
For cross-linking experiments, spheroplasts were prepared as above but not frozen. Instead, they were resuspended in 200 μl of phosphate buffer (25 mm potassium phosphate, pH 7.4, 200 mm sorbitol, 20 mm phenylmethylsulfonyl fluoride, 10× Complete EDTA-free protease inhibitor mixture (Roche Molecular Biochemicals, Indianapolis, IN)) containing 1.5 mm dithiobis(succinimidyl propionate) (Pierce). Suspensions were incubated for 30 min at room temperature. To quench the cross-linker, 200 μl of ice-cold 200 mm Tris-HCl (pH 7.4) were added and tubes were transferred to 4 °C for 5 min. Finally, 1.6 ml of ice-cold dilution solution (187.5 mm KCl, 6.25 mm MgCl2, 1.25% Triton X-100) was added and after Dounce homogenization, protein A affinity isolation was performed as above. After SDS-PAGE and Western blot, membranes were probed with anti-HA, anti-Tlg1 (41), anti-Pep12 (Molecular Probes), or anti-Sed5 (42) antiserum or antibodies.
Miscellaneous Procedures
The analyses of protease sensitivity, Prc1 missorting, cell viability under nitrogen starvation conditions, pexophagy, Pho8Δ60 activity, and electron microscopy using Spurr's resin (Ted Pella, Redding, CA) for embedding were conducted as described previously (7, 23, 27, 43).
RESULTS
The ykr020wΔ, vps52Δ, vps53Δ, and vps54Δ Mutants Block Precursor Ape1 Maturation Only Under Rich Growth Conditions
We identified the YKR020w gene in a genomic approach where nonessential gene deletions in the yeast S. cerevisiae were scored for a defect in precursor Ape1 (prApe1) maturation. The ykr020wΔ cells grown in rich medium accumulate prApe1, indicating a defect in the Cvt pathway (Fig. 1A). Certain apg and cvt mutants that are defective in prApe1 transport under vegetative conditions are able to import the protein into the vacuole when autophagy is induced by starvation. For example, mutants specific for the Cvt pathway such as cvt9, vac8, cvt13, and cvt20 or mutants that block autophagosome expansion but not induction such as aut7 are able to mature prApe1 when shifted to starvation conditions (12, 16, 17, 23). We examined processing of prApe1 in the ykr020wΔ mutant following induction of autophagy. The ykr020wΔ strain displayed normal processing of prApe1 when this strain was shifted to a nitrogen starvation medium (Fig. 1A), indicating that autophagic induction is not impaired.
Fig 1.

The ykr020wΔ, vps52Δ, vps53Δ, and vps54Δ cells have a defect in Cvt vesicle completion. A, The ykr020wΔ, vps52Δ, vps53Δ, and vps54Δ strains have a reversible inhibition of prApe1 processing similar to that of vac8Δ cells. Wild type (WT), ykr020wΔ, vps52Δ, vps53Δ (FRY107), vps54Δ, apg9Δ, and vac8Δ cells in the BY4742 background grown either in YPD or nitrogen starved in SD-N medium for 4 h were trichloroacetic acid precipitated. Acetone-washed proteins were then resolved by SDS-PAGE and prApe1 maturation analyzed by immunoblot. B, Prc1 is mislocalized to the periplasmic space in the ykr020wΔ mutant similarly to vps52Δ cells. Cells from wild type, ykr020wΔ, and vps52Δ strains in the BY4742 background were pulse-labeled for 10 min and chased for 30 min. Internal (I) and external (E) Prc1 was immunoprecipitated and then resolved by SDS-PAGE. C, The ykr020wΔ, vps52Δ, vps53Δ, and vps54Δ strains have a reversible accumulation of GFP-prApe1 in a cytosolic punctate structure. WT, ykr020wΔ, vps52Δ, vps53Δ (FRY107), and vps54Δ strains in the BY4742 background were transformed with a plasmid expressing the N-terminal GFP-tagged prApe1 (pTS466). Transformed cells were grown either in SMD or nitrogen starved for 3 h in SD-N medium and examined with a fluorescence microscope. DIC, differential interference contrast. D, precursor Ape1 is protease-sensitive in the vps52Δ mutant. The vps52Δ, vps54Δ, ykr020wΔ, and ccz1Δ strains in the BY4742 background were grown in YPD until early log phase. The cells were converted to spheroplasts and lysed. The total cells lysates (T) were centrifuged at 13,000 rpm for 5 min to separate the pellet (P13) and the supernatant (S13) fractions. The P13 fraction was resuspended in lysis buffer (27) and then mixed with equal volumes of lysis buffer, 40 μg/ml proteinase K, 0.4% Triton X-100, or proteinase K plus 0.4% Triton X-100, and incubated on ice for 30 min. The reactions were stopped by adding trichloroacetic acid. Samples were resolved by SDS-PAGE and examined by immunoblot with serum to Ape1. The ccz1Δ control cells accumulate completed Cvt vesicles and prApe1 is accessible to proteinase K only in the presence of Triton X-100. Even in the absence of detergent, prApe1 in the vps52Δ strain was digested to its mature form by the same protease indicating that Cvt vesicles were not completely assembled. Essentially identical results were obtained for the vps52Δ and ykr020wΔ strains. The T, S13, and P13 fractions were also probed for the cytosolic marker Pgk1. Recovery of Pgk1 in the S13 fractions indicates efficient lysis of spheroplasts.
A genome-wide approach for the identification of yeast protein complexes by mass spectrometry indicated a putative interaction between Ykr020 and Vps52/Sac2 (44). Vps52 together with Vps53 and Vps54 form the so-called VFT complex, a putative tethering factor required for the retrieval of proteins from the early endosome to the late Golgi (33, 45). The vps52Δ and vps54Δ mutants were also identified in our screen that detected the prApe1 defect in the ykr020wΔ strain. Furthermore, the vps52Δ and vps54Δ cells displayed a similar property in reverting the prApe1 maturation defect after nitrogen starvation (Fig. 1A). In contrast, vps53Δ cells showed normal prApe1 processing in all media. However, further analysis of the commercial vps53Δ haploid strain supplied by ResGen indicated that the deleted gene was not VPS53. To analyze the prApe1 phenotype in the vps53Δ strain, we sporulated the ResGen vps53Δ homozygous diploid strain. The resulting haploid spores showed the same reversible block in prApe1 maturation that was observed for the ykr020wΔ, vps52Δ, and vps54Δ strains (Fig. 1A).
A defect in prApe1 maturation under vegetative conditions can be caused either by a block in the Cvt pathway or by inefficient cleavage of the prApe1 propeptide by proteases after reaching the vacuole lumen. The vps52Δ, vps53Δ, and vps54Δ mutants missort carboxypeptidase Y (Prc1) (46, 47). This mis-sorting is caused by a block in retrieval of Vps10 to the Golgi complex (46); Vps10 is the receptor required for Prc1 transport to the vacuole (48). We examined the sorting of Prc1 in the ykr020wΔ mutant to determine whether it displayed a similar phenotype. Cells from the ykr020wΔ strain were converted to spheroplasts, subjected to pulse-chase labeling, and immuno-precipitated with antiserum to Prc1. In wild type cells, Prc1 was correctly delivered to the vacuole and processed to the mature form; essentially no missorting was observed (Fig. 1B). In agreement with the published data, vps52Δ cells secreted part of Prc1 as the p2 (Golgi-modified) form (Fig. 1B) (46). The result obtained with ykr020wΔ cells was very similar to that seen with the vps52Δ strain (Fig. 1B).
Most vacuolar protein sorting (vps) mutants display pleiotropic defects in the sorting of additional vacuolar hydrolases including Pep4 and Prb1 (49-51). As a result, these strains are severely compromised for vacuolar processing activity. Accordingly, we could not exclude a defect in prApe1 processing because of reduced proteolytic activity of the vacuole. To explore this possibility, we took advantage of a green fluorescent protein (GFP)-tagged version of prApe1 that can be used to monitor defects in the Cvt pathway (38). In wild type cells expressing GFP-Ape1, fluorescence was localized to the vacuole lumen either when cells were grown in rich medium or after nitrogen starvation (Fig. 1C). The vps52Δ, vps53Δ, vps54Δ, and ykr020wΔ strains have fragmented vacuoles (46, 47) (Fig. 4). Nonetheless, in contrast to the wild type strain, we could determine that when vps52Δ, vps53Δ, vps54Δ, and ykr020wΔ cells expressing the same GFP-Ape1 chimera were grown in rich medium, GFP-Ape1 was concentrated at a perivacuolar punctate structure (Fig. 1C). This site may correspond to the PAS (24, 25). The PAS is generally enhanced in mutants with a defect either in the formation of Cvt vesicles or in their fusion with the vacuole (24, 25). This result indicates that the mis-sorting of vacuolar proteases in the vps52Δ, vps53Δ, vps54Δ, and ykr020wΔ strains does not account for the accumulation of prApe1. Rather, this defect is because of a block of prApe1 import into the vacuole lumen. Transport of GFP-Ape1 into the vacuole lumen could be restored after transferring the same cells into medium lacking nitrogen (Fig. 1C), in agreement with the reversibility of the prApe1 processing phenotype.
Fig. 4.
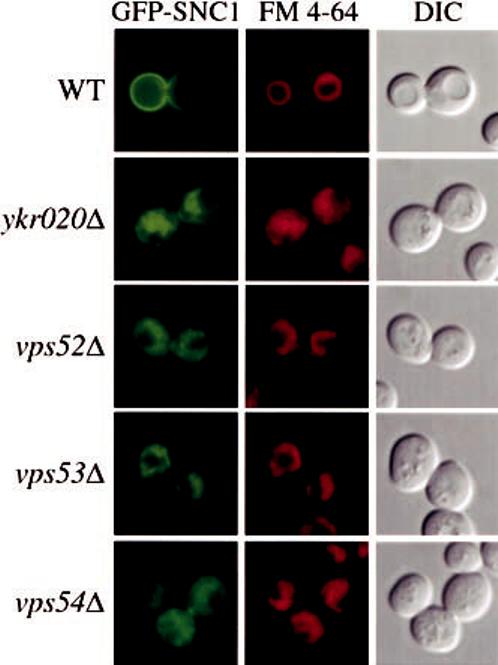
The ykr020wΔ, vps52Δ, vps53Δ, and vps54Δ cells have a defect in Snc1 recycling and display abnormal vacuole morphology. Wild type (WT), ykr020wΔ, vps52Δ, vps53Δ (FRY107), and vps54Δ cells in the BY4742 background were transformed with a plasmid expressing the GFP-Snc1 chimera (pGS416). Transformed cells were grown to a mid-log stage in SMD medium and FM 4-64 was added to the culture medium for 15 min. Cells were then collected, incubated in the same medium without FM 4-64 for an additional 30 min to chase the dye, and finally imaged with a fluorescence microscope. In wild type cells, Snc1 was concentrated on the plasma membrane while it was redirected to the vacuole lumen in the mutants. Pictures represent cells with an average level of GFP-Snc1 expression. In cells where those levels were lower, the staining pattern was primarily cytosolic punctate dots. FM 4-64 staining showed that deletion strains have a tubular, fragmented morphology of the vacuole. DIC, differential interference contrast.
With the previous analysis we could not differentiate between a block in Cvt vesicle formation or fusion of completed vesicles with the vacuole. This question was addressed with a protease sensitivity experiment. Yeast cells were converted to spheroplasts and lysed under conditions that allow lysis of the plasma membrane while retaining the integrity of subcellular compartments (52). The lysates were then treated with proteinase K in the absence or presence of detergent. During Cvt vesicle formation, prApe1 is not yet enclosed within a membrane compartment and consequently, in mutants affecting this process, prApe1 is accessible to exogenous proteases. In strains such as ccz1Δ that have a block in fusion (53, 54), prApe1 is enwrapped by the membrane of the Cvt vesicle and therefore isolated from the cytosol. As shown in Fig. 1D, in ccz1Δ cells prApe1 was protected from proteinase K. In contrast, prApe1 was fully accessible to the proteolytic action of the same proteinase in the vps52Δ strain. Identical results were also obtained with vps54Δ and ykr020wΔ cells (data not shown). We concluded that the vps52Δ, vps54Δ, and ykr020wΔ mutants, and by extension the vps53Δ mutant, are blocked at the formation/completion step of Cvt vesicle biogenesis.
Ykr020, Vps52, Vps53, and Vps54 Are Essential during Starvation but Are Not Required for Autophagy and Pexophagy
Most of the machinery required for the Cvt pathway is also exploited by autophagy and pexophagy (3, 4, 7). Results shown in Fig. 1, A and B, indicated that prApe1 processing was normal in the vps52Δ, vps53Δ, vps54Δ, and ykr020wΔ strains when autophagy was induced by nitrogen starvation. Recently, Ishihara et al. (29) reported that the Cvt pathway is operative and autophagy blocked during nitrogen deprivation in a sec12 mutant. Accordingly, we could not exclude the possibility that prApe1 maturation in the vps52Δ, vps53Δ, vps54Δ, and ykr020wΔ strains under starvation conditions was because of the Cvt pathway. To explore this possibility, we took advantage of vac8Δ, a mutant that is defective for the Cvt pathway but not for autophagy (17). We made a vps52Δ vac8Δ double mutant to see if prApe1 transport still occurred when those cells were starved for nitrogen. Precursor Ape1 processing was analyzed by Western blot in wild type, vps52Δ, vac8Δ, and vps52Δ vac8Δ cells grown either in the presence or absence of nitrogen. As expected, in both vps52Δ and vac8Δ cells prApe1 transport was blocked when the strain was grown in rich medium and restored when cells were shifted to a medium lacking nitrogen (17) (Fig. 2A). A similar result was obtained with the vps52Δ vac8Δ double mutant (Fig. 2A). The ability to transport prApe1 to the vacuole under nitrogen starvation conditions in the absence of Vac8 indicated that in vps52Δ cells this process was performed by autophagy and not by the Cvt pathway.
Fig. 2.
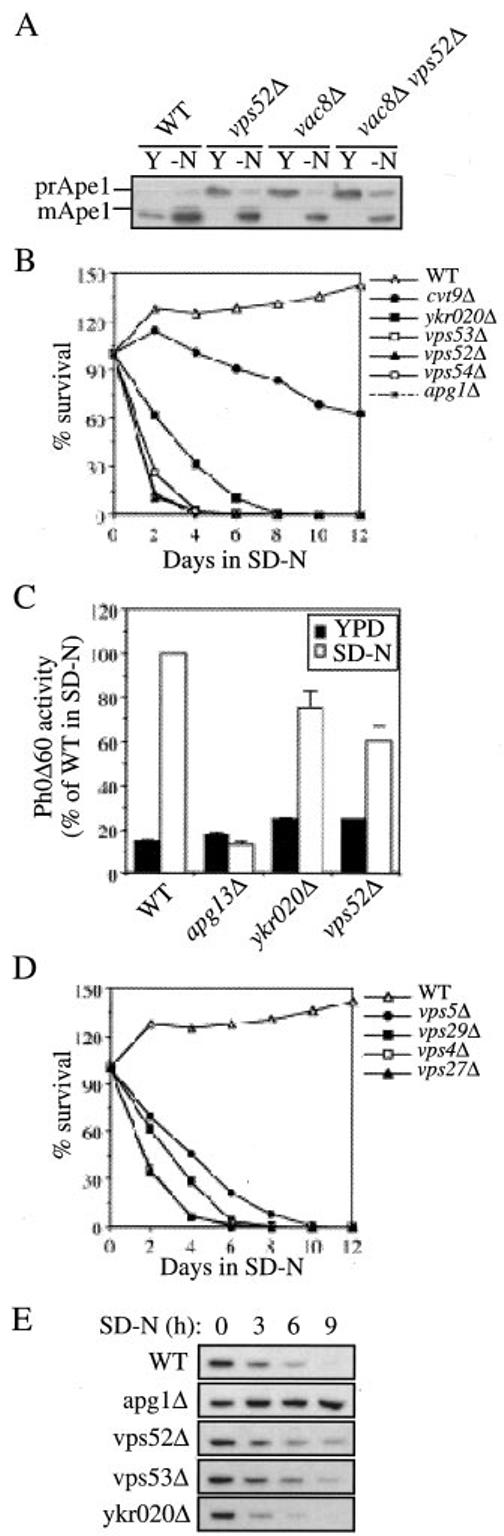
The ykr020wΔ, vps52Δ, vps53Δ, and vps54Δ mutants are specifically defective in the Cvt pathway. A, precursor Ape1 is not imported to the vacuole by the Cvt pathway under starvation conditions in the vps52Δ strain. Wild type (WT), vps52Δ, vac8Δ, and vps52Δ vac8Δ (FRY119) cells in the BY4742 background were analyzed as in Fig. 1A. The vps52Δ vac8Δ double mutant was able to import and mature prApe1. Y, YPD; -N, SD-N. B, the ykr020wΔ and VFT mutant strains are sensitive to nitrogen starvation. Wild type, ykr020wΔ, vps52Δ, vps53Δ (FRY107), vps54Δ, cvt9Δ, and apg1Δ strains in the BY4742 background were grown in YPD medium until early log phase and then shifted to SD-N. At the indicated time points, equal volumes of culture were withdrawn and plated on YPD plates. After 3 days the number of colonies representing the viable cells was counted, and the percentage of survival calculated and plotted against time in starvation medium. C, autophagy is functional in the vps52Δ and ykr020wΔ mutants. Wild type (TN124), ykr020wΔ (FRY122), vps52Δ (FRY122), and apg13Δ (D3Y103) cells expressing Pho8Δ60 were shifted from YPD medium (black bars) to SD-N medium (white bars) for 4 h. Autophagy induction was determined by a Pho8 activity assay. Results were expressed as percentage of the activity measured for the wild type strain starved for nitrogen. Error bars represent the standard deviation from three separate experiments. D, viability of vps mutants is sensitive to nitrogen starvation. Wild type (WT), vps4Δ, vps5Δ, vps27Δ, and vps29Δ strains in the BY4742 background were treated as in panel D. E, the ykr020wΔ, vps52Δ, and vps53Δ strains are not defective in pexophagy. Cells from wild type, apg1Δ, ykr020wΔ, vps52Δ, and vps53Δ (FRY107) strains in the BY4742 background were grown in conditions that induce peroxisomes, washed, and resuspended in SD-N medium for the times indicated. The presence of the peroxisomal thiolase enzyme, Fox3, was detected by immunoblotting.
The observation that prApe1 import in the vps52Δ strain is dependent on autophagy cannot be interpreted as indicating that autophagy is fully functional in this strain; prApe1 can be transported to the vacuole in an autophagy-dependent process even in situations where autophagy is severely impaired (12, 17). For example, in the absence of Aut7, induction of autophagy leads to the formation of abnormally small autophagosomes that can transport prApe1 to the vacuole but that cannot maintain a normal level of autophagy (12). We examined survival of the vps52Δ, vps53Δ, vps54Δ, and ykr020wΔ strains under starvation conditions as another method for assessing the autophagic capacity of these mutants. Strains that are defective in autophagy display limited viability under starvation conditions (55). The wild type, apg1Δ (15), cvt9Δ (16), vps52Δ, vps53Δ, vps54Δ, and ykr020wΔ strains were grown to mid-log phase, shifted to nitrogen starvation conditions, and their viability was determined over time. The wild type strain survived nitrogen starvation for more than 12 days without a significant decrease in viability (Fig. 2B). The cvt9Δ strain, defective primarily in the Cvt pathway, was also relatively resistant to starvation (16). In contrast, the number of viable cells decreased dramatically in the VFT complex and ykr020w deletion strains over the same time period. This rapid loss in viability was similar to that seen with apg1Δ cells, a strain that is extremely sensitive to starvation (55).
The starvation sensitivity suggests that the vps52Δ, vps53Δ, vps54Δ, and ykr020wΔ strains may be partially defective in autophagy. Alternatively, this phenotype could be because of other defects. Under conditions of nutrient stress it becomes necessary for the cell to transport cytoplasm to the vacuole by autophagy. However, subsequent to vacuolar delivery, these components must be degraded to generate an internal supply of nutrients (8). Cells require a fully functional vacuole to degrade and transport the recycled material back to the cytosol. The vps52Δ, vps53Δ, vps54Δ, and ykr020wΔ mutants affect the proper delivery of resident vacuolar hydrolases (46, 47) (Fig. 1). For this reason, it could not be excluded that the loss of viability observed after nitrogen starvation was caused by the inability to degrade the cytoplasmic substrates that had been delivered through autophagy. To directly quantify autophagy, we decided to measure the vacuolar processing of the cytosolic marker protein Pho8Δ60. This truncated form of the vacuolar alkaline phosphatase (Pho8) lacks the transmembrane domain and consequently localizes to the cytosol (56). This protein is delivered to the vacuole exclusively by autophagy. Proteolytic cleavage of the Pho8Δ60 propeptide in the vacuole lumen generates the active form of the enzyme, which can be detected by an activity assay (56). The YKR020w and VPS52 genes were knocked out in a strain where the chromosomal PHO8 gene was replaced with pho8Δ60, and phosphatase activity was determined either before or after nitrogen starvation. In wild type cells, there was low alkaline phosphatase activity when cells were grown in rich medium (Fig. 2C). There was an ∼7-fold increase in activity following the induction of autophagy when cells were shifted to SD-N. Apg13 is essential for autophagy (14, 17). In contrast to wild type cells, there was no increase in alkaline phosphatase activity from Pho8Δ60 when apg13Δ cells were shifted to starvation conditions. As shown in Fig. 2C, autophagy in the ykr020wΔ and vps52Δ strains was reduced to 75 and 60%, respectively, of the wild type levels but was not completely abolished compared with apg13Δ cells. This result indicates that autophagy was at least partially active in the vps52Δ and ykr020wΔ mutants.
There are two explanations for the reduced autophagic activity in the vps52Δ and ykr020wΔ strains. First, these differences may reflect a slower cellular metabolism and as a result, deletion strains form a reduced number of autophagosomes compared with wild type cells during the same time period. In fact, the vps52Δ, vps53Δ, and vps54Δ strains grow substantially slower than wild type cells, whereas the ykr020wΔ mutant has an intermediate doubling rate (data not shown). The second possibility is that in the mutant cells, autophagosomes are smaller and consequently they are able to transport less material into the vacuole. Accordingly, we decided to analyze the morphology of the autophagosomes in the mutant strains by electron microscopy to determine their size and number. Autophagosomes are transient structures that fuse with the vacuole. To stabilize cytosolic autophagosomes we took advantage of a conditional allele of VAM3, a gene coding for a tSNARE required for the fusion of Cvt vesicles and autopha gosomes with the vacuole (57). Cells were grown in YPD medium at 26°C to early log phase and then shifted to 37°C for 3 h either in the same medium or in SD-N medium. All three strains showed the absence of autophagosomes when grown in rich medium (Fig. 3). Under nitrogen starvation conditions, however, vps52Δ and ykr020wΔ cells accumulated smaller autophagosomes than those present in the wild type strain (Fig. 3). The total number of autophagosomes in the mutant strains, however, appeared comparable with that in the wild type strain. We therefore concluded that reduced autophagic activity in the ykr020wΔ, vps52Δ, vps53Δ, and vps54Δ strains is caused by the inability to assemble normal sized autophagosomes.
Fig. 3.

The ykr020wΔ and vps52Δ strains have smaller auto-phagosomes. Wild type (WT, TDY2), ykr020wΔ (FRY124), and vps52Δ (FRY125) cells were grown in YPD medium at 26°C to early log phase. Cultures were split in half and centrifuged. One sample was resus-pended again in YPD medium whereas the other was resuspended in SD-N medium. Cells were then grown at 37°C for 3 h. Permanganate fixation, dehydration, and embedding were carried out as described by Kaiser and Schekman (78). Uranyl acetate-stained sections were observed using a Philips CM10 transmission electron microscope. Examples of autophagosomes are indicated with an arrow. The bar is 1 μm.
Mutations in structural genes encoding vacuolar hydrolases such as PEP4 and PRB1 result in a starvation-sensitive phenotype (8, 58). This finding is in agreement with the requirement for Prb1 (proteinase B) in the breakdown of autophagic bodies, the single membrane subvacuolar vesicles that contain the cytoplasmic cargo resulting from autophagy (8). To our knowledge, however, the viability of vps mutants that are blocked in the biosynthetic delivery of resident hydrolases to the vacuole has not been examined in starvation conditions. For this reason, we decided to analyze the viability under starvation conditions of other deletion strains with a vps defect. We selected four strains that affect different functions of the late endosome without interfering with normal prApe1 transport.2 The vps4Δ, vps5Δ, vps27Δ, and vps29Δ cells all lost viability very quickly, similar to what was observed for vps52Δ, vps53Δ, vps54Δ, and ykr020wΔ cells (Fig. 2D). This result confirmed our hypothesis that normal traffic to the vacuole is required to utilize the potential nutrient pool created by auto-phagy and provides a likely explanation for the starvation sensitivity of strains such as vps52Δ, vps53Δ, vps54Δ, and ykr020wΔ that do not appear to be defective for autophagy.
When cells are shifted from conditions that necessitate peroxisome function (e.g. oleic acid) to glucose, excess peroxisomes are degraded (7, 59, 60). Peroxisome degradation requires most of the machinery that is needed for autophagy (7). The specific degradation of peroxisomes, pexophagy, induced by glucose adaptation or nitrogen starvation can be monitored by following the degradation of the peroxisomal matrix protein Fox3 (7). Growing cells were first shifted for 12 h to a medium containing glycerol, a suboptimal carbon source for yeast. Peroxisome proliferation was then induced by transferring cells for 19 h into a medium with oleic acid as the sole carbon source. Once shifted to SD-N, the excess peroxisomes were delivered to the vacuole and degraded in wild type cells (Fig. 2E). Pexophagy is blocked in mutants such as apg1Δ where autophagy is defective, and consequently Fox3 levels remain unchanged (7) (Fig. 2E). Fox3 degradation in ykr020wΔ, vps52Δ, vps53Δ, and by extension vps54Δ cells was essentially normal with only some minor differences in the degradation rate relative to wild type cells (Fig. 2E). We concluded that the VFT complex and Ykr020 are not required for pexophagy.
Snc1 Recycling Is Blocked in ykr020Δ, vps52Δ, vps53Δ, and vps54Δ Cells
Vps52, Vps53, and Vps54 are subunits of the VFT complex and in their absence the transport step between the early endosome and late Golgi is blocked (40). Snc1 is a member of the vSNARE family and it mediates fusion of exocytic vesicles with the plasma membrane (37, 61). Snc1 is mostly at the cell surface, but it undergoes rapid endocytosis and is transported from the early endosome to the late Golgi where it is reused (37). Mutants that affect early endosome function and transport from this compartment mislocalize Snc1 (37, 40, 45, 62, 63). We transformed wild type, vps52Δ, vps53Δ, vps54Δ, and ykr020wΔ cells with GFP-Snc1 (37) to see if the cycling of this chimera was affected. Cells were also stained with FM 4-64, a lipophilic fluorescent dye that allows visualization of the yeast vacuole (64).
As previously reported, in wild type cells most of the fluorescent staining corresponding to GFP-Snc1 was on the plasma membrane (Fig. 4) (37). A fraction of GFP-Snc1 was also observed in internal cell structures, probably organelles that Snc1 passes through during its recycling pathway (Fig. 4) (37). In contrast, in the four mutants analyzed, GFP-Snc1 was no longer found on the cell surface. Because of the co-localization with FM 4-64, it became evident that Snc1 was mislocalized to the vacuole (Fig. 4). It should be noted that in cells where the GFP-Snc1 levels were lower, the staining pattern was primarily cytosolic punctate dots. These dots may represent vesicles that cannot fuse with the late Golgi compartment; in the absence of the VFT complex and Ykr020, Snc1 retrieval to the Golgi complex is blocked resulting in its delivery to the vacuole via vesicular intermediates. This cytosolic staining pattern is most evident in the vps52Δ and vps54Δ strains (Fig. 4). Default delivery of Snc1 to the vacuole is similar to the fate observed for the Vps10 receptor in VFT mutant cells (46).
Another similarity between ykr020wΔ cells and VFT complex mutants is the structure of the vacuole. Wild type cells typically display a single large vacuole or a multilobed organelle depending on osmotic conditions (65). In contrast, this organelle is highly fragmented in vps52Δ, vps53Δ, and vps54Δ cells (46, 47). FM 4-64 staining and differential interference contrast microscopy along with electron microscopy confirmed the fragmented vacuole morphology of the vps52Δ, vps53Δ, and vps54Δ strains and showed that ykr020wΔ cells have a similar vacuole morphology but with a slightly less severe fragmentation (Figs. 3 and 4). Taken together with the analysis of Prc1 sorting, these data indicate that the ykr020wΔ cells have essentially identical trafficking defects as vps52Δ, vps53Δ, and vps54Δ mutants in regard either to prApe1 transport or retrieval from the early endosome.
Ykr020 Is Part of the VFT Complex but It Is Not Required for Its Stability
The similar trafficking and vacuole morphology phenotypes suggested that Ykr020 might be involved in the same transport step as the VFT complex. In addition, a recent proteomic analysis indicates that Ykr020 directly interacts with Vps52 (44). We decided to examine if Ykr020 was part of the VFT complex. To do so, we fused PA with the N terminus of Ykr020. The functionality of this fusion was demonstrated by complementation of the vacuole fragmentation phenotype of ykr020wΔ cells (not shown). This construct was then used to transform three different strains, each carrying one subunit of the VFT complex tagged at its C terminus with a triple HA epitope. Spheroplasts obtained from those strains were lysed and the PA chimera isolated using IgG-Sepharose beads. Bound complexes were released from the Sepharose by boiling it in sample buffer, and the presence of HA-tagged proteins was tested by immunoblotting. As shown in Fig. 5A, each one of the VFT complex subunits was selectively pulled down by the PAYkr020 fusion indicating that Ykr020 was part of this complex. During the writing of this article a review article was published stating that YKR020w was allelic with VPS51 (33). Accordingly, we decided to name the YKR020w gene VPS51 to avoid future confusion.
Fig. 5.

The YKR020w gene product is a component of the VFT complex. A, Ykr020 binds Vps52, Vps53, and Vps54. Strains expressing Vps52-HA (PSY118), Vps53-HA (PSY119), or Vps54-HA (PSY120) and carrying a plasmid expressing either the PA-Ykr020 construct (pCuPAYKR020(416)) or PA alone (pRS416-CuProtA) were used to prepare detergent-solubilized extracts (Ext) as described under “Experimental Procedures.” IgG-Sepharose beads were used to affinity purify the PA fusions together with the associated proteins (IP). Eluted polypeptides were separated with SDS-PAGE and then visualized by immunoblotting with antiserum to HA. For each experiment 0.2% of the total lysate or 20% of the total eluate were loaded per gel lane. B, there is only one Ykr020 molecule per VFT complex. The experiment performed in panel A was repeated with a strain (PSY116) expressing Ykr020-Myc. C, Ykr020 is not required for the stability of Vps53. Wild type (WT, PSY119), vps52Δ (FRY116), and ykr020wΔ (FRY117) strains expressing Vps53-HA were grown to early log phase and proteins were precipitated with trichloroacetic acid. Proteins were separated by SDS-PAGE and analyzed by Western blot with anti-HA antibodies. Pgk1 was used to verify that the same amount of material was loaded on each gel lane. D, Vps52 is not necessary to maintain normal cellular levels of Ykr020. Wild type (WT, PSY116) and vps52Δ (FRY118) cells expressing Ykr020-Myc were analyzed as in panel C using anti-Myc antibodies.
Having established that Vps51 is part of the VFT complex, we decided to examine the stoichiometry of this protein in the complex. We repeated the PA-Vps51 affinity isolation experiment using a strain carrying a copy of the VPS51 gene tagged on its C terminus with the Myc epitope. PA-Vps51 was unable to pull down Myc-tagged Vps51 indicating that there is only one Vps51 subunit per VFT complex (Fig. 5B).
Vps52, Vps53, and Vps54 depend on each other for stability. That is, in the absence of one of those proteins, the other two are rapidly degraded (46). During our experiments, we noticed that several phenotypes associated with the loss of the VFT complex were less prominent in the vps51Δ strain. For example, the vps51Δ cells grew better, had less fragmented vacuoles (Fig. 4), survived longer in media lacking nitrogen (Fig. 2A), and displayed more rapid pexophagy (Fig. 2C). Accordingly, it seemed possible that Vps51 was dispensable for the stability of the other subunits; in the absence of Vps51, the VFT complex was still able to have a residual activity. To investigate this hypothesis, we disrupted either VPS51 or VPS52 in the strain carrying HA-tagged Vps53. As shown in Fig. 5C, Vps53 levels were dramatically decreased in vps52Δ cells whereas they remained unchanged in the vps51Δ strain. We also decided to analyze the stability of Vps51 in cells lacking one of the three other components. To carry out this analysis we deleted the VPS52 gene in the strain expressing Myc-tagged Vps51. In the absence of Vps52, Vps51 levels were unchanged compared with wild type cells (Fig. 5D). We concluded that Vps51 is not necessary for the stability of the other components of the VFT complex and conversely, the other VFT components are not required for maintaining the appropriate cellular levels of Vps51.
There Is a Single VFT Complex Interacting with Tlg1
It has been shown that the VFT complex transitionally binds the vSNARE Tlg1 during the docking of early endosome-derived vesicles with the late Golgi (45). At present, we could not exclude the presence of two VFT complexes, only one of which contained Vps51. We decided to examine the interaction of the VFT complex containing Vps51 with Tlg1 to determine whether it was functioning similar to the previously analyzed VFT complex (45). Spheroplasts obtained from the strain expressing either PA-Vps51 or Vps52-HA were incubated for 30 min at room temperature in the presence of the amine-reactive cross-linker dithiobis(succinimidyl propionate) prior to lysis. Protein A affinity isolation was then performed and the presence of Tlg1 was examined by immunoblot. Pep12 and Sed5, the tSNAREs of the late endosome and the cis-Golgi complex, respectively (37, 42, 66, 67), were used as controls for nonspecific cross-linking. As can be seen in Fig. 6A, Tlg1 bound the Vps51-containing VFT complex under conditions where the other tSNAREs were not cross-linked.
Fig. 6.

The Vps51-containg VFT complex interacts with Tlg1. A, Tlg1 interacts with the Vps51-containing VFT complex. Spheroplasts from wild type (PSY118) cells expressing Vps52-HA and transformed with either the plasmid expressing PA-Vps51 (pCuPAYKR020(416)) or PA alone (pRS416-CuProtA) were treated with 1.5 mm of the cross-linker (dithiobis(succinimidyl propionate) (DSP), detergent solubilized, and the PA fusions were affinity purified on IgG-Sepharose as described in the legend to Fig. 5A. Samples from the extracts (Ext) or purified fractions (IP) were analyzed by Western blot using anti-HA, Tlg1, Sed5, and Pep12 antibodies or antiserum. A longer film exposure is also shown to demonstrate the total absence of cross-linking between the VFT complex and the control tSNAREs. B, the tlg2Δ strain accumulates prApe1 in YPD medium and this defect is bypassed in nitrogen starvation conditions. Wild type (WT) and tlg2Δ cells in the BY4742 background were essentially treated as in Fig. 1A. C, the tlg2Δ strain has a reversible accumulation of GFP-prApe1 in a cytosolic punctate structure. The same strains used in panel B were transformed with the plasmid carrying the GFP-prApe1 construct (pTS466) and analyzed as described in the legend to Fig. 1C. DIC, differential interference contrast.
Pulse-chase radiolabeling followed by Ape1 immunoprecipitation has shown that under rich growth conditions, Tlg2, Tlg1, and the Sec1 homologue, Vps45, are essential for the formation of Cvt vesicles but not for autophagosome biogenesis (31). This type of experiment demonstrates that those proteins have a direct impact on the vesicle formation process. To show that the steady state conditions used for our investigations were genuinely representative of a direct role of the VFT complex in Cvt vesicle biogenesis, we decided to examine the state of Ape1 in tlg2Δ cells. As expected, prApe1 processing was blocked when tlg2Δ cells were grown in rich medium and was restored after nitrogen starvation (Fig. 6B). This result was confirmed by analyzing the GFP-Ape1 chimera in the same cells under the same growth conditions (Fig. 6C). These results are identical to those that we obtained with the VFT complex components (Fig. 1, A and C). Because Tlg1 and Tlg2 also interact with the VFT complex (45) (Fig. 6A), it is reasonable to assume that all of these proteins are involved in the same fusion event essential for Cvt vesicle biogenesis.
Cvt9 Is Mislocalized and the Ape1-Cvt19 Complex Is No Longer Correctly Transported to the PAS in the Absence of the VFT Proteins
Most of the Cvt and autophagy pathway components are required for the vesicle formation/completion step and localize to a perivacuolar punctate structure (24, 25). Cells lacking the VFT complex are defective in Cvt vesicle biogenesis (Fig. 1D) indicating that probably one or more proteins necessary for this process are missing. To explore this possibility, we checked if the localization pattern of GFP-tagged Apg/Cvt components was altered in vps51Δ and vps52Δ cells. Of the different proteins analyzed, only Cvt9, a protein specific for the Cvt pathway and pexophagy (16), showed a different cellular distribution. Wild type cells transformed with a plasmid expressing a GFP-Cvt9 chimera under the control of the strong CUP1 promoter (68) showed GFP fluorescence at a perivacuolar punc-tate structure (Fig. 7A). In contrast, vps51Δ cells carrying the same construct displayed the presence of several dispersed fluorescent dots (Fig. 7A). This pattern was because of the absence of Vps51 because when the same cells were transformed with a plasmid expressing the PA-Vps51 fusion, the correct Cvt9 localization was restored (Fig. 7A). This phenotype was not caused by the overexpression of the GFP-Cvt9 chimera by the CUP1 promoter because the same fusion under the control of the endogeneous CVT9 promoter gave identical results although the fluorescent signal was fainter (Fig. 7A). After shifting cells for 3 h to a medium lacking nitrogen, GFPCvt9 became a single fluorescent spot in the vps51Δ strain (Fig. 7B). Thus, starvation conditions that bypass the prApe1 block in VFT complex mutants were able to induce the correct targeting of Cvt9. The GFP-Cvt9 chimera displayed similar patterns in vps52Δ, vps53Δ, vps54Δ, and tlg2Δ strains (data not shown).
Fig. 7.

In the absence of the VFT complex, the prApe1-Cvt19-Cvt9 complex is not properly targeted to the PAS. A, Cvt9 is mislocalized in the absence of Vps51. Wild type (WT, SEY6210) and vps51Δ (FRY126) cells were co-transformed with the following pairs of plasmids: pRS414 (empty vector) and pTS495 (promGFP-CVT9); pCuPAYKR020 (414) (pPA-VPS51) and pTS495; pRS414 and pCuGFPCVT9(416) (pCuGFP-CVT9); pCuPAYKR020(414) and pCuGFPCVT9(416). The transformed cells were grown in SMD medium to early log phase and visualized with a fluorescence microscope. It should be noted that there are additional very faint dots in the vps51Δ cells expressing GFP-Cvt9 under the control of its native promoter (promGFP-CVT9) that are easily detected when the strong CUP1 promoter drives expression of the same chimera. B, Cvt9 mislocalization in vps51Δ cells is reversed by nitrogen starvation conditions. The vps51Δ strain in the BY4742 background was transformed with pCuGFPCVT9(416) (pCuGFP-CVT9). Transformed mutants were either grown in SMD medium to early log phase or starved for nitrogen in SD-N medium for 3 h and then analyzed with a fluorescence microscope. Identical results were obtained with tlg2Δ, vps52Δ, vps53Δ, and vps54Δ strains in the BY4742 background (data not shown). C, the prApe1-Cvt19 complex is not correctly targeted to the PAS in the absence of the VFT complex. Wild type (SEY6210) and vps52Δ (PSY113) strains were transformed with the following two pairs of plasmid: pTS470 (CFP-Ape1) and pRS414EYFP-Aut7 (YFP-Aut7); pCVT19CFP(414) (Cvt19-CFP) and pRS414EYFP-Aut7. Transformed cells were either grown in SMD medium to early log phase or starved for nitrogen in SD-N medium for 3 h and then visualized with a fluorescence microscope. The CFP-Ape1 and Cvt19-CFP did not co-localize with the PAS (YFP-Aut7) when cells were grown in SMD medium. Nitrogen starvation conditions induced the correct targeting of CFP-Ape1 and Cvt19-CFP to the PAS because those two chimeras were in the same punctate structure as YFP-Aut7 in all cells. These observations were confirmed by repeating the same experiment in vps51Δ and vps53Δ cells (data not shown). D, Cvt9 is associated with the prApe1-Cvt19 complex away from the PAS in the vps52Δ mutant. Wild type (SEY6210) and vps52Δ (PSY113) strains were transformed with the following two pairs of plasmid: pPS98 (CFP-Cvt9) and pRS414EYFP-Aut7 (YFP-Aut7); pPS97 (YFP-Cvt9) and pCVT19CFP(414) (Cvt19-CFP). Transformed cells were grown in SMD medium to early log stage and analyzed with a fluorescence microscope. The CFP-Cvt9 fluorescent dots were separated from the PAS (YFP-Aut7), whereas co-localization between Cvt19 and Cvt9 was observed in all the images taken. DIC, differential interference contrast.
After synthesis, prApe1 forms a large cytosolic oligomer that binds Cvt19 (69, 70). This association triggers the transport of this complex to the site of Cvt vesicle formation in a process that requires Cvt9 (38). Our examination of GFP-tagged Apg/Cvt proteins in vps51Δ and vps52Δ cells indicated that prApe1 and Cvt19 were restricted to a single punctate structure (Fig. 1C and data not shown). However, this observation could not rule out the possibility that the fluorescent structure observed with GFP-Ape1 or GFP-Cvt19 was not correctly targeted to the site of Cvt vesicle formation, especially considering the observation that Cvt9 is mislocalized in vps51Δ and VFT mutant cells. For this reason we decided to transform the vps52Δ strain with YFP-Aut7 and either Cvt19-CFP or CFP-Ape1. Analysis of Aut7 provides an independent way to mark the site of vesicle formation (24, 71, 72). As predicted, in wild type cells grown in rich medium Cvt19 and prApe1 were both in the same structure as Aut7 (Fig. 7C) (25, 38), whereas in the vps52Δ strain those two proteins no longer co-localized with Aut7 in 40–50% of the cells examined (Fig. 7C). Nitrogen starvation conditions reversed the defect and resulted in localization of the prApe1-Cvt19 complex to the correct destination (Fig. 7C).
Because Cvt9 mediates the correct targeting of the prApe1-Cvt19 complex to the site of Cvt vesicle formation (38), we decided to investigate the localization of this protein relative to Cvt19 and the PAS. Wild type and vps52Δ cells were transformed either with Cvt19-CFP and YFP-Cvt9 or with YFP-Aut7 and CFP-Cvt9. As expected, in wild type cells grown either in rich medium or starved for nitrogen, Cvt19 and Aut7 were both in the same location as Cvt9, e.g. the PAS (Fig. 7D) (25, 38). In contrast, in the absence of VPS52, Cvt9 maintained the co-localization with Cvt19 but not with Aut7 in 30% of the cell population (Fig. 7D). These observations suggest that at least one of the causes of the Cvt pathway block in vps52Δ cells is the inability to correctly target the prApe1-Cvt19-Cvt9 complex to the site of vesicle formation. Nitrogen deprivation was once again able to reverse the defect and direct the prApe1-Cvt19-Cvt9 complex to the Aut7-containing structure (data not shown).
DISCUSSION
Vps51 Is a Subunit of the VFT Complex
The Vps fifty-three (VFT) complex and the rab GTPase Ypt6 are required for the tethering of early endosome-derived vesicles with late Golgi membranes (Fig. 8) (33, 45). Ric1 and Rgp1 form another complex that plays a crucial role during this recognition event. These two proteins localize to the late Golgi compartment where they recruit and catalyze nucleotide exchange on Ypt6 (40). Ypt6:GTP then binds directly to the VFT complex on the incoming vesicles (40). Because the VFT complex is associated with the vSNARE Tlg1, this tethering association brings Tlg1 in proximity with Tlg2, the tSNARE on the late Golgi membranes, leading to the assembly of the SNARE bundle, the core of the membrane fusion reaction (45).
Fig. 8.

Model for VFT complex function in the Cvt pathway. The VFT complex, including Vps51, functions as a tethering factor that is required for retrieval (retrograde traffic) from the early endosome to the Golgi complex (33, 45, 46). In the Cvt pathway, mutations in the VFT proteins result in mislocalization of the PAS component Cvt9, suggesting a tethering role in anterograde transport from the Golgi complex to the PAS. By analogy with its role in retrieval from the endosome, it is also possible that the VFT complex is needed for retrograde recovery of certain components from the PAS. Most of the Apg/Cvt proteins that localize to the PAS are not found in completed Cvt vesicles suggesting that they are excluded from the vesicle and retained at the PAS or that they are recycled back to another compartment such as the Golgi complex. See text for details.
The VFT complex is composed of three subunits: Vps52, Vps53, and Vps54 (40, 46). In a screen designed to find new deletion strains affecting the Cvt pathway, we identified Ykr020 (originally named Cvt22) as the fourth component of this complex and we named it Vps51. Vps51Δ, but also vps52Δ, vps53Δ, and vps54Δ, cells have a defect in prApe1 maturation when grown in rich medium (Fig. 1A). We started to consider the possibility that Vps51 was interacting with the VFT complex when the results of a genome-wide approach for the identification of yeast protein complexes by mass spectrometry appeared indicating a putative interaction between Vps51 and Vps52 (44). We performed three different analyses that confirmed that Vps51 was participating in the same sorting step as the VFT complex. Vps51Δ cells missort a population of Prc1 to the periplasmic space, have a fragmented vacuole, and fail to correctly recycle the vSNARE Snc1, phenotypes that are shared by the vps52Δ, vps53Δ, and vps54Δ strains (46, 47) (Figs. 1A and 4). The Prc1 and vacuolar morphology results are also corroborated by two different genome-wide studies where all nonessential genes necessary for Prc1 sorting and homo-typic vacuole fusion were identified (65, 73). Snc1 needs to be retrieved from the early endosome back to the late Golgi to be reused for exocytosis (37). Tethering/fusion partners of the VFT complex, Tlg1, Tlg2, Ypt6, Ric1, and Rgp1, are essential for this transport route (37, 40). In the present study we show that the VFT complex and Vps51 are also required for this recycling step (Fig. 4).
We obtained direct evidence that Vps51 is part of the VFT complex through an affinity isolation analysis. Vps51 tagged with protein A pulled down each of the other components of the VFT complex (Fig. 5A). Previous work has established that the ratio between each subunit is 1:1:1 (46). Vps51 did not pull down itself indicating that there is only one Vps51 subunit per complex (Fig. 5B). Thus, we concluded that the VFT complex is a heterotetramer with a 1:1:1:1 stoichiometry.
Vps51 is a small 164-amino acid protein that contains 2–3 putative coiled-coil regions. This protein has characteristics that differ from those of the other VFT complex components. For example, Vps52, Vps53, and Vps54 depend on each other to avoid rapid degradation, whereas Vps51 is not necessary for their stability and conversely, these three VFT components are not required for maintaining the appropriate cellular levels of Vps51 (46) (Fig. 5, C and D). The Vps52-Vps53-Vps54 trimer is still present in the absence of Vps51, which probably accounts for a residual activity of the VFT complex that would explain the less severe phenotypes observed in the vps51Δ strain (Figs. 2, A and C, and 4). Another peculiarity is that Vps52, Vps53, and Vps54 have clear mammalian homologues whereas Vps51 does not (33, 74). In a paper submitted in parallel with ours, Siniossoglou and Pelham (73) show that Vps51 is the VFT complex component that binds the N terminus of Tlg1. Because this region and that of the closest mammalian Tlg1 homologue, syntaxin 6, are not conserved, they hypothesize that the mammalian VFT complex possesses a divergent subunit.
The VFT Complex Is Specifically Required for Cvt Vesicle Biogenesis but Not for Autophagy and Pexophagy
Autophagy, pexophagy, and the Cvt pathway utilize a common machinery and only a few factors specific for one route are known (1, 3, 4, 7) (Table II). In this article we demonstrate that the VFT complex is one of those. The Cvt pathway operates during vegetative growth conditions and it assures the delivery of prApe1 from the cytosol to the vacuole lumen (5, 6). Under the same conditions, the absence of the VFT complex causes a block in prApe1 transport and in its consequent processing (Fig. 1, A and C). Interestingly, the same cells are able to correctly carry out autophagy and pexophagy (Figs. 1A and 2, A, C, and E). There are some delays in the kinetic rates of those processes with vps51Δ cells being the most similar to the wild type (Fig. 2, C and E). The missorting of vacuolar proteases in VFT complex mutants is not total, a fraction of those proteins normally reach the vacuole where they are processed and activated because of the correct acidification of this organelle (46, 47) (Fig. 1B). For this reason a reduced hydrolytic activity of the vacuole is not a likely explanation for the slower kinetics of autophagy and pexophagy seen in the vps51Δ and VFT mutant strains. Rather, these differences may reflect a slower cellular metabolism. However, analysis of autophagosomes by electron microscopy revealed that those structures have a reduced size in vps51Δ and vps52Δ cells indicating that the membrane expansion process may be impaired (Fig. 3). This is an interesting finding because it suggests that a specific block in the Cvt pathway reduces autophagosome size indicating that this route may be used to transport membranes to the PAS during nitrogen starvation. Alternatively, the VFT components and Vps51 may play some direct role in membrane delivery for auto-phagosome expansion. Further studies will be needed to fully identify the source membranes for Cvt vesicles and autophagosomes.
Tlg2, Tlg1, and Vps45 are also essential for the formation of Cvt vesicles but not for autophagosome biogenesis (31) (Fig. 6, B and C). It has been shown that the VFT complex interacts directly with Tlg1 and indirectly with Tlg2 (45). Here we demonstrated that indeed, there is a unique VFT complex composed of four subunits that transiently bind Tlg1 (Fig. 6 A). Taking these results together, we can conclude that the VFT complex, Tlg2, and by extension Tlg1 and Vps45, are mediating a transport step essential for the formation/completion of Cvt vesicles.
The absence of the VFT complex results in a block of prApe1 import into the vacuole under vegetative conditions (Fig. 1C). This defect is because of an incomplete Cvt vesicle because prApe1 is accessible to exogenous proteinase K (Fig. 5D). The arrest of the vesicle formation process can be caused either by the lack of one or more components necessary for Cvt vesicle biogenesis or by the failure to deliver membranes that are needed for the formation process. We find that Aut7 is correctly lipidated and localized to the PAS in vps51Δ and VFT mutant strains (Fig. 7, C and D).3 The Aut7 targeting process requires the presence and the correct functioning of the two conjugating systems, the autophagy-specific PtdIns 3-kinase complex and Apg9 (20, 21, 24, 76). The proper modification and localization of Aut7 in the absence of the VFT complex indicates that all of these Apg/Cvt factors are still fulfilling their functions. Investigating the cellular distribution of the remaining Apg/Cvt proteins, we found that in the absence of either the VFT complex or Tlg2, GFP-Cvt9 is dispersed in several fluorescent dots (Fig. 7, A and B, data not shown). Cvt9 plays an essential role in targeting the prApe1-Cvt19 complex to the site of Cvt vesicle formation (16, 38). In agreement with this model, we found that in the same cells, prApe1 and Cvt19, together with Cvt9, were no longer able to reach the PAS (Fig. 7, C and D). The cause of Cvt9 mislocalization in the VFT mutant cells is still unclear.
Cvt9 is a specific factor required for the Cvt pathway and pexophagy but not for autophagy (16) (Table II). Because Cvt9 is mislocalized in VFT complex mutants, one would expect that pexophagy would be impaired in the same cells. An interesting observation is that nitrogen deprivation triggers the correct targeting of Cvt9 to the PAS in the VFT mutants, furnishing an explanation as to why the prApe1 block is rapidly bypassed during autophagy (Fig. 7B). As mentioned, Cvt9 itself is not required for autophagy, but its proper re-localization may reflect the correct positioning of an associated factor that is essential for this pathway (16) (Table II). Nitrogen starvation conditions are also employed to induce peroxisome degradation (see “Experimental Procedures”). Thus, under those circumstances, Cvt9 is probably correctly localized so that this catabolic process proceeds normally (Fig. 2E). That leads us to conclude that prApe1-Cvt19 and peroxisomes need a common element, Cvt9, to be targeted to the PAS, but those two pathways have different trafficking requirements with pexophagy probably using the same membrane source as autophagy.
The great specificity in vesicular traffic is achieved by the partnership between SNAREs and tethering factors (33, 34). These proteins can be involved in one or several different fusion events, but the combination between them creates a specific assortment utilized only for a unique vesicular trafficking step. For this reason, Tlg2, Tlg1, and the VFT complex are probably participating in one or more retrieval steps back to the Golgi (Fig. 8). Two hypotheses can explain the role of this fusion machinery in Cvt vesicle assembly. The first possibility is that there is a retrograde transport route from the PAS for specific proteins. One possible cargo molecule of this recycling pathway is Apg9. This transmembrane protein is localized to the PAS but it is not found on complete autophagosomes, indicating that it is retrieved prior to the fusion of those vesicles with the vacuole (24, 25, 77). The second hypothesis is that in the absence of this fusion machinery, the proper homeostasis of either the early endosome or Golgi complex is severely compromised, and that has an indirect effect on Cvt vesicle completion by altering the sorting of specific transmembrane proteins. Tlg2 also localizes to Cvt vesicles (31). For this reason, at the moment we cannot exclude that this tSNARE is playing additional roles in the homotypic fusion that leads to Cvt vesicle completion or in the transport of intermediate vesicles to the PAS that are required for Cvt vesicle formation.
Future work will help to connect together all the specific proteins required for Cvt vesicle assembly and will help to elucidate in better detail the mechanism that leads to the formation of the PAS. Autophagy, however, employs factors not required for the Cvt pathway to supply the PAS with membranes suggesting that there are fundamental differences between the mechanism of formation of Cvt vesicles and autophagosomes. The comparison between the trafficking requirements for PAS formation during vegetative growth with those during starvation will improve our understanding of the reorganization of these trafficking routes in different environmental situations.
Acknowledgments
We thank Drs. Hugh Pelham, Yoshinori Ohsumi, and Lois Weisman for plasmids and antibodies. We are also very grateful to Drs. Symeon Siniossoglou and Hugh Pelham for communication of results before publication and Dorothy Roak Sorenson for help with electron microscopy.
Footnotes
This work was supported by National Institutes of Health Public Health Service Grant GM53396 (to D. J. K.) and by a European Molecular Biology Organization long-term fellowship (to F. R.). The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: Cvt, cytoplasm to vacuole targeting; Apg, autophagy; CFP, cyan fluorescent protein; GFP, green fluorescent protein; HA, hemagglutinin; PA, protein A; PAS, preautophagosomal structure; PtdIns, phosphatidylinositol; tSNARE, target soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor; VFT, Vps fifty-three; vps, vacuolar protein sorting; vSNARE, vesicle SNARE; YFP, yellow fluorescent protein; SMD, synthetic minimal medium; MES, 4-morpholineethanesulfonic acid.
F. Reggiori and D. J. Klionsky, unpublished observations.
J. Guan and D. J. Klionsky, unpublished observations.
REFERENCES
- 1.Reggiori F, Klionsky DJ. Euk. Cell. 2002;1:11–21. doi: 10.1128/EC.01.1.11-21.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klionsky DJ, Emr SD. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harding TM, Hefner-Gravink A, Thumm M, Klionsky DJ. J. Biol. Chem. 1996;271:17621–17624. doi: 10.1074/jbc.271.30.17621. [DOI] [PubMed] [Google Scholar]
- 4.Scott SV, Hefner-Gravink A, Morano KA, Noda T, Ohsumi Y, Klionsky DJ. Proc. Natl. Acad. Sci. U. S. A. 1996;93:12304–12308. doi: 10.1073/pnas.93.22.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scott SV, Baba M, Ohsumi Y, Klionsky DJ. J. Cell Biol. 1997;138:37–44. doi: 10.1083/jcb.138.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klionsky DJ, Cueva R, Yaver DS. J. Cell Biol. 1992;119:287–299. doi: 10.1083/jcb.119.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hutchins MU, Veenhuis M, Klionsky DJ. J. Cell Sci. 1999;112:4079–4087. doi: 10.1242/jcs.112.22.4079. [DOI] [PubMed] [Google Scholar]
- 8.Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. J. Cell Biol. 1992;119:301–311. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baba M, Osumi M, Scott SV, Klionsky DJ, Ohsumi Y. J. Cell Biol. 1997;139:1687–1695. doi: 10.1083/jcb.139.7.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baba M, Osumi M, Ohsumi Y. Cell Struct. Funct. 1995;20:465–471. doi: 10.1247/csf.20.465. [DOI] [PubMed] [Google Scholar]
- 11.Baba M, Takeshige K, Baba N, Ohsumi Y. J. Cell Biol. 1994;124:903–913. doi: 10.1083/jcb.124.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abeliovich H, Dunn WA, Jr., Kim J, Klionsky DJ. J. Cell Biol. 2000;151:1025–1034. doi: 10.1083/jcb.151.5.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. J. Cell Biol. 2000;150:1507–1513. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Funakoshi T, Matsuura A, Noda T, Ohsumi Y. Gene (Amst.) 1997;192:207–213. doi: 10.1016/s0378-1119(97)00031-0. [DOI] [PubMed] [Google Scholar]
- 15.Matsuura A, Tsukada M, Wada Y, Ohsumi Y. Gene (Amst.) 1997;192:245–250. doi: 10.1016/s0378-1119(97)00084-x. [DOI] [PubMed] [Google Scholar]
- 16.Kim J, Kamada Y, Stromhaug PE, Guan J, Hefner-Gravink A, Baba M, Scott SV, Ohsumi Y, Dunn WA, Jr., Klionsky DJ. J. Cell Biol. 2001;153:381–396. doi: 10.1083/jcb.153.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott SV, Nice DC, III, Nau JJ, Weisman LS, Kamada Y, KeizerGunnink I, Funakoshi T, Veenhuis M, Ohsumi Y, Klionsky DJ. J. Biol. Chem. 2000;275:25840–25849. doi: 10.1074/jbc.M002813200. [DOI] [PubMed] [Google Scholar]
- 18.Wang YX, Catlett NL, Weisman LS. J. Cell Biol. 1998;140:1063–1074. doi: 10.1083/jcb.140.5.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuma A, Mizushima N, Ishihara N, Ohsumi Y. J. Biol. Chem. 2002;277:18619–18625. doi: 10.1074/jbc.M111889200. [DOI] [PubMed] [Google Scholar]
- 20.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, Noda T, Ohsumi Y. Nature. 2000;408:488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 21.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 22.Kihara A, Noda T, Ishihara N, Ohsumi Y. J. Cell Biol. 2001;152:519–530. doi: 10.1083/jcb.152.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nice DC, Sato TK, Stromhaug PE, Emr SD, Klionsky DJ. J. Biol. Chem. 2002;277:30198–30207. doi: 10.1074/jbc.M204736200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. EMBO J. 2001;20:5971–5981. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim J, Huang W-P, Stromhaug PE, Klionsky DJ. J. Biol. Chem. 2002;277:763–773. doi: 10.1074/jbc.M109134200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noda T, Suzuki K, Ohsumi Y. Trends Cell Biol. 2002;12:231–235. doi: 10.1016/s0962-8924(02)02278-x. [DOI] [PubMed] [Google Scholar]
- 27.Wang C-W, Kim J, Huang W-P, Abeliovich H, Stromhaug PE, Dunn WA, Jr., Klionsky DJ. J. Biol. Chem. 2001;276:30442–30451. doi: 10.1074/jbc.M102342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abeliovich H, Zhang C, Dunn WA, Jr., Shokat KM, Klionsky DJ. Mol. Biol. Cell. 2003 doi: 10.1091/mbc.E02-07-0413. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishihara N, Hamasaki M, Yokota S, Suzuki K, Kamada Y, Kihara A, Yoshimori T, Noda T, Ohsumi Y. Mol. Biol. Cell. 2001;12:3690–3702. doi: 10.1091/mbc.12.11.3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunn WA., Jr. J. Cell Biol. 1990;110:1935–1945. doi: 10.1083/jcb.110.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abeliovich H, Darsow T, Emr SD. EMBO J. 1999;18:6005–6016. doi: 10.1093/emboj/18.21.6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wurmser AE, Emr SD. J. Cell Biol. 2002;158:761–772. doi: 10.1083/jcb.200112050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whyte JR, Munro S. J. Cell Sci. 2002;115:2627–2637. doi: 10.1242/jcs.115.13.2627. [DOI] [PubMed] [Google Scholar]
- 34.Pelham HRB. Trends Cell Biol. 2001;11:99–101. doi: 10.1016/s0962-8924(01)01929-8. [DOI] [PubMed] [Google Scholar]
- 35.Longtine MS, McKenzie A, III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 36.Sikorski RS, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis MJ, Nichols BJ, Prescianotto-Baschong C, Riezman H, Pelham HR. Mol. Biol. Cell. 2000;11:23–38. doi: 10.1091/mbc.11.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shintani T, Huang W-P, Stromhaug PE, Klionsky DJ. Dev. Cell. 2002;3:825–837. doi: 10.1016/s1534-5807(02)00373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reggiori F, Black MW, Pelham HR. Mol. Biol. Cell. 2000;11:3737–3749. doi: 10.1091/mbc.11.11.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siniossoglou S, Peak-Chew SY, Pelham HR. EMBO J. 2000;19:4885–4894. doi: 10.1093/emboj/19.18.4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holthuis JC, Nichols BJ, Dhruvakumar S, Pelham HR. EMBO J. 1998;17:113–126. doi: 10.1093/emboj/17.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Banfield DK, Lewis MJ, Rabouille C, Warren G, Pelham HR. J. Cell Biol. 1994;127:357–371. doi: 10.1083/jcb.127.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seaman MN, Marcusson EG, Cereghino JL, Emr SD. J. Cell Biol. 1997;137:79–92. doi: 10.1083/jcb.137.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti-Furga G. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 45.Siniossoglou S, Pelham HRB. EMBO J. 2001;20:5991–5998. doi: 10.1093/emboj/20.21.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Conibear E, Stevens TH. Mol. Biol. Cell. 2000;11:305–323. doi: 10.1091/mbc.11.1.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Conboy MJ, Cyert MS. Mol. Biol. Cell. 2000;11:2429–2443. doi: 10.1091/mbc.11.7.2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marcusson EG, Horazdovsky BF, Cereghino JL, Gharakhanian E, Emr SD. Cell. 1994;77:579–586. doi: 10.1016/0092-8674(94)90219-4. [DOI] [PubMed] [Google Scholar]
- 49.Robinson JS, Klionsky DJ, Banta LM, Emr SD. Mol. Cell. Biol. 1988;8:4936–4948. doi: 10.1128/mcb.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bankaitis VA, Johnson LM, Emr SD. Proc. Natl. Acad. Sci. U. S. A. 1986;83:9075–9079. doi: 10.1073/pnas.83.23.9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rothman JH, Stevens TH. Cell. 1986;47:1041–1051. doi: 10.1016/0092-8674(86)90819-6. [DOI] [PubMed] [Google Scholar]
- 52.Harding TM, Morano KA, Scott SV, Klionsky DJ. J. Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meiling-Wesse K, Barth H, Thumm M. FEBS Lett. 2002;526:71–76. doi: 10.1016/s0014-5793(02)03119-8. [DOI] [PubMed] [Google Scholar]
- 54.Wang C-W, Stromhaug PE, Shima J, Klionsky DJ. J. Biol. Chem. 2002;277:47917–47927. doi: 10.1074/jbc.M208191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tsukada M, Ohsumi Y. FEBS Lett. 1993;333:169–174. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- 56.Noda T, Matsuura A, Wada Y, Ohsumi Y. Biochem. Biophys Res. Commun. 1995;210:126–132. doi: 10.1006/bbrc.1995.1636. [DOI] [PubMed] [Google Scholar]
- 57.Darsow T, Rieder SE, Emr SD. J. Cell Biol. 1997;138:517–529. doi: 10.1083/jcb.138.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teichert U, Mechler B, Muller H, Wolf DH. J. Biol. Chem. 1989;264:16037–16045. [PubMed] [Google Scholar]
- 59.Evers ME, Hohfeld J, Kunau WH, Harder W, Veenhuis M. EMS Microbiol. Lett. 1991;69:73–78. doi: 10.1016/0378-1097(91)90649-u. [DOI] [PubMed] [Google Scholar]
- 60.Chiang H-L, Schekman R, Hamamoto S. J. Biol. Chem. 1996;271:9934–9941. doi: 10.1074/jbc.271.17.9934. [DOI] [PubMed] [Google Scholar]
- 61.Protopopov V, Govindan B, Novick P, Gerst JE. Cell. 1993;74:855–861. doi: 10.1016/0092-8674(93)90465-3. [DOI] [PubMed] [Google Scholar]
- 62.Galan JM, Wiederkehr A, Seol JH, Haguenauer-Tsapis R, Deshaies RJ, Riezman H, Peter M. Mol. Cell. Biol. 2001;21:3105–3117. doi: 10.1128/MCB.21.9.3105-3117.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Panek HR, Conibear E, Bryan JD, Colvin RT, Goshorn CD, Robinson LC. J. Cell Sci. 2000;113:4545–4555. doi: 10.1242/jcs.113.24.4545. [DOI] [PubMed] [Google Scholar]
- 64.Vida TA, Emr SD. J. Cell Biol. 1995;128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seeley ES, Kato M, Margolis N, Wickner W, Eitzen G. Mol. Biol. Cell. 2002;13:782–794. doi: 10.1091/mbc.01-10-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Becherer KA, Rieder SE, Emr SD, Jones EW. Mol. Biol. Cell. 1996;7:579–594. doi: 10.1091/mbc.7.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burd CG, Peterson M, Cowles CR, Emr SD. Mol. Biol. Cell. 1997;8:1089–1104. doi: 10.1091/mbc.8.6.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Labbé S, Thiele DJ. Methods Enzymol. 1999;306:145–153. doi: 10.1016/s0076-6879(99)06010-3. [DOI] [PubMed] [Google Scholar]
- 69.Scott SV, Guan J, Hutchins MU, Kim J, Klionsky DJ. Mol. Cell. 2001;7:1131–1141. doi: 10.1016/s1097-2765(01)00263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim J, Scott SV, Oda MN, Klionsky DJ. J. Cell Biol. 1997;137:609–618. doi: 10.1083/jcb.137.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang W-P, Scott SV, Kim J, Klionsky DJ. J. Biol. Chem. 2000;275:5845–5851. doi: 10.1074/jbc.275.8.5845. [DOI] [PubMed] [Google Scholar]
- 72.Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, Noda T, Ohsumi Y. J. Cell Biol. 1999;147:435–446. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bonangelino CJ, Chavez EM, Bonifacino JS. Mol. Biol. Cell. 2002;13:2486–2501. doi: 10.1091/mbc.02-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Whyte JR, Munro S. Dev. Cell. 2001;1:527–537. doi: 10.1016/s1534-5807(01)00063-6. [DOI] [PubMed] [Google Scholar]
- 75.Siniossoglou S, Pelham HRB. J. Biol. Chem. 2002;277:48318–48324. doi: 10.1074/jbc.M209428200. [DOI] [PubMed] [Google Scholar]
- 76.Kim J, Huang W-P, Klionsky DJ. J. Cell Biol. 2001;152:51–64. doi: 10.1083/jcb.152.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Noda T, Kim J, Huang W-P, Baba M, Tokunaga C, Ohsumi Y, Klionsky DJ. J. Cell Biol. 2000;148:465–480. doi: 10.1083/jcb.148.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kaiser CA, Schekman R. Cell. 1990;61:723–733. doi: 10.1016/0092-8674(90)90483-u. [DOI] [PubMed] [Google Scholar]