

Abstract
The abundant cell surface glycolipid lipophosphoglycan (LPG) was implicated in many steps of the Leishmania infectious cycle by biochemical tests. The presence of other abundant surface or secreted glycoconjugates sharing LPG domains, however, has led to uncertainty about the relative contribution of LPG in vivo. Here we used an Leishmania major lpg1- mutant, which lacks LPG alone and shows attenuated virulence, to dissect the role of LPG in the establishment of macrophage infections in vivo. lpg1- was highly susceptible to human complement, had lost the ability to inhibit phagolysosomal fusion transiently, and was oxidant sensitive. Studies of mouse mutants defective in relevant defense mechanisms confirmed the role of LPG in oxidant resistance but called into question the importance of transient inhibition of phagolysosomal fusion for Leishmania macrophage survival. Moreover, the limited lytic activity of mouse complement appears to be an ineffective pathogen defense mechanism in vitro and in vivo, unlike human hosts. In contrast, lpg1- parasites bound C3b and resisted low pH and proteases normally, entered macrophages efficiently and silently, and continued to inhibit host-signaling pathways. These studies illustrate the value of mechanistic approaches focusing on both parasite and host defense pathways in dissecting the specific biological roles of complex virulence factors such as LPG.
Keywords: phosphoglycans, trypanosomatid protozoan parasite, oxidant resistance, inhibition of macrophage activation, adhesin
The protozoan parasite Leishmania is an intracellular pathogen that resides within an acidified phagolysosome of vertebrate host macrophages and is transmitted by biting phlebotomine sand flies. Within the phagolysosome, the parasite must resist the hydrolytic environment and avoid macrophage activation. People infected with Leishmania can develop diseases ranging from mild to disfiguring to fatal, depending on the species of parasite and the host factors. Current chemotherapy is inadequate, and although vaccination is thought to be feasible, no clinically effective vaccine exists.
Leishmania promastigotes are covered with a dense surface glycocalyx, composed largely of molecules attached by glycosylphosphatidylinositol (GPI) anchors (1). These GPI-anchored molecules include proteins such as the parasite surface protease gp63 and proteophosphoglycans (PPGs), as well as short GPI-anchored glycosylinositolphospholipids (GIPLs). The most abundant constituent is a large GPI-anchored phosphoglycan called lipophosphoglycan (LPG) (2–4). In all Leishmania species, the GPI anchor of LPG is composed of a 1-O-alkyl-2-lysophosphatidylinositol lipid anchor and a heptasaccharide core, to which is joined a long phosphoglycan (PG) polymer composed of 15–30 [Galβ1,4Manα1-PO4] repeating units (substituted with other sugars in some species) and which is terminated by a capping oligosaccharide. These domains are shared with other molecules; the PG repeating units and caps occur on secreted proteins such as PPG or secreted acid phosphatase, and the core GPI anchor domains have similarities with those present in both GIPLs and GPI-anchored proteins (1, 5, 6).
After vertebrate infection, infective metacyclic Leishmania must resist the action of complement, attach and enter macrophages, resist host defenses such as oxidants and hydrolytic enzymes, inhibit macrophage activation, and differentiate to the amastigote stage, which is adapted for long-term survival and replication within an acidified phagolysosome. LPG has been implicated in many steps required for establishment of macrophage infections and for survival in the insect vector (3, 4, 7–10). LPG does not play a role in the amastigote stage, however, because this parasite stage synthesizes little or no LPG and does not require LPG for virulence. However, amastigotes continue to make structurally related glycoconjugates (3, 4, 7, 9, 11).
Typically, LPG roles were studied with purified LPG and sophisticated biochemical and cellular assays. However, concerns have been raised: LPG may be applied in routes and amounts that may not be physiologically relevant, and the sharing or similarity of LPG domains to those of other parasite molecules discussed above raises the issue of specificity and the possibility of cross-activity. For example, many of the functions attributed to LPG above have been ascribed also to PPG, GIPLs, and/or GPI-anchored proteins (3, 6, 9, 12–14). Thus, teasing out the specific contributions of LPG within the complex milieu of the parasite glycocalyx remains a significant challenge.
Here we describe studies of a Leishmania major mutant specifically lacking LPG, which is defective in its ability to infect sand flies, mice, and macrophages (11, 15). The lpg1- mutant was obtained by targeted inactivation of a putative galactofuranosyltransferase necessary for synthesis of the LPG glycan core; the lpg1- mutant was otherwise normal in PG, GPI-anchored proteins, GIPLs, and metacyclic gene expression (11, 16, 17). We also made use of mouse mutants defective in relevant host defenses to extend and confirm our findings on the role(s) of LPG. Overall, the studies establish a role for LPG in many but not all of the steps previously identified in macrophage invasion and survival and, in some cases, raise questions about the relevance of several aspects of the interaction of Leishmania with its hosts assumed previously to be important.
Materials and Methods
Leishmania Culture. L. major LV39 clone 5 promastigotes (Rho/SU/59/P) (18) were grown in medium 199 (M199) (19). The LPG1 mutant (LPG1::HYG/LPG1::PAC), referred to as lpg1-, and its LPG1-restored derivative (LPG1::HYG/LPG1::PAC [pSNBR-LPG1]) referred to as lpg1-/+LPG1 or the LPG1 “add-back,” were grown briefly in the absence of selective agents before use (11). Metacyclics were prepared from day 4 stationary-phase cultures by an LPG-independent density-gradient centrifugation method (17).
Mouse Strains. BALB/c mice were obtained from Charles River Breeding Laboratories. C57BL/6-Cybbtm1 null mutant (phox-), C57BL/6-Nos2tm1Lau inducible NO synthase null mutant, C57BL/6, C5-deficient B10.D2-H2d H2-T18c Hc0/oSnJ, and C5-sufficient control B10.D2-H2d H2-T18c Hc1/nSnJ mice were obtained from The Jackson Laboratory.
Mouse Infections. Virulence was assessed after inoculation of mouse footpads (20). Groups were injected s.c. into the footpad with 105 metacyclic parasites per mouse. Infections were monitored by comparing the thickness of the injected and uninjected footpads with a Vernier caliper. Parasites were enumerated in the infected tissue by a limiting-dilution assay (21).
Macrophage Infections. Infections of mouse peritoneal exudate macrophages (PEMs) were performed and relative survival was calculated by normalizing values to the initial parasite burden at 2 h postinfection (11, 17, 22). Typically 104 macrophages were seeded in 24-well plates and incubated with 105 parasites in 500 μl. For synchronous infections, parasites were incubated at 4°C for 20 min for attachment, free parasites were removed by washing, and cultures were shifted to 37°C. Macrophages were activated with 100 ng/ml lipopolysaccharide (LPS; Escherichia coli serotype O26:B6) and 100 units/ml IFN-γ (Sigma). All media were endotoxin-free by the Pyrotell Limulus amoebocyte lysate test (Associates of Cape Cod).
Opsonization. For infections, parasites were incubated for 30 min with 4% C5-deficient mouse serum in DMEM (22). For flow cytometry, parasites were incubated with 25% normal or heat-inactivated mouse serum for 20 min at room temperature (RT). C3b binding was revealed with goat anti-C3b (clone H11, 1:100 dilution; Research Diagnostics, Flanders, NJ) and Alexa Fluor 488-conjugated anti-goat IgG. IgM binding was revealed with Alexa Fluor 488-conjugated anti-mouse IgM (1:100 dilution; Molecular Probes).
Measurement of Oxidant, pH, and Protease Resistance. The xanthine–xanthine oxidase system (23, 24) was used to generate oxidants in vitro. Logarithmic-phase promastigotes were washed with PBS and suspended in 150 μM xanthine in PBS at 5 × 106 cells per ml, and 0.5–5 units of xanthine oxidase per ml of cells. After 1 h at RT, cells were allowed to recover for 2 h in M199, plated at various densities (103 to 104 cells) in duplicate on M199 semisolid media plates, and incubated for 7–14 days to allow colony formation. Colony numbers were normalized to controls (plating efficiencies ranged between 10% and 30%), an “EC50” was calculated (typically 2 units/ml for WT), and the results were expressed as fold resistance relative to WT cells (Fig. 5B). Limiting-dilution assays were used to test the susceptibility of parasites to proteases (2.5% trypsin in DMEM for 30 min at RT), and pH effects were tested in growth assays in M199 at pH 5.5.
Fig. 5.

Intact LPG is not required for quiet entry (without macrophage activation) or for inhibition of macrophage activation. (A) lpg1- promastigotes do not activate PEMs. PEMs were infected with purified C3b-opsonized WT (open bars) or lpg1- (filled bars) metacyclics. As indicated, the PEMs were incubated 12 h before infection with 100 μM NMMA, 100 ng/ml LPS plus 100 units/ml IFN-γ, or NMMA plus LPS plus IFN-γ. NO production was measured after 24 h and parasite survival was monitored; only the 5-day survival data are shown. Two independent experiments were performed; bars show standard deviations of a representative triplicate experiment. (B) lpg1- parasites retain the ability to inhibit PEM activation. Purified serum-opsonized WT (open bars) or lpg1- (filled bars) metacyclics were allowed to infect PEMs for 6 h, and LPS plus IFN-γ were added for another 24 h. Controls (gray bars) were mock-infected PEMs. NO-derived nitrite in culture supernatants was determined by the Griess reaction (46). IL-12 p40 levels were determined in the PEM culture supernatants by an ELISA capture method (PharMingen). Two experiments were performed; the average and standard deviation for one with triplicate samples are shown.
Phagolysosomal Fusion. PEMs were seeded in 12-well plates onto 18-mm glass coverslips (3 × 105 cells per ml) and incubated overnight (at least 12 h) in DMEM supplemented with 10% FCS and 2.5 mg/ml FITC-conjugated dextran (10 kDa, lysine fixable; Molecular Probes). Cells were washed vigorously and synchronous uptake of either zymosan (106 particles per ml) or parasites [multiplicity of infection (moi) of 10 for WT, or of 3 and 10 for lpg1- low- and high-infectivity infections, respectively] was achieved as described above. Fusogenic FITC-positive phagosomes were quantified over a period of 3 h after uptake by fluorescence microscopy on paraformaldehyde-fixed preparations.
Complement Lysis. Human serum was obtained from healthy volunteers. After incubation of blood to allow clotting for 10 min at RT, serum was recovered by centrifugation for 10 min at 6,000 × g at 4°C. C5-depleted human serum and human C5 were obtained from Sigma. Mouse serum was obtained by heart puncture of anesthetized animals. Washed parasites (106 cells in 500 μl of DMEM with 40 μg/ml propidium iodide, lacking FBS) were incubated at RT with human serum for 30 min, and fluorescence (lysis) was measured by flow cytometry.
Results
lpg1- Parasites Are Sensitive to Lysis by Human but Not Mouse Serum. Purified metacyclic promastigotes were incubated with various amounts of fresh human serum, and the motility and integrity of the parasites were assessed. lpg1- parasites were lysed rapidly by 1% serum, whereas WT were resistant up to 10% serum (data not shown). Quantitation of lysis by flow cytometry confirmed that WT and the LPG1 add-back controls were resistant, whereas lpg1- parasites were lysed (Fig. 1A). This lysis was due to complement (and not other) serum factors, because C5-deficient human serum was inactive unless C5 was added (data not shown).
Fig. 1.

lpg1- metacyclic promastigotes are sensitive to human but not mouse complement. (A) Flow cytometric analysis of lysis by human serum. Metacyclic WT, lpg1-, and lpg1-/+LPG1 lines were incubated in the presence (shaded) or absence (open) of 2% human serum for 30 min in the presence of propidium iodide and then subjected to flow cytometry. (B) Infections of normal and C5-deficient mice. WT (○) or lpg1- (▵) metacyclics (105) were inoculated into the footpad of isogenic WT (Upper) or C5-deficient (Lower) mice, and lesion formation was followed. The average and standard deviation are shown for groups of five mice; this experiment is representative of three independent tests.
In contrast, incubations with up to 60% fresh mouse serum (BALB/c or C57BL/6) failed to show detectable lysis of the lpg1- parasite (data not shown). Isogenic C57BL/10Sn WT and C5-deficient mice were infected with WT and lpg1- parasites. The lpg1- parasites showed a delay in lesion progression in the C57BL background similar to that seen in susceptible BALB/c mice (11) (Fig. 1B; as expected, the magnitude of the lesion was reduced in this genetically resistant strain). Second, attenuation of the lpg1- parasite did not differ in the C5-sufficient or deficient C57BL mice (Fig. 1B). Together these data suggest that the lytic activity of mouse complement plays little role against Leishmania in vitro or in vivo.
lpg1- Parasites Enter Macrophages Normally. Parasites were incubated with normal or heat-inactivated mouse serum, labeled with anti-C3b antiserum, and analyzed by flow cytometry. Both WT and lpg1- parasites bound similar levels of C3b, which suggests that LPG is not required for opsonization (Fig. 2A). Similar results were obtained for IgM binding to both WT and lpg1- (data not shown). We then examined the ability of the opsonized cells to enter cultured PEMs over a 2-h period as a function of the multiplicity of infecting parasites per PEM (Fig. 2B). WT and the control add-back parasites entered macrophages identically, whereas lpg1- parasites showed a 4-fold increase (Fig. 2B). We have noticed a tendency for this and other LPG-deficient parasites in virulent Leishmania backgrounds to form aggregates in culture, which could account for the apparent increase. These data argue that LPG is not an essential adhesin.
Fig. 2.

lpg1- metacyclic promastigote parasites are opsonized efficiently by C3b and can invade macrophages. (A) Opsonization by C3b. WT (Left)or lpg1- (Right) metacyclics were treated with normal (shaded) or heat-inactivated (open) mouse serum, and deposition of C3b was quantitated by flow cytometry. Controls in which anti-C3b was omitted gave results similar to that obtained with heat-inactivated serum (data not shown). (B) Attachment and entry of opsonized metacyclics into macrophages (mφ). Metacyclics were opsonized with C5-deficient mouse serum and incubated with cultured PEMs (3 × 105 per well; WT, ○; lpg1-, ▵; and lpg1-/+LPG1, •) at the indicated moi. For attachment (Left), parasites were incubated for 20 min at 4°C; for uptake (Right) parasites were incubated for 2 h more at 37°C. Free parasites were removed by washing with cold PBS, and cells were fixed with 3.5% paraformaldehyde, stained, and counted. The bars show the standard deviations of three replicates and are representative of at least three independent experiments.
lpg1- Parasites Are Sensitive to Oxidants.
Serum-opsonized WT and lpg1- parasites induced similar
levels of 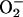 production in PEMs,
≈50% of that induced by the opsonized zymosan control
(Fig. 3A
Left). This finding suggested that neither LPG nor L. major
down-regulate the oxidative burst during macrophage invasion. The
lpg1- cells showed a significant 2-fold increase in their
sensitivity to oxidants generated by the xanthine–xanthine oxidase
system; the control add-backs showed similar sensitivity as WT
(Fig. 3A
Right).
production in PEMs,
≈50% of that induced by the opsonized zymosan control
(Fig. 3A
Left). This finding suggested that neither LPG nor L. major
down-regulate the oxidative burst during macrophage invasion. The
lpg1- cells showed a significant 2-fold increase in their
sensitivity to oxidants generated by the xanthine–xanthine oxidase
system; the control add-backs showed similar sensitivity as WT
(Fig. 3A
Right).
Fig. 3.

lpg1- promastigotes are susceptible to oxidative stress. (A) lpg1- and WT Leishmania induce comparable PEM oxidative bursts (Left). PEMs were treated with zymosan or incubated with 4-day stationary-phase WT or lpg1- promastigotes for 90 min at 37°C, and superoxide production was determined (45). The average and standard deviation of triplicate determinations are shown, representative of two independent experiments. lpg1- parasites are more susceptible to oxidative stress (Right). The relative resistance of WT, lpg1-, and lpg1-/+LPG1 promastigotes to oxidants generated by the xanthine–xanthine oxidase system is shown. An EC50 was calculated, data were normalized to WT, and the average and standard deviation are shown. (B) lpg1- survives normally in infections of phox- macrophages. Infection efficiency (% infected cells, Top), intracellular growth (parasites per infected cell, Middle), and parasite survival (parasites per 100 cells, Bottom) of C3b-opsonized WT and lpg1- metacyclics in PEMs derived from C57BL/6 control (phox+) or isogenic oxidant-deficient (phox-) mice are shown. Three independent experiments were performed; bars show the standard deviations of one representative triplicate experiment.
To assess the significance of LPG-dependent oxidant resistance in vivo, PEMs were prepared from resistant C57BL/6 control and phox- mice (defective in the phagocyte oxidase system) (25), and infected by WT and lpg1- metacyclics (Fig. 3B). As for BALB/c PEMs (11), WT survived well and lpg1- parasites showed considerable destruction within 2 days (Fig. 3B Left). In contrast, the host phox- mutation was able to compensate for the LPG-dependent oxidant sensitivity, as WT and lpg1- parasites showed comparable abilities to survive in infections of phox- PEMs during the first 2 days (Fig. 3B Right). Thus, LPG plays an important role in oxidant resistance under physiologically relevant conditions.
Curiously, if the infection was extended to 5 days, a time at which LPG is normally absent, lpg1- survival was decreased in the phox- macrophages as well (Fig. 3B Right). The significance of this finding is not obvious.
Release of PGs from L. major During Macrophage Invasion and Inhibition of Phagolysosomal Fusion. We performed synchronous macrophage infections of serum-opsonized metacyclic WT and lpg1- parasites, and we monitored the release of PGs with the monoclonal antibody WIC 79.3 (Fig. 4A). Immediately after infection, PGs were confined to the parasite, but after 1 h, PG labeling was seen throughout WT-infected macrophages with evident punctate and diffuse localization (Fig. 4A and data not shown). Only a weaker punctate labeling pattern was evident with lpg1- parasites (Fig. 4A), reflecting the deposition of other less abundant PGs (such as PPGs; ref. 11) within a vesicular compartment. The kinetics of macrophage PG deposition was similar for the WT and lpg1- parasites, and by 1 h most macrophages were labeled (data not shown).
Fig. 4.

Release and effects of PGs in infected macrophages. (A) Release of PGs. PEMs were incubated (20 min at 4°C) with serum-opsonized metacyclic WT or lpg1- promastigotes (moi of 10 or 3, respectively, to normalize for initial uptake; see Fig. 2B) and then incubated for up to3hat37°C. Cells were fixed with 3.5% paraformaldehyde (5 min at RT), permeabilized with 100% ethanol (15 min at 4°C), incubated for 20 min at 37°C with anti-PG antibody (1:500 dilution of WIC79.3) and anti-tubulin antibody (1:2,000 dilution of anti-tubulin primary antibodies), washed with PBS, and incubated further with an FITC-conjugated goat anti-mouse IgG and Texas-red-conjugated donkey anti-rabbit IgG (each at 1:200 dilution; Jackson Immunochemicals). Exposures were 0.66 s for WT-infected macrophages and 0.25 s for lpg1--infected macrophages (because PG levels are much higher in WT Leishmania). (Scale bar = 5 μm.) (B and C) lpg1- metacyclic parasites are defective in inhibition of phagolysosomal fusion and survival. Phagolysosomal fusion was quantitated by labeling of parasitophorous vacuoles with FITC-dextran, loaded previously into the lysosomal compartment (see Materials and Methods). The ratio of infecting lpg1- to PEMs was varied, and the effect on phagolysosomal fusion (B, 3 h postinfection) or parasite survival (C) was determined in triplicate. Purified C3-opsonized metacyclic lpg1- promastigotes were infected at an moi of 3 (low) or 10 (high); at the 2-h time point, 1.4 or 7.4 intracellular parasites per infected macrophage were obtained, respectively. The bars indicate standard deviations; one experiment is shown, which is representative of three independent experiments.
A fluorescence microscopy assay was used to measure inhibition of phagolysosomal fusion after invasion of C3-opsonized Leishmania. Within 3 h, most control zymosan-containing phagosomes showed fusion with the FITC-dextran-labeled lysosomes, whereas only 20% of the phagosomes containing WT or lpg1-/+LPG1 had fused (Fig. 4B). We compared the ability of lpg1- parasites to inhibit phagolysosomal fusion and survive in WT PEMs as a function of moi; in these experiments, the actual intracellular moi was determined. At low mois (1–2 parasites per infected macrophage), lpg1- parasites failed to inhibit phagosomal fusion and did not survive (Fig. 4 B and C, “lpg1- low”).
Later studies, however, questioned the role of phagolysosomal fusion in parasite survival. At high mois (>6 parasites per infected macrophage), the lpg1- parasites showed inhibition of phagosomal fusion comparable to WT (Fig. 4B, “lpg1- high”), possibly resulting from the low levels of residual PGs in the lpg1- parasite. Despite their ability to inhibit phagosomal fusion at high mois (Fig. 4C), the lpg1- parasites were eliminated (Fig. 4C). Similarly, survival of the lpg1- parasites in phox- macrophages occurred despite the fact that these macrophages showed fusogenicity identical to that of control macrophages when infected with either WT (17% vs. 14.5% fusion for phox- vs. WT PEMs) or lpg1- (64.5% vs. 55% fusion) parasites. Last, lpg1- parasites showed no difference from WT in their resistance to low pH or treatment with proteases (data not shown).
lpg1- Parasites Retain the Ability to Inhibit Macrophage Activation Pathways. Purified LPG inhibits macrophage signaling and activation in vitro (13, 26, 27), as revealed by monitoring key effectors such as NO and IL-12. However, infection of BALB/c PEMs by lpg1- did not lead to significant induction of either effector, despite destruction of >70% of the invading parasites (Fig. 5A). Addition of the NO synthase (NOS) inhibitor NG-monomethyl-L-arginine (NMMA) did not improve lpg1- survival, confirming the lack of involvement of NO in lpg1- destruction (Fig. 5A). That these macrophages were competent to kill Leishmania in an NO-dependent manner was shown in infections of PEMs previously activated by treatment with LPS + IFN-γ. These PEMs synthesized abundant NO and destroyed WT and lpg1- parasites completely, whereas inhibition of NOS activity with NMMA restored survival to that seen in nonactivated macrophages (30%; Fig. 5A). The lack of involvement of NO in the elimination of lpg1- parasites was supported by infections of PEMs derived from inducible NOS-knockout mice, where lpg1- parasites were destroyed also (data not shown).
We then asked whether infection by lpg1- parasites rendered infected macrophages resistant to subsequent activation. Parasites were allowed to infect PEMs for 6 h and were treated with LPS + IFN-γ for 24 h. As expected, control uninfected macrophages produced NO and IL-12, whereas macrophages infected with WT parasites showed inhibition of both NO and IL-12 production (Fig. 5B). Again, the lpg1- parasites resembled WT in that production of both IL-12 and NO was also inhibited despite considerably decreased survival (Fig. 5B). Thus, LPG is not required for the Leishmania parasite to enter the host cell quietly (without macrophage activation) or to inhibit the ability of macrophages to become activated by other stimuli.
Discussion
The availability of the L. major lpg1- mutant specifically defective in LPG synthesis (11, 17) allowed us to assess the contribution of LPG to specific steps implicated in parasite survival and establishment of infection in its mammalian host. In support of previous studies, LPG-deficient L. major were highly sensitive to human complement, more susceptible to oxidants generated by a xanthine–xanthine oxidase system, and unable to inhibit phagolysosomal fusion immediately after invasion (Figs. 1, 3, and 4). In these properties, the in vitro and in vivo results were concordant.
The contribution of these LPG-dependent functions to parasite virulence in vivo was assessed with the aid of mutant macrophages or mice defective in specific defense mechanisms. The increased susceptibility of the lpg1- parasite to oxidants was relatively modest, albeit comparable to that seen in other pathogens defective in several well-studied oxidant defense systems (28, 29). Interestingly, WT and lpg1- parasites induced similar levels of oxidant production in normal macrophages (Fig. 3A). Its importance was shown in infections of oxidative-burst-defective phox- mouse macrophages, where the LPG-deficient parasites survived as well as WT during the first 2 days when LPG is present. This finding established from both the parasite's and the host's perspective in vivo the importance of LPG in resistance to oxidative defenses.
A different perspective emerged from our studies showing that loss of LPG conferred susceptibility to lysis by human complement (Fig. 1; ref. 30). This result occurred in the presence of the abundant surface protease gp63, which has also been implicated in susceptibility to complement lysis in several Leishmania species (31, 32). Because complement resistance in Leishmania arises by prevention of binding of the membrane attack complex (8), these data suggest that any perturbation leading to disruption of the dense parasite glycocalyx will confer complement sensitivity.
We were surprised to find that complement lytic activity was not an effective defense mechanism against Leishmania in the inbred mice commonly used as virulence models. This finding arose from direct tests of mouse serum and mice genetically lacking the C5 component of complement (Fig. 2B). Although common inbred strains of laboratory mice were known to have vanishingly low lytic complement activity (33, 34), the relevance to murine models of microbial pathogenesis has not been widely appreciated. Our data suggest that for Leishmania and perhaps other pathogens, inbred mice may not be good models for probing the role of the lytic functions of complement relevant to human infectious diseases (in contrast to their utility in studying other aspects of the complement pathway) (8).
Whereas Leishmania amastigotes reside in acidified, fusogenic phagosomes (35), metacyclic L. major promastigotes transiently inhibited phagolysosomal fusion in an LPG-dependent manner (Fig. 4B), as seen in Leishmania donovani (36). Transient fusion inhibition was thought to be required for protection while the parasite completes its differentiation to the amastigote stage within the first 2 days. Several predictions of this model, however, are not supported. First, infections of phox- macrophages restored survival of the LPG-deficient parasite nearly to that of WT in the first 2 days (when LPG is normally present), yet the lpg1- parasites resided in fusogenic phagosomes in the phox- macrophages (Fig. 3). One would not expect phox- rescue if fusion inhibition was important for survival during this time. Second, infection of macrophages with high numbers of parasites, which restored inhibition of phagolysosomal fusion by the lpg1- parasites, did not reverse parasite destruction (Fig. 4). Third, lpg1- parasites were as resistant as WT to proteases, low pH conditions, and, previously, to sand fly gut proteases in vivo (15). Last, in Leishmania infections of macrophages of STAT1-knockout mice, which show a defect in phagosome acidification, rescue of lpg1- survival relative to WT was not observed (G.F.S., P. Schlesinger, R. Schreiber, and S.M.B., unpublished work).
Thus, the data collectively lead to the remarkable conclusion that although transient LPG-mediated phagolysosomal fusion inhibition occurs after parasite uptake, it does not play a prominent role in the ability of Leishmania to establish macrophage infections. Possibly, inhibition of fusion is just an ancillary effect of LPG peripheral to its other roles in parasite virulence, as seen with amphipathic molecules with structural features resembling LPG in a variety of contexts (37). Alternatively, transient inhibition of fusion may lead to effects downstream of the “early” acidification or hydrolytic processes, as suggested by the finding that lpg1- parasites survive poorly in the phox- macrophages after 5 days, a time when LPG has largely disappeared (Fig. 3B). Other possibilities include steps that are unrelated to parasite survival in macrophages in vitro but that are important in later steps in pathogenesis in mice, such as antigen presentation (38).
Our data also did not provide support for a prominent role for LPG in several processes required for macrophage infection. One involves parasite uptake and entry; studies have shown that LPG serves as a major adhesin mediating binding and entry of Leishmania into macrophages, probably after opsonization by C3 (39). However, C3b deposition and uptake of the lpg1- mutant were not altered, suggesting that Leishmania possess an abundance of alternative C3 acceptors (Fig. 2). Similar results were obtained with IgM binding, which has also been implicated in Leishmania entry (40).
Interestingly, ligation of complement receptors after uptake of opsonized parasites has been suggested to mediate deactivation of macrophage signaling pathways relevant to infection, such as those leading to NO and IL-12 synthesis (8, 40). This hypothesis could explain why the lpg1- parasite retained the ability to enter macrophages without stimulation of NO or IL-12 production and to inhibit these pathways after establishment of infection (Fig. 5). However, many macrophage receptors have been implicated in Leishmania uptake with no clear consensus on which (if any) are specifically required for survival (41). The lpg1- mutant synthesizes a residual LPG glycan core-phosphatidylinositol anchor (11), related to the abundant heterogeneous parasite GIPLs also implicated in modulation of host cell signaling (14, 27, 42). Possibly, the role of LPG in macrophage deactivation could be redundant with GIPLs or other parasite molecules such as PPG, which has also been implicated in parasite virulence (13, 14, 43). To resolve these questions, it will be necessary to generate other L. major mutants defective in individual or multiple classes of candidate molecules.
In contrast to the importance of LPG to the virulence of L. major, lpg1- mutants of Leishmania mexicana, an agent of cutaneous leishmaniasis in the New World, show very little effect on parasite virulence in infections of mice or macrophages (12). This discrepancy extends to other LPG genes (refs. 5 and 44; G.F.S., L.-F. Lye, H. Segawa, D. L. Sacks, S.J.T., and S.M.B., unpublished work). These data and studies of the host immune response suggest that different Leishmania species place different emphasis on the importance of canonical virulence determinants, including LPG (5). Our mechanistic studies of the defects of LPG-deficient L. major not only provide confirmation of its importance in this species but also will serve as a guide for future work concerning the differences among species. In this regard, limitations in the ability of murine models to assess candidate Leishmania virulence factors relevant to human infection (such as lysis by complement) may contribute to the perceived differences among species. Regardless, our findings firmly establish the importance of LPG in several key steps of the infectious cycle of L. major and emphasize the importance of mechanistic approaches involving both the host and the pathogen.
Acknowledgments
We thank D. Russell and E. Brown for antisera, J. Atkinson for advice on mouse complement, S. Hickerson for complement tests of various Leishmania species, and D. E. Dobson and J. Vogel for reading this manuscript and for discussions. This work was supported by National Institutes of Health Grant AI31078, the Deutsche Akademische Austauschdienst, and the Human Frontiers Science Program (to G.F.S.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: GIPLs, glycosylinositolphospholipids; GPI, glycosylphosphatidylinositol; LPG, lipophosphoglycan; LPS, lipopolysaccharide; moi, multiplicity of infection; NMMA, NG-monomethyl-l-arginine; PEMs, peritoneal exudate macrophages; PG, phosphoglycan; PPGs, proteophosphoglycans; RT, room temperature.
References
- 1.McConville, M. J., Mullin, K. A., Ilgoutz, S. C. & Teasdale, R. D. (2002) Microbiol. Mol. Biol. Rev. 66, 122-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beverley, S. M. & Turco, S. J. (1998) Trends Microbiol. 6, 35-40. [DOI] [PubMed] [Google Scholar]
- 3.Ilg, T. (2001) Med. Microbiol. Immunol. 190, 13-17. [DOI] [PubMed] [Google Scholar]
- 4.Ilgoutz, S. C. & McConville, M. J. (2001) Int. J. Parasitol. 31, 899-908. [DOI] [PubMed] [Google Scholar]
- 5.Turco, S. J., Späth, G. F. & Beverley, S. M. (2001) Trends Parasitol. 17, 223-226. [DOI] [PubMed] [Google Scholar]
- 6.Ilg, T., Handman, E. & Stierhof, Y. D. (1999) Biochem. Soc. Trans. 27, 518-525. [DOI] [PubMed] [Google Scholar]
- 7.Descoteaux, A. & Turco, S. J. (1999) Biochim. Biophys. Acta 1455, 341-352. [DOI] [PubMed] [Google Scholar]
- 8.Mosser, D. M. & Brittingham, A. (1997) Parasitology 115, S9-S23. [DOI] [PubMed] [Google Scholar]
- 9.Handman, E. (1999) Adv. Parasitol. 44, 1-39. [DOI] [PubMed] [Google Scholar]
- 10.Sacks, D. & Kamhawi, S. (2001) Annu. Rev. Microbiol. 55, 453-483. [DOI] [PubMed] [Google Scholar]
- 11.Späth, G. F., Epstein, L., Leader, B., Singer, S. M., Avila, H. A., Turco, S. J. & Beverley, S. M. (2000) Proc. Natl. Acad. Sci. USA 97, 9258-9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ilg, T. (2000) EMBO J. 19, 1953-1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Proudfoot, L., Nikolaev, A. V., Feng, G. J., Wei, W. Q., Ferguson, M. A., Brimacombe, J. S. & Liew, F. Y. (1996) Proc. Natl. Acad. Sci. USA 93, 10984-10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Proudfoot, L., O'Donnell, C. A. & Liew, F. Y. (1995) Eur. J. Immunol. 25, 745-750. [DOI] [PubMed] [Google Scholar]
- 15.Sacks, D. L., Modi, G., Rowton, E., Späth, G. F., Epstein, L., Turco, S. J. & Beverley, S. M. (2000) Proc. Natl. Acad. Sci. USA 97, 406-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang, C. & Turco, S. J. (1993) J. Biol. Chem. 268, 24060-24066. [PubMed] [Google Scholar]
- 17.Späth, G. F. & Beverley, S. M. (2001) Exp. Parasitol. 99, 97-103. [DOI] [PubMed] [Google Scholar]
- 18.Marchand, M., Daoud, S., Titus, R. G., Louis, J. & Boon, T. (1987) Parasite Immunol. (Oxf.) 9, 81-92. [DOI] [PubMed] [Google Scholar]
- 19.Kapler, G. M., Coburn, C. M. & Beverley, S. M. (1990) Mol. Cell. Biol. 10, 1084-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Titus, R. G., Muller, I., Kimsey, P., Cerny, A., Behin, R., Zinkernagel, R. M. & Louis, J. A. (1991) Eur. J. Immunol. 21, 559-567. [DOI] [PubMed] [Google Scholar]
- 21.Titus, R. G., Marchand, M., Boon, T. & Louis, J. A. (1985) Parasite Immunol. (Oxf.) 7, 545-555. [DOI] [PubMed] [Google Scholar]
- 22.Racoosin, E. L. & Beverley, S. M. (1997) Exp. Parasitol. 85, 283-295. [DOI] [PubMed] [Google Scholar]
- 23.Ager, A. (1982) Agents Actions 11, Suppl., 73-81. [PubMed] [Google Scholar]
- 24.Chan, J., Fujiwara, T., Brennan, P., McNeil, M., Turco, S. J., Sibille, J. C., Snapper, M., Aisen, P. & Bloom, B. R. (1989) Proc. Natl. Acad. Sci. USA 86, 2453-2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pollock, J. D., Williams, D. A., Gifford, M. A., Li, L. L., Du, X., Fisherman, J., Orkin, S. H., Doerschuk, C. M. & Dinauer, M. C. (1995) Nat. Genet. 9, 202-209. [DOI] [PubMed] [Google Scholar]
- 26.Descoteaux, A., Turco, S. J., Sacks, D. L. & Matlashewski, G. (1991) J. Immunol. 146, 2747-2753. [PubMed] [Google Scholar]
- 27.McNeely, T. B., Rosen, G., Londner, M. V. & Turco, S. J. (1989) Biochem. J. 259, 601-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsolis, R. M., Baumler, A. J. & Heffron, F. (1995) Infect. Immun. 63, 1739-1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seyler, R. W., Jr., Olson, J. W. & Maier, R. J. (2001) Infect. Immun. 69, 4034-4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puentes, S. M., Sacks, D. L., da Silva, R. P. & Joiner, K. A. (1988) J. Exp. Med. 167, 887-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brittingham, A., Morrison, C. J., McMaster, W. R., McGwire, B. S., Chang, K. P. & Mosser, D. M. (1995) J. Immunol. 155, 3102-3011. [PubMed] [Google Scholar]
- 32.Joshi, P. B., Kelly, B. L., Kamhawi, S., Sacks, D. L. & McMaster, W. R. (2002) Mol. Biochem. Parasitol. 120, 33-40. [DOI] [PubMed] [Google Scholar]
- 33.Ong, G. L. & Mattes, M. J. (1989) J. Immunol. Methods 125, 147-158. [DOI] [PubMed] [Google Scholar]
- 34.Atkinson, J. P., McGinnis, K. & Shreffler, D. (1980) J. Immunol. Methods 33, 351-368. [DOI] [PubMed] [Google Scholar]
- 35.Russell, D. G., Xu, S. & Chakraborty, P. (1992) J. Cell Sci. 103, 1193-1210. [DOI] [PubMed] [Google Scholar]
- 36.Desjardins, M. & Descoteaux, A. (1997) J. Exp. Med. 185, 2061-2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miao, L., Stafford, A., Nir, S., Turco, S. J., Flanagan, T. D. & Epand, R. M. (1995) Biochemistry 34, 4676-4783. [DOI] [PubMed] [Google Scholar]
- 38.Moll, H. (2000) Adv. Exp. Med. Biol. 479, 163-173. [DOI] [PubMed] [Google Scholar]
- 39.Mosser, D. M. & Rosenthal, L. A. (1993) Semin. Cell Biol. 4, 315-322. [DOI] [PubMed] [Google Scholar]
- 40.McDowell, M. A. & Sacks, D. L. (1999) Curr. Opin. Microbiol. 2, 438-443. [DOI] [PubMed] [Google Scholar]
- 41.Alexander, J., Satoskar, A. R. & Russell, D. G. (1999) J. Cell Sci. 112, 2993-3002. [DOI] [PubMed] [Google Scholar]
- 42.Frankenburg, S., Leibovici, V., Mansbach, N., Turco, S. J. & Rosen, G. (1990) J. Immunol. 145, 4284-4289. [PubMed] [Google Scholar]
- 43.Piedrafita, D., Proudfoot, L., Nikolaev, A. V., Xu, D., Sands, W., Feng, G. J., Thomas, E., Brewer, J., Ferguson, M. A., Alexander, J. & Liew, F. Y. (1999) Eur. J. Immunol. 29, 235-244. [DOI] [PubMed] [Google Scholar]
- 44.Ilg, T., Demar, M. & Harbecke, D. (2001) J. Biol. Chem. 276, 4988-4997. [DOI] [PubMed] [Google Scholar]
- 45.Johnston, R. B., Jr. (1978) Fed. Proc. 37, 2759-2764. [PubMed] [Google Scholar]
- 46.Green, L. C., Wagner, D. A., Glogowski, J., Skipper, P. L., Wishnok, J. S. & Tannenbaum, S. R. (1982) Anal. Biochem. 126, 131-138. [DOI] [PubMed] [Google Scholar]