

Abstract
Covalent attachment of palmitate to proteins is a post-translational modification that exerts diverse effects on protein localization and function. The three key technical approaches required for an investigator to determine the role of palmitoylation of Your Favorite Palmitoylated Protein (YFPP) are methods to: 1) detect YFPP palmitoylation; 2) alter or inhibit palmitoylation of YFPP; 3) determine the functional significance of altered YFPP palmitoylation. Here, I describe experimental methods to address these three issues. Both radioactive (radiolabeling with 3H palmitate or 125I-IC16 palmitate) and non-radioactive (chemical labeling and mass spectrometry) methods to detect palmitoylated proteins are presented. Next, techniques to inhibit protein palmitoylation are described. These include site specific mutagenesis, and treatment of cells with inhibitors of protein palmitoylation, including 2-bromopalmitate, cerulenin, and tunicamycin. Alternative methods to replace palmitate with other fatty acids are also presented. Finally, general approaches to determining the effect of altered palmitoylation status on YFPP association with membranes and lipid rafts, as well as signal transduction, are described.
Keywords: palmitate, 2-bromopalmitate, lipid rafts, plasma membrane, fatty acylation, polyunsaturated fatty acids, cerulenin, tunicamycin
1. Introduction
Increasing numbers of proteins have been shown to be covalently modified by the 16 carbon saturated fatty acid palmitate [1–3]. Attachment of palmitate to proteins occurs primarily via thio-ester linkage to cysteine (S-palmitoylation). Palmitoylated proteins can be categorized into five general classes. One group consists of transmembrane proteins that are S-acylated on cysteines at or near the transmembrane domain. A second group is typified by the Ras family. S-Palmitoylation occurs within the C-terminal region and is dependent on prior prenylation of the cysteine residue within the C-terminal “CAAX” box. A third class of proteins is S-palmitoylated at one or more cysteines near the N- or C-terminus. Members of the Src family of tyrosine protein kinases typify the fourth set of proteins. Seven of the nine Src family members, as well as several α subunits of heterotrimeric G proteins, contain a consensus sequence for dual acylation within their N-terminal SH4 domain: Met-Gly-Cys. Gly-2 is N-myristoylated and Cys-3 is S-palmitoylated [4,5]. N-myristoylation is required for subsequent palmitoylation, most likely because the presence of a myristate moiety enhances accessibility to a membrane bound palmitoyl acyl transferase. Recently, a fifth class of palmitoylated proteins, consisting of Hedgehog and the Gαs subunit, was identified [6,7]. These proteins contain palmitate covalently bound via amide-linkage to an N-terminal cysteine residue (N-palmitoylation).
Protein palmitoylation exerts diverse effects on the modified proteins. For example, attachment of this long chain fatty acid increases protein hydrophobicity and thereby increases membrane association of the modified protein. Palmitoylation also promotes protein targeting to lipid rafts, membrane microdomains that are enriched in cholesterol and glycosphingolipids [8,9]. Raft association enhances the protein/protein and protein/lipid interactions that are important for efficient signal transduction. The lipids in rafts primarily contain saturated fatty acid chains, allowing the lipid molecules to pack tightly together and form a “liquid ordered” phase. The saturated nature of the palmitoyl chain promotes insertion of the fatty acylated protein into liquid ordered membrane domains [10]. Palmitoylation also influences intracellular protein trafficking and targeting to specific subcellular membranes, protein-protein interactions and protein activity [3,11].
Given the diverse functions attributable to palmitoylated proteins, it is important to have appropriate techniques that can be readily applied to analyzing the functional significance of palmitate attachment. In this report, I describe multiple methods that can be used to monitor palmitoylation of Your Favorite Palmitoylated Protein (YFPP) and its functional significance. The basic assumption is that the investigator has available a cDNA clone encoding YFPP, and/or anti-YFPP antibody, and/or an epitope-tagged YFPP cDNA clone. Methods are first described to enable the investigator to confirm that YFPP is indeed palmitoylated. Next, I outline and describe in detail the use of analogs and inhibitors to block and/or alter protein palmitoylation. Finally, general methods to assess the functional significance of YFPP palmitoylation are outlined.
2. Methods to detect palmitoylation of proteins
There are two choices of methods for detecting palmitate incorporation into proteins: radioactive and non-radioactive. As outlined below, each method has its own distinct advantages and disadvantages. It is recommended that cells be transfected with a mammalian expression vector encoding YFPP. The advantage of this approach is that the transfected, exogenous protein is typically overexpressed several-fold compared to levels of endogenous YFPP, thus facilitating detection of palmitate incorporation. Commonly used laboratory cell lines that are readily transfectable include COS-1 cells, NIH 3T3 cells, MDCK cells, and 293T cells; these cell lines are available from the ATCC (American Type Culture Collection; Manassas, VA). In addition to the calcium phosphate method of transfection [12], there are a number of pre-made commercially available transfection reagents that can be used, such as Lipofectamine 2000 (Invitrogen, Carlsbad, CA), or FuGENE 6 (Roche Diagnostics, Indianapolis, IN). Cells are typically assayed 24–36 hrs after transient transfection. Stable cell lines that have been selected for stable expression of YFPP can also be used. Alternatively, if no cDNA to YFPP is available, but anti-YFPP antibody has been generated, palmitoylation of endogenous YFPP can be monitored.
2.1 Radiolabeling with 3H Palmitate
To perform the radiolabeling, cells growing in 100 mm dishes are starved for 1 h at 37°C in medium (eg DME) containing 2% dialyzed fetal bovine serum and radiolabeled for 4 hrs at 37°C in the same medium containing 25–100 μCi/ml 9,10-[3H] palmitic acid (Perkin Elmer Life and Analytical Sciences, Boston, MA). The cell monolayers are then rinsed with cold STE (100 mM NaCl, 10 mM Tris, pH 7.4, 1 mM EDTA) and lysed in 2 ml cold lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 2 mM EDTA, 5 mM NaF). Lysates are clarified by centrifugation at 100,000xg for 15 min at 4°C and then immunoprecipitated with antibody to YFPP. The immunoprecipitates are analyzed by SDS-PAGE. To detect the 3H emission signal by fluorography, gels are soaked in H20 for 30 minutes and then 1 M NaSalicylate for 30 minutes. Dried gels are exposed to X-ray film, BioMax MR or X-omat AR (Eastman Kodak Co, Rochester, NY) and then developed and fixed.
2.2 Radiolabeling with 125I-IC16 Palmitate
IC16 palmitate (16-iodo-hexadecanoic acid) is a palmitate analog containing a molecule of iodine at the ω carbon (Figure 1). This compound can be synthesized by published methods [13,14]; small aliquots are also available by request from this author. Radio-iodination is accomplished by incubation with Na125I-Iodide to generate 125I-IC16 palmitate (16-125I-iodohexadecanoic acid) [14]. Use of 125I-IC16 palmitate for labeling palmitoylated proteins provides two distinct advantages over [3H]-palmitate. First, the IC16 analog exhibits minimal metabolic interconversion [13]. Second, the gamma radiation emitted from 125I can be readily detected by phosphorimaging technology with vastly reduced exposure times, compared to fluorography with 3H. For example, detection of palmitate incorporation into Fyn, a Src family kinase, required 1–2 weeks of exposure for the fluorogram with 3H palmitate vs 1 day (24 hr) exposure for 125I-IC16 palmitate labeling.
Figure 1.
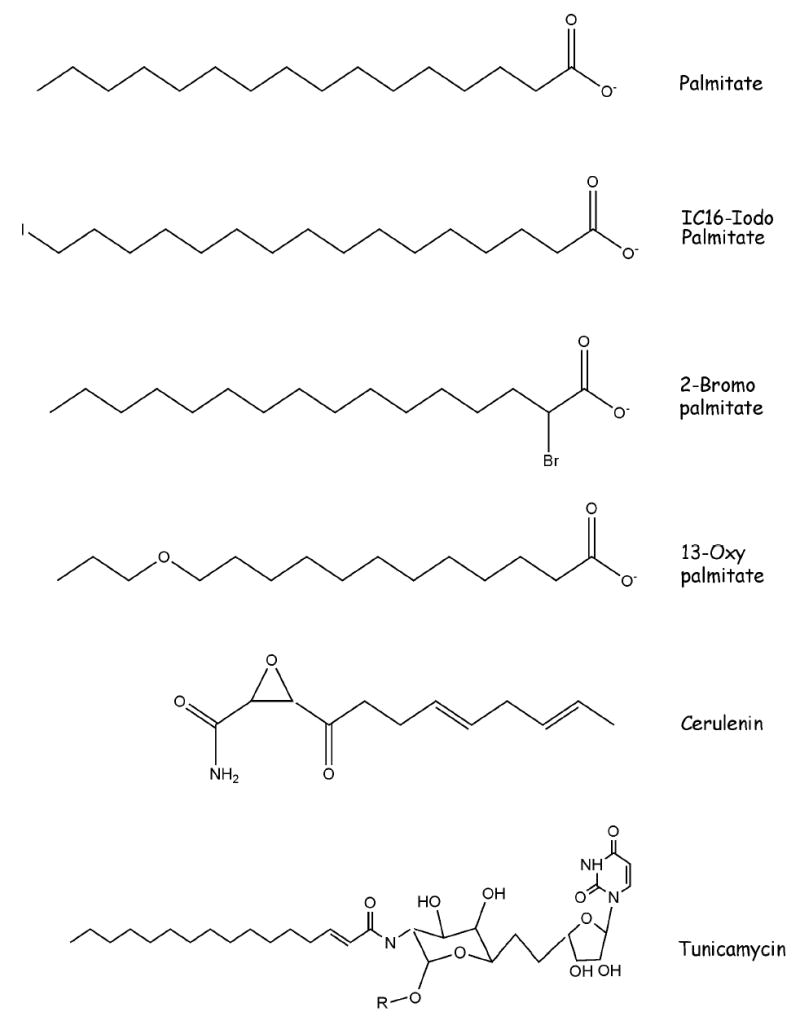
Chemical Structures of Palmitate Analogs and Inhibitors
To perform the radiolabeling, cells are starved for 1 h at 37°C in medium (eg DME) containing 2% dialyzed fetal bovine serum and radiolabeled for 4 hrs at 37°C in the same medium containing 25–50 μCi/ml 125I-IC16. Cells are lysed and immunoprecipitated as described above for [3H]palmitate labeling. The final pellet is resuspended in SDS-PAGE sample buffer containing 0.1 M dithiothreitol instead of β-mercaptoethanol, and the samples are not boiled. After SDS-PAGE, the gels are dried and exposed directly to a phosphorimaging screen. Quantitation of the signal can be accomplished using the imaging software provided with the phosphorimager system (eg, we use ImageQuant software with a Molecular Dynamics Phosphorimager, (Molecular Dynamics, Sunnyvale, CA)).
The investigator must take care to utilize appropriate precautions when working with 125I, a potent γ emitter. All containers must be shielded with lead, which blocks nearly all γ emissions. We typically wrap plexiglass tissue culture trays and test tube racks with thin pieces of lead sheeting and handle all 125I-labeled samples behind lead impregnated plexiglass shields.
2.3 Chemical Labeling of the Palmitoylation Site
An non-radioactive alternative to monitoring incorporation of radiolabeled palmitate is to chemically label the palmitoylation site [15,16]. This method is described in detail in Chapter 1 of this volume. Immunoprecipitates containing YFPP are first treated with N-ethyl maleimide, to block free sulhydryl groups. Incubation with 1 M hydroxylamine is used to cleave the palmitoyl-thioester bond, thereby generating a free sulfhydryl group. The samples are then incubated for 2 hr at room temperature with 1 μM of the sulfhydryl-specific reagent biotin-conjugated 1-biotinamido-4-[4′-(maleimidomethyl)cyclohexanecarboxamido] butane (Btn-BMCC), which quantitatively labels the newly exposed palmitoylation sites. Labeled proteins are detected by Western blotting with streptavidin-conjugated HRP.
2.4 Mass Spectrometric Detection of Palmitoylated Proteins
MALDI-TOF MS (matrix-assisted laser desorption ionization time-of-flight mass spectrometry) can be used to examine the fatty acid(s) attached to proteins. The advantages of MS are that it allows one to estimate the stoichiometry of palmitoylation of a given protein and it provides the exact mass of the modifying group [10,17,18]. It is recommended that the investigator collaborate with a microchemistry laboratory that is highly skilled in analyzing hydrophobic peptides by MS. The protein of interest needs to be purified and then digested with protease (typically trypsin). The tryptic peptides are then analyzed in the mass spectrometer in reflection mode using standard instrument settings. The masses of the isolated peptides are then compared to the calculated monoisotopic mass of the equivalent unmodified peptides.
We have found that it is important to minimize oxidation of free sulfhydryl groups by air during protein purification. Cells can be treated with 5 mM N-ethylmaleimide (NEM), a small membrane-permeable reagent that covalently modifies free sulfhydryl groups, prior to and during protein isolation.
The prevalence of various fatty acylated peptide species can be estimated based on relative ion intensities in the MS analysis. However, it is important to note that many factors can influence MALDI-TOF ion intensities (peak heights), especially peptide composition. In addition, a peptide modified with two mono-unsaturated fatty acids cannot be distinguished from a peptide containing one di-unsaturated fatty acid and one saturated fatty acid since the molecular masses of the two species are the same. Moreover, the thio-ester linked palmitate is susceptible to alkali hydrolysis and thus can readily be lost. Finally, palmitoylated peptides are notoriously “sticky” and it is easy to lose them along the way during peptide preparation.
3 Methods to alter protein palmitoylation
Several different methods can be utilized to block protein palmitoylation, including site directed mutagenesis, pharmacologic inhibitors of protein palmitoylation, and depalmitoylation with palmitoyl thioesterase. Since each method has its own inherent “side effects”, the use of at least two approaches to inhibit palmitoylation of a given protein is recommended.
3.1 Inhibition of Protein Palmitoylation by Site Directed Mutagenesis of the Acylated Cysteine Residue
The most common and direct approach to inhibiting palmitoylation is to use site specific mutagenesis to generate a cDNA construct encoding a mutant form of YFPP with the modified cysteine residue mutated to alanine or serine. Cells are transfected with cDNA constructs encoding either the mutant or wild type forms of the protein, and palmitate incorporation into each form is determined as described above. The total amount of YFPP should also be assayed by either Western blotting or immunoprecipitation of 35S-methionine labeled YFPP. This is important to ensure that wild type and mutant forms of YFPP are being expressed to equivalent levels and that mutation of the cysteine residue(s) did not alter protein stability and/or abundance. The limitation of this method is that impairment of protein function may be due to loss of the specific cysteine residue rather than loss of the palmitate moiety. In addition, this method relies on overexpression of wild type and mutant forms of YFPP, and thus does not directly measure palmitoylation of endogenous YFPP.
3.2 Pharmacologic Inhibitors of Protein Palmitoylation
3.2.1 Inhibition of Protein Palmitoylation with 2-Bromopalmitate (2BP)
A method to inhibit protein palmitoylation that has gained increasing popularity over the past few years is to treat cells with 2-bromopalmitate (2BP) (Fig 1), a non-metabolizable palmitate analog that blocks palmitate incorporation into proteins [19]. The validity of this compound as an inhibitor of protein palmitoylation has been verified for at least 2 dozen palmitoylated proteins, including Src family kinases, Rho family proteins, H-Ras, PSD95 and transmembrane receptors such as the Nicotinic α7 Receptor and CCR5 [19–24]. The advantages of this approach are threefold: 1) the reagent is simple and inexpensive to use; 2) the function of endogenous YFPP can be studied without the need to generate and overexpress mutants; 3) the modified cysteine residue is left intact.
2BP is available from Sigma-Aldrich (St. Louis, MO) as 2-bromo-hexadecanoic acid. A 100 mM stock is prepared in DMSO and then diluted 1:1000 into media for a final concentration of 100 μM. Cells expressing YFPP are incubated with or without 100 μM 2BP overnight prior to determination of protein palmitoylation and/or function. The expression level of YFPP should be determined in the presence and absence of 2BP. Note that some cells, eg Jurkat T cells, are more sensitive to 2BP treatment and cannot withstand overnight treatment. Shorter (3–4 hr) incubation times with 2BP have been shown to be effective for these cells.
It is important to note that 2BP has long been used as an in vitro and in vivo marker for free fatty acids. 2BP binds to serum albumin and fatty acid binding protein with an affinity similar to that of palmitate [25]. Once inside the cell, 2BP is converted to 2BP-CoA. The presence of the 2-bromo group prevents breakdown via β-oxidation, thus making 2BP a non-metabolizable analog of palmitate. Since 2BP resembles palmitate, it is not surprising that there are multiple effects of this compound on lipid metabolism. 2BP binds to and inhibits carnitine palmitoyl transferase-1. This reaction prevents long chain free fatty acids from entering mitochondria and thereby inhibits fatty acid β-oxidation [26,27]. 2BP also inhibits other enzymes involved in lipid metabolism, including triacylglycerol biosynthesis, Fatty Acid CoA ligase, and Glycerol–3-phosphate acyltransferase [27]. Inhibitory effects of 2BP on NADPH cytochrome c reductase and glucose-6-phosphatase have also been reported. In pre-adipocytes, 2BP treatment induces gene expression and differentiation via activation of peroxisome proliferator-activated receptors (PPARs) [28,29]. Thus, it is important to keep in mind that 2BP treatment exerts pleiotropic effects on cellular metabolism.
The mechanism responsible for 2BP mediated inhibition of protein palmitoylation is not known. The presence of the 2-bromo group likely confers some degree of specificity, because other palmitate analogs, such as 2-hydroxypalmitate, 16-hydroxypalmitate, and palmitoleic acid, have no effect on palmitoylation of the Src family kinase Fyn [19]. Several possibilities could explain the ability of 2BP to block protein palmitoylation. Binding of 2BP-CoA to a palmitoyl transferase could result in formation of an inhibitor:enzyme complex if the presence of the halogen prevents transfer of 2BP to the acceptor protein. Alternatively, transfer of 2BP might occur, but the increased hydrophilicity of the bromine atom would reduce binding of the modified protein to the lipid bilayer. Another possibility is that 2BP treatment induced alterations in cellular lipid metabolism reduce the concentration of intracellular palmitoyl-CoA pools available for protein palmitoylation. A recent study with a related halogen substituted palmitate analog, 2-fluoropalmitate, concluded that this compound inhibited uptake of [3H]palmitate into brain cells, perhaps by competing with palmitate for cellular entry. As a result, 2-fluoropalmitate treatment leads to decreases in [3H]palmitoyl CoA levels and decreased fatty acylation of phospholipids and myelin proteolipid protein [30].
3.2.2 Inhibition of Protein Palmitoylation with Cerulenin
Cerulenin (2,3-epoxy-4-oxo-7,10 dodecadienoylamide) (Fig 1) and cerulenin analogs have also been shown to function as inhibitors of protein palmitoylation [31]. Cerulenin is a natural product antibiotic that inhibits fatty acid synthesis by alkylating β-ketoacyl acyl-carrier protein synthase. Cerulenin treatment inhibits palmitoylation of myelin proteolipid protein [30] and CD36 [32]. A palmitoyl analog of cerulenin has been synthesized and has been shown to block palmitoylation of p21Ras without inhibiting fatty acid synthesis. It has been proposed that this analog inhibits protein palmitoylation by alkylating protein palmitoyltransferase [33]. Indeed, a recent study successfully used a cerulenin-based inhibitor to partially purify palmitoyl acyltransferase activity [34].
Cerulenin can be purchased from Sigma-Aldrich (St. Louis, MO) or Calbiochem-EMD-Biosciences (La Jolla, CA); a stock solution of 5 mg/ml in ethanol is stored in the dark at 4°C. Cells are incubated with 5–100 μg/ml cerulenin for 4 hours prior to assaying for palmitate incorporation [31,35]. The expression level of YFPP should be determined in the presence and absence of cerulenin.
It is important to consider that cerulenin exerts additional effects on cellular lipid metabolism. Cerulenin binds to β-keto-acyl-ACP synthase and thereby inhibits fatty acid biosynthesis. The compound also inhibits HMG-CoA synthetase activity, resulting in reduced biosynthesis of cholesterol and other sterols. When tissue culture cells were treated with 20 μg/ml cerulenin, incorporation of [3H]acetate into cellular lipids was inhibited by 60%, compared to 10% inhibition of [3H]palmitate incorporation into cellular lipids [35]. These findings suggest that cerulenin has a minimal effect on the incorporation of exogenously added long chain fatty acid into cellular lipids. However, treatment with higher (> 1mM) concentrations of cerulenin result in reduced palmitate incorporation into phosphatidylcholine, but not other phospholipids [30]. No changes in uptake of labeled palmitate or its conversion to palmitoyl CoA were observed with cerulenin.
The mechanism responsible for inhibition of protein palmitoylation by cerulenin is not known. Two groups have suggested that the compound chemically modifies sulfhydryl groups [30,33]. Thus, the ability of cerulenin to inhibit protein palmitoylation may be due to the formation of chemical adducts with critical thiols on either the palmitoyl acyltransferase or the protein acceptor substrate.
3.2.3 Inhibition of Protein Palmitoylation with Tunicamycin
Tunicamycin (Fig 1) is a nucleoside antibiotic that is best known for its ability to inhibit protein N-linked glycosylation. The structural similarity between tunicamycin and palmitoyl CoA led to the finding that tunicamycin can also inhibit protein palmitoylation [36]. Inhibitory effects of tunicamycin on palmitoylation of GAP-43, N-type Ca++ channels, estrogen receptor α variant, and myelin proteolipid protein have been documented [30,36–38].
Detailed methods for the use of tunicamycin in inhibition of protein palmitoylation have been described by Patterson and Skene [39]. Tunicamycin can be purchased from Sigma-Aldrich (St. Louis, MO) or Calbiochem- EMD-Biosciences (La Jolla, CA); a stock solution of 10mg/ml is made in DMSO and stored at 4°C. Cell monolayers should be washed with serum-free media and then incubated in serum-free media containing 0.01% fatty acid-free BSA. Freshly diluted tunicamycin at concentrations from 5–30 μM is then added to the cells for 1–2 hr at 37°C. The use of serum-free conditions is recommended because serum contains fatty acids that could compete with tunicamycin for inhibition of palmitoylation. Likewise, BSA should only be added to concentrations no greater that 0.01% because tunicamycin can bind to BSA and thereby be prevented from entering cells.
Several technical considerations can be used to separate the inhibitory effects of tunicamycin on glycosylation vs palmitoylation. First, the use of short incubation times minimizes the drug’s inhibitory effects on protein glycosylation. Second, cells can be treated tunicamycin in the presence of a protein synthesis inhibitor such as cycloheximide (100 μg/ml for 1 hr at 37°C). The rationale for this approach is that N-linked glycosylation occurs co-translationally and is thus tightly linked to protein synthesis, whereas protein palmitoylation primarily occurs post-translationally. Effects of tunicamycin that are seen in the presence of cycloheximide are likely due to inhibition of palmitoylation. Third, it has been suggested that tunicamycin competes with palmitoyl CoA for binding to PAT. Addition of increasing amounts of exogenous palmitate (1–100 μM for 1 hr), to generate intracellular palmitoyl CoA, should blunt the inhibitory effect of tunicamycin on palmitoylation. Stock solutions of palmitate in DMSO should be freshly diluted into serum-free tissue culture media and sonicated briefly prior to addition to the cells.
3.3 Depalmitoylation with protein palmitoyl thioesterase
Another approach to inhibiting protein palmitoylation is to use a protein palmitoyl thioesterase to deacylate the fatty acylated protein. APT1 has been shown to depalmitoylate eNOS, Gαs, and H-Ras but not caveolin-1 [40–42]. The ability of YFPP to be depalmitoylated by APT1 can be tested by co-expressing the two proteins in cultured cells, or by incubating cell lysates or membranes with APT1 purified from recombinant bacteria. The loss of radiolabeled palmitate from YFPP is then monitored in the presence or absence of APT1.
3.4 Replacement of Palmitate with other Fatty Acids
Palmitoylation of proteins provides not only increased membrane binding affinity but also often promotes partitioning of the palmitoylated protein into membrane rafts. In order to distinguish between these two roles, it is necessary to develop methods that inhibit raft partitioning without affecting membrane binding. Techniques that block protein palmitoylation prevent both membrane and raft association and thus cannot distinguish between these two events. Instead, acylation with alternative fatty acids can be used. Many “palmitoylated” proteins have been shown to be S-acylated with fatty acids other than palmitate, including oleate (18:1), stearate (18:0), arachidonate (20:4) and eicosapentanoate (20:5). In general, attachment of these unsaturated or polyunsaturated fatty acids, in place of palmitate, reduces membrane raft association of the modified protein while maintaining plasma membrane localization [10,18,19]. Thus, one approach to determining the role of palmitoylation in directing membrane raft association of YFPP is to substitute palmitate with other fatty acids, by feeding cells a “diet” consisting of media supplemented with an unsaturated or polyunsaturated fatty acid (PUFA).
Fatty acids are purchased from Sigma-Aldrich; 100 mM stocks are prepared in either ethanol or DMSO and stored at −20°C. Note that PUFAs such as arachidonic acid or eicosapentaenoic acid are light and air sensitive. Stock solutions in ethanol or DMSO are made within the original vial, and the vials are flushed with nitrogen after each use, resealed and wrapped with aluminum foil, and stored at −20°C. The vials should be stored in the dark, and opened vials should be re-used only 1–2 times before discarding them. Cells are incubated with 50–100 μM fatty acid (Sigma) in media containing 2% dialyzed FBS, 0.25% defatted BSA for 12–15 hrs at 37°C. For PUFA treatment, work in the dark and add the PUFA to media containing 0.25% defatted BSA (Sigma) immediately before adding to the tissue culture plate. These conditions have been shown to result in incorporation of the exogenous fatty acid into Src family kinases expressed in transfected COS-1 cells, and displacement of the Src kinases modified with unsaturated fatty acids from membrane rafts [10,19]. It should be noted that longer treatment times, eg 2 days of treatment of cells with 50 μM polyunsaturated fatty acids, results in incorporation of the polyunsaturated fatty acids into the lipids in membrane rafts and thereby markedly changes the lipid composition of the rafts [43]. Confirmation that YFPP incorporated the exogenous fatty acid is achieved by using radiolabeled fatty acids or mass spectrometry (see Section 2.1). The ability of the modified YFPP to associate with membranes, membrane rafts, and to promote signal transduction can then be assessed.
At least two additional methods are available that maintain plasma membrane assocation while blocking raft association. One approach is to use a heteroatom substituted palmitate analog, 13-oxypalmitate (13-OP) [44]. 13-OP (Fig 1) has a similar chain length to palmitate, but has reduced hydrophobicity. Treatment of cells with 200 μM 13-OP for 2 hr in media containing 0.1% fatty acid free BSA has been shown to reduce raft association of and signaling by the Src family kinase Lck, while maintaining Lck localization to the plasma membrane [44]. An alternative approach is to fuse a non-acylated mutant form of YFPP to another membrane bound protein. This can be accomplished using chimeras of YFPP fused to the transmembrane and extracellular domains of a transmembrane protein [45,46] or tagging with the C-terminus of K-Ras, a farnesylated plasma membrane bound protein [47].
4. Assessing the functional significance of altered protein palmitoylation
There are many potential outcomes resulting from alteration or inhibition of protein palmitoylation. Depending on the function of YFPP, it is possible that changes in YFPP trafficking, subcellular localization, enzymatic activity, or adaptor/scaffolding function might occur. Below I will outline three general criteria that could be tested after YFPP palmitoylation levels have been altered.
4.1 Membrane binding
A simple method to determine whether membrane association of YFPP has been perturbed is to generate membrane (P100) and cytosolic (S100) fractions from cells expressing YFPP, as follows.
Aspirate off the media from the plate.
Rinse the plate with 5–10 ml of cold STE (100 mM NaCl/10 mM Tris pH 7.4/ 1 mM EDTA).
Scrape or pipet the cells off the dish with STE (5–10 mls). Spin down the cells at 1,000 x g for 5 min at 4°C.
To the cell pellet add 0.8 ml Hypotonic buffer (10 mM Tris, pH 7.4/ 0.2 mM MgCl2). Gently vortex and incubate on ice for 15 min.
Break open the cells by 30 up and down strokes in a Dounce homogenizer, being careful not to cause foaming. More strokes may be needed for some cell types.
Add 200 μl 5X Sucrose (1.25 M = 21.39 g to 50 mls with dH2O) and 2 μl 0.5 M EDTA.
Spin the lysate at 100,000xg for 15–30 min at 4°C.
Remove the supernatant (S100). Resuspend pellet (P100) in appropriate buffer (0.5 – 1.0 ml) and Dounce homogenize.
Assay for YFPP by Western blotting of samples from the S100 and P100 fractions. In general, one expects to observe decreased YFPP in the P100 fraction and increased amounts in the S100 fraction after palmitoylation has been inhibited.
4.2 Lipid Raft Association
This method uses Optiprep (Iodixanol) (Axis-Shield, Norton, MA) gradients to monitor raft associated and soluble proteins derived from non-ionic detergent treated cell lysates [48]. Decreased palmitoylation has been shown to lead to decreased lipid raft association.
Prepare the cell pellet as in steps 1–3 above.
Resuspend the cell pellet in 300 μl TNET buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.4, 5 mM EDTA, 0.5% Triton X-100) containing protease inhibitors, and incubate on ice for 20–30 minutes.
Homogenize the lysate in a Dounce homogenizer and adjust to 35% (w/v) Iodixanol.
Place 240 μl of lysate at the bottom of an SW55 centrifuge tube (Beckman), overlay with 3.5 ml 30% (w/v) iodixanol in TNET buffer, followed by 200 μl of TNET buffer. Centrifuge the sample for 4 hr at 170,000xg at 4°C.
After centrifugation, collect five 800 μl fractions from the top of the gradient and assay for YFPP. Fraction 1, derived from the top of the gradient, contains the raft fraction. Soluble and non-raft associated proteins fractionate to the bottom of the gradient (fractions 4 and 5).
4.3 Alterations in Signal Transduction: activation of ERK/MAPK
Many palmitoylated signaling molecules participate in downstream signal transduction pathways leading to activation of ERK/MAPK. If YFPP happens to fall within this class, it is relatively straightforward to determine if alteration of YFPP palmitoylation levels leads to an alteration of ERK/MAPK activation. Cell monolayers are rinsed in STE and lysed directly in SDS-PAGE. The samples are Western blotted with anti-phosphoERK antibody, and the blots are then stripped and reprobed for total ERK/MAPK (SantaCruz Biotechnology, Santa Cruz, CA).
5. Concluding Remarks
In this article, multiple techniques for analyzing palmitate incorporation into YFPP, altering palmitoylation of YFPP, and determining the functional significance of palmitoylation of YFPP have been presented. The use of analogs and inhibitors, especially 2BP, has facilitated the technical aspects of studying protein palmitoylation for cell and molecular biologists. The investigator is encouraged to use multiple methods to inhibit YFPP palmitoylation and to determine their effects on YFPP localization and function with both biochemical and cytological (ie confocal imaging) readouts. It is likely that many more palmitoylated proteins remain to be discovered, and the search for these proteins will continue to fuel the ever expanding field of protein fatty acylation.
Acknowledgments
Work in the author’s laboratory was supported by NIH Grants GM57966 and CA72309.
References
- 1.Resh MD. Biochim Biophys Acta. 1999;1451:1–16. doi: 10.1016/s0167-4889(99)00075-0. [DOI] [PubMed] [Google Scholar]
- 2.Linder ME, Deschenes RJ. Biochemistry. 2003;42:4311–20. doi: 10.1021/bi034159a. [DOI] [PubMed] [Google Scholar]
- 3.Smotrys JE, Linder ME. Annu Rev Biochem. 2004;73:559–87. doi: 10.1146/annurev.biochem.73.011303.073954. [DOI] [PubMed] [Google Scholar]
- 4.Resh MD. Cell. 1994;76:411–413. doi: 10.1016/0092-8674(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 5.Alland L, Peseckis SM, Atherton RE, Berthiaume L, Resh MD. J Biol Chem. 1994;269:16701–16705. [PubMed] [Google Scholar]
- 6.Mann RK, Beachy PA. Annu Rev Biochem. 2004;73:891–923. doi: 10.1146/annurev.biochem.73.011303.073933. [DOI] [PubMed] [Google Scholar]
- 7.Kleuss C, Krause E. EMBO J. 2003;22:826–32. doi: 10.1093/emboj/cdg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galbiati F, Volonte D, Meani D, Milligan G, Lublin DM, Lisanti MP, Parenti M. J Biol Chem. 1999;274:5843–5850. doi: 10.1074/jbc.274.9.5843. [DOI] [PubMed] [Google Scholar]
- 9.Zacharias DA, Violin JD, Newton AC, Tsien RY. Science. 2002;296:913–6. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 10.Liang X, Nazarian A, Erdjument-Bromage H, Bornmann W, Tempst P, Resh MD. J Biol Chem. 2001;276:30987–94. doi: 10.1074/jbc.M104018200. [DOI] [PubMed] [Google Scholar]
- 11.el-Husseini A-D, Bredt DS. Nat Rev Neurosci. 2002;3:791–802. doi: 10.1038/nrn940. [DOI] [PubMed] [Google Scholar]
- 12.Sambrook J, Russell DW. Molecular Cloning. Third Edition. Cold Spring Harbor: Laboratory Press; 2001. [Google Scholar]
- 13.Peseckis SM, Deichaite I, Resh MD. J Biol Chem. 1993;268:5107–5114. [PubMed] [Google Scholar]
- 14.Berthiaume L, Peseckis SM, Resh MD. Meth Enzymol. 1995;250:454–466. doi: 10.1016/0076-6879(95)50090-1. [DOI] [PubMed] [Google Scholar]
- 15.Berzat AC, Buss JE, Chenette EJ, Weinbaum CA, Shutes A, Der CJ, Minden A, Cox AD. J Biol Chem. 2005;280:33055–33065. doi: 10.1074/jbc.M507362200. [DOI] [PubMed] [Google Scholar]
- 16.Drisdel RC, Green WN. Biotechniques. 2004;36:276–285. doi: 10.2144/04362RR02. [DOI] [PubMed] [Google Scholar]
- 17.Liang X, Lu Y, Neubert TA, Resh MD. J Biol Chem. 2002;277:33032–40. doi: 10.1074/jbc.M204607200. [DOI] [PubMed] [Google Scholar]
- 18.Liang X, Lu Y, Wilkes M, Neubert TA, Resh MD. J Biol Chem. 2004;279:8133–9. doi: 10.1074/jbc.M311180200. [DOI] [PubMed] [Google Scholar]
- 19.Webb Y, Hermida-Matsumoto L, Resh MD. J Biol Chem. 2000;275:261–270. doi: 10.1074/jbc.275.1.261. [DOI] [PubMed] [Google Scholar]
- 20.Chenette EJ, Abo A, Der CJ. J Biol Chem. 2005;280:13784–13792. doi: 10.1074/jbc.M411300200. [DOI] [PubMed] [Google Scholar]
- 21.Chen HQ, Tannous M, Veluthakal R, Amin R, Kowluru A. Biochem Pharmacol. 2003;66:1681–1694. doi: 10.1016/s0006-2952(03)00549-5. [DOI] [PubMed] [Google Scholar]
- 22.El-Husseini Ael-D, et al. Cell. 2002;108:849–863. doi: 10.1016/s0092-8674(02)00683-9. [DOI] [PubMed] [Google Scholar]
- 23.Drisdel RC, Manzana E, Green WN. J Neurosci. 2004;24:10502–10510. doi: 10.1523/JNEUROSCI.3315-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Percherancier Y, et al. J Biol Chem. 2001;276:31936–31944. doi: 10.1074/jbc.M104013200. [DOI] [PubMed] [Google Scholar]
- 25.Oakes ND, Furler SM. Ann N Y Acad Sci. 2002;967:158–175. doi: 10.1111/j.1749-6632.2002.tb04273.x. [DOI] [PubMed] [Google Scholar]
- 26.Chase JF, Tubbs PK. Biochem J. 1972;129:55–65. doi: 10.1042/bj1290055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coleman RA, Rao P, Fogelsong RJ, Bardes ES. Biochim Biophys Acta. 1992;1125:203–209. doi: 10.1016/0005-2760(92)90046-x. [DOI] [PubMed] [Google Scholar]
- 28.Brandes R, Arad R, Bar-Tana J. Biochem Pharmacol. 1995;50:1949–1951. doi: 10.1016/0006-2952(95)02082-9. [DOI] [PubMed] [Google Scholar]
- 29.Bastie C, Luquet S, Holst D, Jehl-Pietri C, Grimaldi PA. J Biol Chem. 2000;275:38768–38773. doi: 10.1074/jbc.M006450200. [DOI] [PubMed] [Google Scholar]
- 30.DeJesus G, Bizzozero OA. Neurochem Res. 2002;27:1669–1675. doi: 10.1023/a:1021643229028. [DOI] [PubMed] [Google Scholar]
- 31.Lawrence DS, Zilfou JT, Smith CD. J Med Chem. 1999;42:4932–4941. doi: 10.1021/jm980591s. [DOI] [PubMed] [Google Scholar]
- 32.Jochen AL, Hays J, Mick G. Biochim Biophys Acta. 1995;1259:65–72. doi: 10.1016/0005-2760(95)00147-5. [DOI] [PubMed] [Google Scholar]
- 33.De Vos ML, Lawrence DS, Smith CD. Biochem Pharmacol. 2001;62:985–995. doi: 10.1016/s0006-2952(01)00739-0. [DOI] [PubMed] [Google Scholar]
- 34.Hiol A, Caron JM, Smith CD, Jones TL. Biochim Biophys Acta. 2003;1635:10–19. doi: 10.1016/j.bbalip.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 35.Schlesinger MJ, Malfer C. J Biol Chem. 1982;257:9887–9890. [PubMed] [Google Scholar]
- 36.Patterson SI, Skene JHP. J Cell Biol. 1994;124:521–536. doi: 10.1083/jcb.124.4.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hurley JH, Cahill AL, Currie KP, Fox AP. Proc Natl Acad Sci U S A. 2000;97:9293–9298. doi: 10.1073/pnas.160589697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li L, Haynes MP, Bender JR. Proc Natl Acad Sci U S A. 2003;100:4807–4812. doi: 10.1073/pnas.0831079100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patterson SI, Skene JH. Methods Enzymol. 1995;250:284–300. doi: 10.1016/0076-6879(95)50079-0. [DOI] [PubMed] [Google Scholar]
- 40.Duncan JA, Gilman AG. J Biol Chem. 1998;273:15830–15837. doi: 10.1074/jbc.273.25.15830. [DOI] [PubMed] [Google Scholar]
- 41.Yeh DC, Duncan JA, Yamashita S, Michel T. J Biol Chem. 1999;274:33148–33154. doi: 10.1074/jbc.274.46.33148. [DOI] [PubMed] [Google Scholar]
- 42.Duncan JA, Gilman AG. J Biol Chem. 2002;277:31740–31752. doi: 10.1074/jbc.M202505200. [DOI] [PubMed] [Google Scholar]
- 43.Stulnig TM, Huber J, Leitinger N, Imre EM, Angelisova P, Nowotny P, Waldhausl W. J Biol Chem. 2001;276:37335–37340. doi: 10.1074/jbc.M106193200. [DOI] [PubMed] [Google Scholar]
- 44.Hawash IY, Hu XE, Adal A, Cassady JM, Geahlen RL, Harrison ML. Biochim Biophys Acta. 2002;1589:140–150. doi: 10.1016/s0167-4889(02)00165-9. [DOI] [PubMed] [Google Scholar]
- 45.Kabouridis PS, Magee AI, Ley SC. EMBO J. 1997;16:4983–4998. doi: 10.1093/emboj/16.16.4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prabhakar P, Cheng V, Michel T. J Biol Chem. 2000;275:19416–19421. doi: 10.1074/jbc.M001952200. [DOI] [PubMed] [Google Scholar]
- 47.van’t Hof W, Resh MD. J Cell Biol. 1999;145:377–389. doi: 10.1083/jcb.145.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lindwasser OW, Resh MD. J Virol. 2001;75:7913–7924. doi: 10.1128/JVI.75.17.7913-7924.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]