

Abstract
Carboxyl-terminal binding protein (CtBP) is a transcriptional corepressor originally identified through its ability to interact with adenovirus E1A. The finding that CtBP–E1A interactions were regulated by the nicotinamide adeninine dinucleotides NAD+ and NADH raised the possibility that CtBP could serve as a nuclear redox sensor. This model requires differential binding affinities of NAD+ and NADH, which has been controversial. The structure of CtBP determined by x-ray diffraction revealed a tryptophan residue adjacent to the proposed nicotinamide adenine dinucleotide binding site. We find that this tryptophan residue shows strong fluorescence resonance energy transfer to bound NADH. In this report, we take advantage of these findings to measure the dissociation constants for CtBP with NADH and NAD+. The affinity of NADH was determined by using fluorescence resonance energy transfer. The binding of NADH to protein is associated with an enhanced intensity of NADH fluorescence and a blue shift in its maximum. NAD+ affinity was estimated by measuring the loss of the fluorescence blue shift as NADH dissociates on addition of NAD+. Our studies show a >100-fold higher affinity of NADH than NAD+, consistent with the proposed function of CtBP as a nuclear redox sensor. Moreover, the concentrations of NADH and NAD+ required for half-maximal binding are approximately the same as their concentrations in the nuclear compartment. These findings support the possibility that changes in nuclear nicotinamide adenine dinucleotides could regulate the functions of CtBP in cell differentiation, development, or transformation.
An emerging theme in gene regulation is the dependence of transcriptional coregulators on molecules linked to cellular respiration. Acetyl–CoA is required by the histone acetyltransferase coactivators (1) and is perhaps the most central of all intermediary metabolites, bridging the major catabolic and anabolic processes to the Kreb's cycle, where its two carbons are converted to the respiratory product, CO2. ATP is important for essentially all cellular processes requiring energy, including the chromatin remodeling proteins involved in modifying nucleosomal structure (2). The pervasive involvement of acetyl–CoA and ATP in cellular processes generally obscures recognition of their specific contributions to transcriptional regulation, however. In addition, the relatively small changes in acetyl–CoA and ATP that occur during metabolism may not be suitable for regulating the activities of the relevant enzymes.
The breakdown of carbon sources is also associated with the reduction of the nicotinamide adenine dinucleotide NAD+ to NADH. NADH serves as an electron carrier that transports reducing equivalents to the electron transport chain, where ATP is synthesized. The synthesis of ATP involves oxidative phosphorylation, wherein NADH is oxidized to NAD+ and molecular oxygen is reduced to water. These roles of NAD+ and NADH provide additional, albeit somewhat indirect, connections between energy homeostasis and gene regulation. A more direct link was recognized when it became apparent that nicotinamide adenine dinucleotides were capable of interacting with certain enzymes involved in transcriptional silencing. Perhaps the best example of this regulation is the NAD+-dependence of the histone deacetylase, Sir2 (3, 4). The deacetylase activity of Sir2 and related proteins has been shown to contribute importantly to gene repression and the maintenance of heterochromatin (5). Recent data indicate that the regulation of Sir2 by NAD+ may explain the interaction between dietary factors and longevity in yeast and other species (6).
The vast majority of the cellular nicotinamide adenine dinucleotide is bound to protein and, consequently, cannot play a regulatory role in controlling transcription factors involved in gene regulation. Presumably, only the free pool of nicotinamide adenine dinucleotides can subserve these functions and, in most instances, the relevant free pool is in the nuclear compartment. The concentration of free NAD+, at least in the cytoplasm, greatly exceeds that of NADH. Because unbound nicotinamide adenine dinucleotides should pass freely through nuclear pores, this difference in the levels of free NAD+ and NADH should pertain to the nucleus as well. Furthermore, because the free cytoplasmic (and presumably nuclear) NAD+/NADH ratio is ≈700:1 (7), conversion of NAD+ to NADH causes a much larger relative change in the NADH level. This observation implies that the levels of free NADH are likely to more accurately reflect metabolic events than the levels of NAD+. Thus, the ability of a transcriptional regulator to sense levels of free nuclear NADH rather than NAD+ would seem to provide a more sensitive mechanism for linking metabolic events to transcription. Relatively little is known about the functions of NADH in the nucleus, however.
Two nuclear transcription factors have been proposed to be regulated by NADH. The first, NPAS2, is a DNA-binding protein involved in the maintenance of mammalian circadian rhythms. Studies by Rutter et al. (8) suggested that the binding of the NPAS2:BMAL heterodimer to control elements in target genes was regulated by NADH and NADPH. The concentrations of NADH and NADPH required for stimulating binding were in the millimolar range, levels that are considerably higher than the concentrations of the free nucleotides. Thus, whether NPAS2 is sensitive to physiologically relevant levels of NADH or NADPH remains to be determined.
The second NADH-regulated factor is the carboxyl-terminal binding protein, CtBP. CtBP was named for its ability to bind to the carboxyl terminus of the adenovirus E1A oncoprotein (9). Like other E1A-binding proteins, CtBP interacts with a wide variety of cellular factors, including several transcriptional repressors. Thus, CtBP has been categorized as a transcriptional corepressor. Studies in Drosophila as well as mammalian systems have shown that the corepressor function of CtBP is critical for cell differentiation, transformation, and development (10). Precisely how CtBP mediates gene repression has not been entirely resolved, however. Several reports have indicated that CtBP binds to histone deacetylases (11–13), but the importance of this interaction is controversial. Other proteins shown to interact with CtBP include co-REST, histone methyltransferases, and a polyamine oxidase (14). Interactions between CtBP and the polycomb repressors have also been reported, but CtBP was not a component of the polycomb complex isolated by Levine et al. (15). A recent study showed that the association of CtBP with the polycomb protein PC2 led to its sumoylation (16), but how this modification alters CtBP function is unknown. Finally, the high homology between CtBP and 3-phosphoglycerate dehydrogenase led to the suggestion that an oxido-reductase activity might also contribute to its gene repression effects (17). However, the observation that this activity is dispensable in the regulation of endogenous target genes in CtBP-knockout mouse embryo fibroblasts casts doubt on this hypothesis (18).
Zhang et al. (19) proposed that NAD+ and NADH regulate CtBP binding to certain DNA-binding transcriptional repressors, thereby targeting its corepressor functions to particular gene promoters. Although the ability of nicotinamide adenine dinucleotides to stimulate CtBP binding to E1A and other proteins has been confirmed by several laboratories (17, 20), the differential efficacy of NAD+ and NADH is controversial. Zhang et al. (19) showed that NADH was two to three orders of magnitude more effective than NAD+ in stimulating CtBP binding and proposed that this differential effect might link CtBP-mediated repression to the redox state of the nuclear compartment. In support of this model, they determined that the concentration of free nuclear NADH was ≈100 nM, similar to that required for half-maximal CtBP-E1A binding. The free nuclear NAD+ concentration cannot be measured directly but was estimated to be ≈70 μM, also approximately the half-maximal level for stimulating CtBP binding. In contrast, however, Kumar et al. (17) and Balasubramanian et al. (20) found that NAD+ and NADH were equally effective in stimulating CtBP binding. These results would not be consistent with metabolic regulation of CtBP function. Although Balasubramanian et al. did not examine levels of NAD+ and NADH that were low enough to discern a differential effect, Kumar et al. tested a full range of nicotinamide adenine dinucleotide concentrations. Moreover, they concluded on the basis of the CtBP crystal structure that the single proton difference between NAD+ and NADH could not result in a two to three order of magnitude difference in binding.
Because the potential for CtBP to serve as a redox sensor depends on its differential affinity for NAD+ and NADH, we decided to investigate these binding events directly. The structure of CtBP determined by Kumar et al. (17) showed that one of the three tryptophan residues in the protein lies within four angstroms of the nicotinamide in NAD+. Thus, we predicted that we could directly measure the binding of NADH by measuring the transfer of energy from the adjacent tryptophan. We showed, by using single tryptophan mutants, that tryptophan 318 is the major contributor to the fluorescence resonance energy transfer (FRET) to NADH. NAD+ affinity was estimated by competition assays. Binding of NADH to protein enhances the intensity of the NADH fluorescence and shifts the fluorescence peak to a shorter wavelength (blue shift). We estimated the NAD+ affinity by measuring its ability to displace NADH, as measured by the decrease in NADH fluorescence and blue shift. Our studies show that the Kd for NADH is significantly lower than that for NAD+ and support the idea that CtBP is indeed suited to serve as a metabolic sensor through its ability to interact differentially with reduced and oxidized nicotinamide adenine dinucleotides.
Materials and Methods
Plasmids. The plasmid pRcCMV-hCtBP1 was a generous gift from G. Chinnadurai (St. Louis University Health Sciences Center, St. Louis). CtBP was cloned by using PCR into pET24 in frame with the carboxyl-terminal His tag. Tryptophan to phenylalanine mutations were generated by using the Quikchange (Novagen) method after subcloning the CtBP cDNA into pBluescript II KS. All CtBP vectors were confirmed by DNA sequencing.
Proteins. His-tagged CtBP proteins were expressed in BL21(DE3) and purified by Ni-NTA affinity (Qiagen, Valencia, CA). Cultures (1 liter) were grown at room temperature to an optical density of 0.4–0.6, induced with IPTG (100 mg/liter) for 4–6 h, pelleted by centrifugation (20 min, 3,000 × g) and resuspended in 30 ml buffer A (25 mM Hepes/KOH, pH 7.6/50 mM KCl/10 mM BME/15% glycerol). Cells were lysed twice by using a French press under a constant pressure of 11,000 psi. Cell debris was removed by centrifugation (25 min, 20,000 × g), and the supernatant was slowly added to a Ni-NTA column (5 ml slurry) equilibrated with buffer A. The column was then washed sequentially with 150 ml of buffer A, 100 ml of pyruvate (0.25 mM), a glycerol gradient (15–40%, 250 ml), 50 ml of buffer A, a KCl gradient (50–350 mM, 250 ml), and 50 ml of buffer A. The column was then treated with a 50 ml of ATP (1 mM) and MgCl2 (1 mM) wash followed by 50 ml of imidazole (30 mM). The protein was eluted with an imidazole gradient (0–250 mM, 200 ml). Fractions were analyzed by SDS/PAGE and silver staining. Fractions of highest purity were pooled, concentrated in a centricon 30 matrix, and dialyzed in 4 liters of buffer B (25 mM Hepes/KOH, pH 7.2/50 mM KCl/5 mM DTT/10% glycerol). Purified protein was aliquoted and stored at –80°C. Protein concentration was determined by the Bradford method by using a BSA standard and confirmed by amino acid analysis (Texas A&M University Protein Chemistry Laboratory, College Station).
Fluorescence Assays. Fluorescence studies were carried out by using a Photon Technologies QM-1 steady state fluorescence spectrophotometer. NADH was excited directly at 340 nm or through FRET by exciting tryptophan at 285 nm. Emission spectra were collected from 300 to 500 nm. Titrations of NADH and NAD+ were performed in 1 ml volumes, at constant temperature (25°C) and stirring with excitation/emission slit widths of 2 nm and 3 nm, respectively. Assays were performed in 10 mM Tris (pH 8.0) by using 300–600 nM CtBP. NADH and NAD+ were obtained from Sigma in preweighed vials. The increase in fluorescence at 425 nm (excitation 285 nm) was used to follow NADH binding. The displacement of NADH (100–500 nM) by NAD+, observed as a decrease in fluorescence at 410 nm (excitation 340 nm), was used to measure NAD+ binding. Binding constants determined at various protein concentrations were in agreement. Inner filtering effects were determined to be negligible at the NADH concentrations used (≤500 nM) by measuring the absorbance of NADH at the excitation wavelength (285 nm).
Data Analysis. The data were analyzed by using the computer program kaleidagraph (Abelbeck Software, Reading, PA). The fluorescent signal ΔF was plotted against the concentration of free NADH. Because of the high affinity of CtBP for NADH, the protein concentration must be considered when determining the free NADH concentration. This was done by using the relationships in Eq. 1a–c below. The Kd was determined by using Eq. 2 (rectangular hyperbola) to fit the binding data.
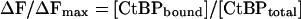 |
[1a] |
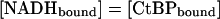 |
[1b] |
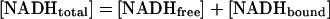 |
[1c] |
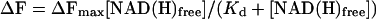 |
[2] |
The binding of NAD+ was measured by monitoring the displacement of NADH as a decrease in fluorescence at 410 nm. A plot of the change in fluorescence versus the NAD+ concentration was fit with Eq. 2 to determine the Kd(NAD+,app). The Kd(NAD+) was calculated by using Eq. 3, where NAD+ and NADH compete for the same site.
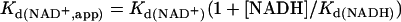 |
[3] |
Results
Crystal Structure of CtBP Reveals the Potential for FRET. The hypothesis that CtBP serves as a nuclear redox sensor relies on the observation that NADH is more effective than NAD+ in stimulating its binding to transcriptional repressors. A significant difference in the efficacy of NAD+ and NADH was observed by Zhang et al. (19) but was not seen by other investigators in subsequent studies (17, 20). The CtBP in these studies was expressed by using different techniques, however, and the proteins were purified to varying degrees. Additionally, the methods used, GST-pulldown and protease protection assays, are somewhat indirect and have limitations in their sensitivity. To examine the binding of NAD+ and NADH more directly, we performed fluorescence measurements on highly purified bacterially expressed protein. Purification of CtBP was monitored by silver staining and the concentrations of the protein, determined by Bradford assay, were confirmed by amino acid analysis. Occasional CtBP preparations contained various amounts of contaminating NADH that could be detected by its fluorescence at 425 nm after excitation at 340 nm (Fig. 1, middle trace). This residual NADH could be detected despite extensive dialysis, suggestive of an extremely low off-rate. Addition of saturating amounts of NADH (Fig. 1, top trace) revealed that ≈10% of the binding sites for NADH were occupied before addition of exogenous ligand in this particular CtBP preparation. We hypothesize that varying amounts of contaminating NADH might account in part for the inconsistency in the binding assays among various laboratories. To overcome this problem, we treated the purified protein preparations with pyruvate, which has been determined to be a substrate for the CtBP dehydrogenase activity (17). Although pyruvate is a relatively poor substrate, its addition caused a release of the bound NADH, presumably after conversion to NAD+. NAD+ binds to CtBP at relatively low affinity (see below) and can be removed by dialysis. The addition of pyruvate (Fig. 1, bottom trace) caused a complete loss of NADH fluorescence. Only samples that were free of contaminating NADH were used for binding studies.
Fig. 1.

Removal of bound NADH with pyruvate. Excitation of bacterially expressed purified CtBP at 340 nm results in an emission with a peak at 425 nm (middle trace). Saturating levels of NADH demonstrate fully bound CtBP (top trace). The addition of pyruvate abolished NADH fluorescence (bottom trace).
As described by Kumar et al. (17), the crystal structure of CtBP bound to NAD+ supports the prediction that CtBP is structurally similar to 3-phosphoglycerate dehydrogenase. An additional feature revealed by the structure was the location of a tryptophan residue adjacent to the nicotinamide moiety of NAD+. This tryptophan (W318) is ideal for detecting FRET because the nicotinamide portion of NAD+ is within four angstroms (Fig. 2). FRET requires that the donor and acceptor fluorophores are within the Förster radius, determined to be 25 Å for tryptophan to NADH energy transfer (21). NADH absorbs at 340 nm (the emission maximum of tryptophan); thus, excitation of the tryptophan at 285 nm would be expected to cause fluorescence from NADH at ≈425 nm.
Fig. 2.

NAD+ binding site of CtBP, taken from Kumar et al. (17). Several contacts of CtBP residues and NAD+ are shown. The distance between W318 and the nicotinamide moiety of NAD+ is within the Förster distance observed for tryptophan to NADH energy transfer (21). This structure is deposited in the Protein Data Bank (accession number 1MX3; ref. 17).
NADH Binding Constant Determined by FRET. When CtBP was excited at 285 nm, a typical tryptophan fluorescence spectrum was observed (Fig. 3a, bold trace). Titration of NADH resulted in the loss of fluorescence at 340 nm and an increase in fluorescence at 425 nm, demonstrating the transfer of resonance energy from an excited tryptophan to the bound NADH.
Fig. 3.

FRET signal of CtBP. (a) The excitation of CtBP at 285 nm resulted in a typical tryptophan fluorescence emission with a peak at 340 nm (bold trace). The titration of NADH resulted in a decrease of tryptophan fluorescence at 340 nm and an increase in NADH fluorescence at 425 nm. (b) Single tryptophan mutants were generated by mutating two of the three tryptophan residues to phenylalanines. The emission scan of each single tryptophan mutant excited at 285 nm is shown. Each mutant has a typical emission scan with a peak at 340 nm. (c) The addition of 200 nM NADH resulted in a strong energy transfer observed only with W318 (bold trace).
There are three tryptophan residues in CtBP: W145, W318, and W362. To determine the contribution of each tryptophan to the FRET signal, we generated proteins containing single tryptophan residues by mutating two of the three tryptophans to phenylalanine. Presumably, mutation of tryptophan to phenylalanine will cause only minimal perturbations of CtBP structure. A blue shift (see below) of NADH fluorescence was observed with each single tryptophan mutant, suggesting that the ability to bind NADH was retained. We measured the contribution of each tryptophan to the FRET signal by exciting each mutant at 285 nm (Fig. 3b). Of the three mutants, only W318 demonstrated substantial FRET on the addition of NADH (Fig. 3c, bold trace). This result agrees with the structural information suggesting that W318 is in position to excite the bound NADH (Fig. 2). A relatively small decrease in 340 nm fluorescence was observed when NADH was added to the W145 and W362 mutants. This may be the result of a conformational change induced by NADH binding and likely contributes to the quenching of wild-type CtBP at 340 nm in the presence of NADH.
To avoid potential artifacts caused by the phenylalanine substitutions, we performed the NADH binding experiments using wild-type CtBP. The increase in fluorescence at 425 nm (ΔF) was measured at various NADH concentrations. The concentration of free NADH at each titration point was calculated by using the relationships described in Materials and Methods. A plot of ΔF versus the free NADH was then fit by using the standard binding equation for a rectangular hyperbola and gave a Kd of 66 ± 14 nM (Fig. 4). This affinity is consistent with the levels of NADH found to stimulate the CtBP–E1A interaction and the concentration of free NADH found in the nucleus under basal conditions.
Fig. 4.

Plot of free NADH versus ΔF. Data were fit with Eq. 2.
Estimation of the Affinity of NAD+ by Measurement of the NADH Blue Shift. NAD+ does not fluoresce, therefore its binding to CtBP cannot be measured directly by fluorescence techniques. Theoretically, one could monitor the ability of NAD+ to compete for the NADH binding site by measuring a loss in FRET signal, but the absorbance spectrum of NAD+ overlaps to some degree with that of tryptophan. Because high concentrations of NAD+ are required for competition, the inner filtering effect of NAD+ complicates approaches that depend on excitation at 285 nm. To estimate the affinity of NAD+, we took advantage of the change that occurs in NADH fluorescence on binding to protein. Free NADH excited at 340 nm has an emission maximum at 455 nm (Fig. 5a, lower trace). Addition of CtBP causes a dramatic increase in NADH fluorescence as well as a shift in the emission maximum to 425 nm (Fig. 5a, upper trace). This change, termed a blue shift, occurs when a fluorescent molecule enters a more hydrophobic environment (22). Because this measurement relies on absorption at 340 nm and not 285 nm, inner filtering is less problematic. By measuring the disappearance of the NADH blue shift as a decrease in fluorescence intensity, we were able to monitor NAD+ binding and determine its dissociation constant (Fig. 5b). The emission maximum of displaced NADH at saturating NAD+ concentrations is identical to that of free NADH (Fig. 5a), indicating that the binding of NAD+ and NADH is mutually exclusive and that the loss of fluorescence at 425 nm is caused by removal of bound NADH and not to quenching. Eq. 3 was used to calculate the Kd for NAD+ to be 8–11 μM. The apparent Kd of CtBP for NAD+ is a function of the Kd for the competing substrate, NADH. At an NADH concentration of 100 nM, the apparent Kd for NAD+ was 16 ± 1 μM (Fig. 5c).
Fig. 5.

(a) Blue shift and enhanced fluorescence of bound NADH. Free NADH has a peak emission at 455 nm when excited at 340 nm (lower trace). Addition of CtBP results in a shift of the emission peak to 425 nm and an increase in the quantum yield (upper trace). (b) NAD+ displacement of NADH. The emission scan of CtBP bound with NADH excited at 340 nm (top trace). The titration of NAD+ resulted in a loss of NADH fluorescence. At saturating levels of NAD+, an emission scan typical of free NADH was observed. (c) Plot of NAD+ versus ΔF. Data were fit with Eq. 2. See Materials and Methods for calculation of Kd.
Discussion
Studies of the histone deacetylase Sir2 first uncovered the connection between the nicotimamide adenine dinucleotides and gene regulation. In the case of Sir2, the related nucleotides NADH, NADP+, and NADPH, cannot replace NAD+ as an essential cofactor for enzymatic activity (5). The finding that NAD+ regulates Sir2 function has led to a far-reaching series of studies linking nutrition, gene silencing, and longevity in a variety of species. Presumably, nuclear NAD+ is predominantly bound to protein and only the free fraction would be expected to be relevant for the regulation of Sir2 activity. It is difficult to quantitate this small pool of free nuclear NAD+ directly, but estimates have suggested that the concentration is in the 10–100 μM range (19). Interestingly, this range corresponds to measurements of the KM of Sir2 and the related Hst proteins for NAD+ (23). Alterations in the energy status of the cell are associated with changes in the ratio of free NAD+ to NADH, however, raising the possibility that NADH levels could be equally important for controlling some transcriptional events. Indeed, it is probably inappropriate to view a protein with a single binding site as detecting a ratio of two ligands; although the binding of one ligand can compete with that of another, the binding events are technically two independent events. Thus, NAD+ and NADH can be considered to be two interdependent ligands capable of recognizing a single binding site. Because the free NADH pool is so much smaller, metabolic events that alter the NAD+/NADH ratio cause a much larger change in the levels of free NADH than NAD+. Thus, NADH can be considered to provide a more sensitive reflection of alterations in cellular energy status.
Unlike NAD+, NADH can be detected directly in various cellular compartments by measurement of its fluorescence. Quantitation of the fraction of nuclear NADH that is not bound to protein can be determined by using fluorescence lifetime measurements, but requires a few assumptions (including an estimation of the ratio of NADH to NADPH, because these two molecules cannot be distinguished by their fluorescence properties). With these caveats, Zhang et al. proposed that the concentration of free nuclear NADH is ≈100 nM (19). Interestingly, this value is very close to the level of NADH reported to stimulate half-maximal binding of CtBP to E1A. Because other investigators could not confirm these findings, however, we attempted to measure NADH binding to CtBP directly. Although it does not necessarily hold that the affinities of CtBP for NAD+ and NADH will correspond exactly to the concentrations required for the effects on E1A binding, a difference in affinity for the two ligands would be expected.
Our FRET studies indicate that NADH has an affinity of ≈66 nM for CtBP. This value is surprisingly close to the concentration of free nuclear NADH determined by using two-photon fluorescence microscopy and fluorescence lifetime measurements. Thus, we predict that alterations in the free NADH concentration could regulate NADH-driven changes in CtBP conformation. How the free nuclear NADH concentration changes in vivo in response to hypoxia or other metabolic stresses has not been determined, however. It is likely that the nuclear levels of free NADH would follow those in the cytoplasm, which vary under a variety of physiological conditions (see below). Determination of the affinity of NAD+ is somewhat more problematic because the use of the NADH blue shift and its competition by NAD+ is less established than FRET as a quantitative measure. Consequently, our determination that half-maximal NAD+ binding occurs at a concentration of 8–11 μM must be considered an estimate. Additionally, because the concentration of free nuclear NAD+ cannot be determined directly, it is difficult to compare this value with the calculated dissociation constant. Interestingly, however, the affinities of NADH and NAD+ for CtBP are very reminiscent of those determined for 3-phosphoglycerate dehydrogenase >40 years ago (24). Because NAD+ and NADH appear to cause similar changes in CtBP binding, albeit at different concentrations, it will be important to determine what fraction of CtBP is occupied by these ligands in vivo.
We suspect that the disparate results of various laboratories on the differential binding of CtBP to NAD+ and NADH reflects the fact that NADH binds tightly and cannot easily be removed. It is important to account for protein concentration when determining the concentration of free ligand in binding experiments. Once the free NADH concentration is calculated and the contaminating NADH is removed, the higher affinity of NADH over NAD+ is readily apparent. Although NAD+ and NADH differ by only a single proton and two electrons, the structures of the nicotinamide moieties are vastly different. In NAD+, the nicotinamide has a planar structure, whereas it is puckered in NADH (25). This marked difference probably explains the differential binding of the two nucleotides and suggests that the nicotinamide moiety plays an important role in mediating NADH binding.
Balasubramanian et al. (20) found that NAD(H) doubled the apparent molecular mass of CtBP, suggesting that its addition promoted dimerization or tetramerization (depending on whether the 65- to 70-kDa species is a monomer or dimer, respectively). It is possible, therefore, that this ability of NAD(H) to stimulate CtBP oligomerization could contribute to the enhanced binding of E1A and other transcriptional repressors. We determined the stoichiometry of NADH binding and found that each monomer subunit of CtBP bound one molecule of NADH (data not shown) by using the fluorescence technique recently described (26) for determining the stoichiometry of nicotinamide adenine dinucleotide binding to 3-phosphoglycerate dehydrogenase. The 1:1 binding that we observed suggests that most of the CtBP used in our assay was properly folded and able to bind nucleotide. The lack of cooperativity in nucleotide binding further suggests that each monomer subunit binds NADH independently.
The homology between CtBP and 3-phosphoglycerate dehydrogenase is striking but the biological significance of the CtBP dehydrogenase activity is unresolved. Three reports have demonstrated that CtBP has weak dehydogenase activity (14, 17, 20), as measured by the conversion of NADH to NAD+, in the presence of pyruvate. However, the turnover rate of CtBP is 30,000-fold less than that of lactate dehydrogenase (unpublished data, ref. 27), which is probably why it has been overlooked or ignored. The structure of CtBP, determined by Kumar et al. (17), also supports the idea that CtBP is a functional dehydrogenase. According to these investigators, the weak enzymatic activity reflects the likelihood that pyruvate is not the appropriate substrate. It is also possible that CtBP simply has very little dehydrogenase activity. This view is supported by the finding that the enzymatic activity, which can be blocked by mutation of a histidine residue essential for catalysis, is not required for repression of target genes in CtBP-knockout mouse embryo fibroblasts (18). It remains possible that the dehydrogenase activity is required for some, as yet undescribed, CtBP function. One possibility, for example, would be that the dehydrogenase activity could contribute to the termination of repression. In this scenario, conversion of NADH to NAD+ would allow the nucleotide to dissociate and thereby disrupt the interaction between CtBP and transcriptional repressor. This process would be somewhat reminiscent of the regulated binding events seen with the Ras family of GTPases, wherein the hydrolysis of GTP to GDP results in the release of downstream effectors. Regardless of its biological role, the dehydrogenase activity was useful in our studies because it provided a method to convert the tightly bound NADH to NAD+, which could then be removed by dialysis.
The regulation of CtBP by nuclear nicotinamide adenine dinucleotides places this factor in the category of molecules that link transcription to cellular metabolism and respiration. The importance of CtBP as a sensor of nuclear redox state in vivo has yet to be determined. Large changes in the free NAD+/NADH ratio occur at birth (28), in response to ethanol (29), and in certain metabolic disorders such as diabetes (7). Hypoxia also has a profound effect on free NADH levels, the relief of which may be responsible for the large change at parturition. Knockout of CtBP in mouse embryonic fibroblast cells causes up-regulation of genes involved in cell adhesion and apoptosis (18). It is possible, therefore, that increased levels of NADH could result in augmented CtBP function, which would be correlated with decreased cell adhesion and protection from apoptosis. These effects could be relevant in certain disease states, such as malignancy, where the relative hypoxia of cancer cells could contribute to metastasis and escape from mechanisms that lead to cell death. Alternatively, or in addition, the dramatic decrease in free NADH levels at birth could result in the relief of repression of CtBP-regulated genes. The relationship between these events, and the role of CtBP, will become more evident as we gain understanding of gene expression in cells where energy homeostasis is challenged.
Acknowledgments
We thank J. Kaplan, J. M. Denu, D. L. Farrens, and R. W. Hanson for helpful discussions, G. Chinnadurai for reagents, and M. G. Rosenfeld and A. Aggarwal for providing the CtBP coordinates. This work was supported by grants from the National Institutes of Health.
Abbreviations: FRET, fluorescence resonance energy transfer; CtBP, carboxyl-terminal binding protein.
Note Added in Proof. Nardini et al. (30) recently determined the structural basis for the increased affinity of CtBP for NADH. They propose that nucleotide binding induces a conformational change that promotes CtBP dimerization, which is essential for corepressor activity.
References
- 1.Sterner, D. E. & Berger, S. L. (2000) Microbiol. Mol. Biol. Rev. 64, 435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peterson, C. L. (2002) Curr. Biol. 12, R245–R247. [DOI] [PubMed] [Google Scholar]
- 3.Landry, J., Sutton, A., Tafrov, S. T., Heller, R. C., Stebbins, J., Pillus, L. & Sternglanz, R. (2000) Proc. Natl. Acad. Sci. USA 97, 5807–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Imai, S., Armstrong, C. M., Kaeberlein, M. & Guarente, L. (2000) Nature 403, 795–800. [DOI] [PubMed] [Google Scholar]
- 5.Denu, J. M. (2003) Trends Biochem. Sci. 28, 41–48. [DOI] [PubMed] [Google Scholar]
- 6.Lin, S. J., Defossez, P. A. & Guarente, L. (2000) Science 289, 2126–2128. [DOI] [PubMed] [Google Scholar]
- 7.Williamson, D. H., Lund, P. & Krebs, H. A. (1967) Biochem. J. 103, 514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rutter, J., Reick, M., Wu, L. C. & McKnight, S. L. (2001) Science 293, 510–514. [DOI] [PubMed] [Google Scholar]
- 9.Schaeper, U., Boyd, J. M., Verma, S., Uhlmann, E., Subramanian, T. & Chinnadurai, G. (1995) Proc. Natl. Acad. Sci. USA 92, 10467–10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nibu, Y., Zhang, H. & Levine, M. (1998) Science 280, 101–104. [DOI] [PubMed] [Google Scholar]
- 11.Bertos, N. R., Wang, A. H. & Yang, X. J. (2001) Biochem. Cell Biol. 79, 243–252. [PubMed] [Google Scholar]
- 12.Dressel, U., Bailey, P. J., Wang, S. C., Downes, M., Evans, R. M. & Muscat, G. E. (2001) J. Biol. Chem. 276, 17007–17013. [DOI] [PubMed] [Google Scholar]
- 13.Subramanian, T. & Chinnadurai, G. (2003) FEBS Lett. 540, 255–258. [DOI] [PubMed] [Google Scholar]
- 14.Shi, Y., Sawada, J., Sui, G., Affar el, B., Whetstine, J. R., Lan, F., Ogawa, H., Po-Shan Luke, M. & Nakatani, Y. (2003) Nature 422, 735–738. [DOI] [PubMed] [Google Scholar]
- 15.Levine, S. S., Weiss, A., Erdjument-Bromage, H., Shao, Z., Tempst, P. & Kingston, R. E. (2002) Mol. Cell. Biol. 22, 6070–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kagey, M. H., Melhuish, T. A. & Wotton, D. (2003) Cell 113, 127–137. [DOI] [PubMed] [Google Scholar]
- 17.Kumar, V., Carlson, J. E., Ohgi, K. A., Edwards, T. A., Rose, D. W., Escalante, C. R., Rosenfeld, M. G. & Aggarwal, A. K. (2002) Mol. Cell 10, 857–869. [DOI] [PubMed] [Google Scholar]
- 18.Grooteclaes, M., Deveraux, Q., Hildebrand, J., Zhang, Q., Goodman, R. H. & Frisch, S. M. (2003) Proc. Natl. Acad. Sci. USA 100, 4568–4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang, Q., Piston, D. W. & Goodman, R. H. (2002) Science 295, 1895–1897. [DOI] [PubMed] [Google Scholar]
- 20.Balasubramanian, P., Zhao, L. J. & Chinnadurai, G. (2003) FEBS Lett. 537, 157–160. [DOI] [PubMed] [Google Scholar]
- 21.Steinberg, I. Z. (1971) Annu. Rev. Biochem. 40, 83–114. [DOI] [PubMed] [Google Scholar]
- 22.Wolff, E. C., Wolff, J. & Park, M. H. (2000) J. Biol. Chem. 275, 9170–9177. [DOI] [PubMed] [Google Scholar]
- 23.Tanner, K. G., Landry, J., Sternglanz, R. & Denu, J. M. (2000) Proc. Natl. Acad. Sci. USA 97, 14178–14182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugimoto, E. & Pizer, L. I. (1968) J. Biol. Chem. 243, 2090–2098. [PubMed] [Google Scholar]
- 25.Meijers, R., Morris, R. J., Adolph, H. W., Merli, A., Lamzin, V. S. & Cedergren-Zeppezauer, E. S. (2001) J. Biol. Chem. 276, 9316–9321. [DOI] [PubMed] [Google Scholar]
- 26.Grant, G. A., Hu, Z. & Xu, X. L. (2002) J. Biol. Chem. 277, 39548–39553. [DOI] [PubMed] [Google Scholar]
- 27.Holbrook, J. J., Liljas, A., Steindel, S. J. & Rossman, M. G. (1975) in The Enzymes, ed. Boyer, P. D. (Academic, New York), Vol. 11, Part A, pp. 191–268. [Google Scholar]
- 28.Philippidis, H. & Ballard, F. J. (1969) Biochem. J. 113, 651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stubbs, M., Veech, R. L. & Krebs, H. A. (1972) Biochem. J. 126, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nardini, M., Spano, S., Cericola, C., Pesce, A., Massen, A., Millo, E., Luini, A., Corda, D. & Bolognesi, M. (2003) EMBO J. 22, 3122–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]