

Summary
Asymmetric localization of PAR proteins is a hallmark of polarized cells, but the mechanisms that create PAR asymmetry are not well understood. In the C. elegans zygote, PAR asymmetry is initiated by a transient actomyosin contraction, which sweeps the PAR-3/PAR-6/PKC-3 complex toward the anterior pole of the egg. The RING finger protein PAR-2 accumulates in a complementary pattern in the posterior cortex. Here we present evidence that PAR-2 participates in a feedback loop to stabilize polarity. PAR-2 is a target of the PKC-3 kinase and is excluded from the anterior cortex by PKC-3-dependent phosphorylation. The RING domain of PAR-2 is required to overcome inhibition by PKC-3 and stabilize PAR-2 on the posterior cortex. Cortical PAR-2 in turn prevents PAR-3/PAR-6/PKC-3 from returning to the posterior, in a PAR-1- and PAR-5-dependent manner. Our findings suggest that reciprocal inhibitory interactions among PAR proteins stabilize polarity by reinforcing an initial asymmetry in PKC-3.
Introduction
The PAR proteins are conserved regulators of cell polarity, which function in many cell types, including eggs, epithelial cells, and neuroblasts (Macara, 2004a). A conserved property of PAR proteins is to localize to specific cortical domains within polarized cells. For example, in Caenorhabditis elegans zygotes and Drosophila oocytes, PAR-3/Bazooka, PAR-6, and the kinase PKC-3/aPKC form a complex enriched in the anterior half of the egg cortex, whereas the kinase PAR-1 localizes in a complementary pattern in the posterior half of the egg cortex (Betschinger and Knoblich, 2004). Similarly in epithelial cells, the PAR-3/PAR-6/PKC-3 complex localizes to apical surfaces (in Drosophila and C. elegans) or tight junctions (in mammalian epithelial cells), whereas PAR-1 localizes to lateral surfaces (Macara, 2004b). These nonoverlapping domains are thought to depend on inhibitory interactions between PAR proteins residing in different domains. For example, in Drosophila oocytes and epithelial cells, PAR-1 phosphorylates PAR-3 at two sites, disrupting PAR-3's ability to oligomerize and interact with aPKC (Benton and St Johnston, 2003). A PAR-3 mutant lacking the two PAR-1 phosphorylation sites invades the lateral cortex (in epithelial cells) or posterior cortex (in oocytes) normally occupied by PAR-1 (Benton and St Johnston, 2003). Conversely, in mammalian epithelial cells, aPKC has been found to phosphorylate PAR-1 and inhibit its kinase activity and cortical localization (Hurov et al., 2004; Suzuki et al., 2004). These studies suggest that reciprocal inhibitory interactions help maintain PAR asymmetry in polarized cells. Whether such interactions also function to delineate PAR domains as cells initially become polarized is not known.
The sorting of PAR proteins into distinct domains can be visualized in real time in newly fertilized C. elegans zygotes (Cuenca et al., 2003). Polarization is initiated after fertilization when the sperm centrosome contacts the actin-rich cortex at one end of the embryo (Gonczy and Rose, 2005). By an unknown mechanism, this interaction triggers a massive asymmetric contraction of the actin cytoskeleton away from the sperm (Munro et al., 2004). Examination of live zygotes undergoing contraction has revealed that PAR-6 flows away from the sperm with the same kinetics as the myosin NMY-2, suggesting that the PAR-3/PAR-6/PKC-3 complex is mobilized by cortical flow (Munro et al., 2004). As PAR-3/PAR-6/PKC-3 becomes enriched on the cortex opposite the sperm (anterior), PAR-1 and the RING finger protein PAR-2 become enriched in a reciprocal pattern on the cortex nearest the sperm (posterior) (Boyd et al., 1996; Cuenca et al., 2003; Cheeks et al., 2004). Analysis of embryos lacking PAR-2 has revealed that PAR-2, although not essential to initiate cortical flow and PAR-3/PAR-6/PKC-3 asymmetry, is essential to prevent a subsequent return flow, which redistributes PAR-3/PAR-6/PKC-3 throughout the cortex in par-2(−) zygotes (Cuenca et al., 2003; Munro et al., 2004). These observations have suggested that PAR-2's primary function is to stabilize the polarity induced by the sperm centrosomes.
To investigate how PAR-2 localizes to the posterior cortex and maintains polarity, we have identified the domains in PAR-2 critical for localization and function. We present evidence that PAR-2's localization to the cortex is inhibited by PKC-3-dependent phosphorylation. The RING finger of PAR-2 is required to overcome cortical exclusion by PKC-3 and establish a stable domain of PAR-2 on the posterior cortex. PAR-2 on the cortex in turn antagonizes the cortical localization of PAR-3 in a manner that depends on PAR-1 and PAR-5. Our findings suggest that PAR-2 participates in a feedback loop that stabilizes a transient change in PKC-3 levels.
Results
Identification of a Minimal PAR-2 Localization Domain
PAR-2 contains two recognizable domains: a RING finger, typical of E3 ubiquitin ligases, and a consensus ATP binding domain (Levitan et al., 1994). To identify the domain(s) in PAR-2 important for localization to the cortex, we created GFP:PAR-2 fusions containing different fragments of the par-2 open reading frame. The fusions were constructed in the pID3.01 vector, which uses the pie-1 promoter to drive expression of transgenes in oocytes (maternal expression). Transgenes were introduced into worms by biolistic transformation (Praitis et al., 2001), and their expression was examined in wild-type embryos. We identified a single, 235 amino acid region (PAR-2[178–412]) sufficient for targeting green fluorescent protein (GFP) to the cortex in oocytes (data not shown) and to the posterior cortex in wild-type zygotes (Table 1). Time-lapse movies of wild-type zygotes expressing PAR-2(178–412) (Figure 1; see Movie S1 in the Supplemental Data available with this article online) confirmed that this domain accumulates in the posterior cortex with dynamics similar to those observed for full-length GFP:PAR-2 (Cuenca et al., 2003). To test whether the posterior localization of PAR-2(178–412) is dependent on anterior PARs, we examined the distribution of PAR-2(178–412) in embryos depleted for par-6 or pkc-3 by RNA-mediated interference (par-6[RNAi] and pkc-3[RNAi] embryos). PAR-2(178–412) was found throughout the cortex of par-6(RNAi) and pkc-3(RNAi) embryos (data not shown and Figure 2D). We conclude that PAR-2(178–412) is sufficient for cortical localization and exclusion by the anterior PAR complex in par-2(+) zygotes.
Table 1.

Identification of a Localization Domain in PAR-2
Figure 1.

Localization Dynamics of the PAR-2 Localization Domain Nomarski and fluorescence photomicrographs from a time-lapse movie of a wild-type zygote expressing GFP:PAR-2(178–412) (see Movie S1). Time in min:s is indicated to the left.
Figure 2.

- Alignment of the PAR-2 localization domain comparing C. elegans and C. briggsae sequences. Predicted PKC sites are highlighted.
- In vitro kinase assay. Recombinant human PKCζ was incubated with 32P-labeled ATP and MBP:PAR-2 partially purified from E. coli (Experimental Procedures). Upper panel, 32P incorporation; middle panel, Coomassie staining of the same gel to control for loading; bottom panel, a gel loaded with 1:200 dilution of the reaction (no 32P-labeled ATP) and blotted with anti-MBP antibody.
- Western blot of anti-GFP immunoprecipitated embryonic extracts probed with anti-GFP antibody and run on a 7% Tris-acetate gel. The immunoprecipitates were incubated with or without calf intestinal alkaline phosphatase (CIAP). Phosphorylated PAR-2 is indicated by an asterisk. For GFP:PAR-2(178–412), an extra two lanes (lanes 3 and 4) were loaded with 1.5× sample to reveal the unphosphorylated band and for quantification purposes. Band intensities were quantified using NIH Image 1.63. The percent phosphorylated protein was calculated by dividing upper band values by the sum of upper and lower band values. The pkc-3(RNAi) treatment used to obtain the extracts used in lanes 7 and 8 was ∼66% effective as judged by GFP:PAR-2(178–412) localization (20/30 hermaphrodites contained embryos with ectopic GFP).
- Fluorescence micrographs of mitotic zygotes expressing the indicated GFP:PAR-2 fusions. A minimum of five mitotic zygotes were examined for each genotype, and 100% showed the GFP:PAR-2 pattern presented in the figure. The cortical/cytoplasmic ratio (Experimental Procedures) of wild-type GFP:PAR-2 was 1.73 ± 0.13, compared to 1.17 ± 0.11 for GFP:PAR-27S/E and 0.81 ± 0.01 for GFP:PAR-27S/E in par-2(−).
- Confocal micrographs of HEK293 cells expressing the indicated PAR-2 (green) and PKC-3 (red) fusions. Cells in left panels are expressing PAR-2 only, cells in right panels are expressing both PAR-2 and PKC-3.
Phosphorylation of PKC Sites in PAR-2 Excludes PAR-2 from the Cortex
GFP:PAR-2(178–412) does not include the RING finger or ATP binding domain of PAR-2. Instead, this domain contains seven serine residues that match the consensus for phosphorylation by atypical protein kinase C (S-X-(R/K)) (Figure 2A). Six of the seven putative PKC sites are conserved in C. briggsae PAR-2, raising the possibility that PAR-2 may be a substrate for PKC-3.
To test this hypothesis, we began by investigating whether recombinant PKCζ, a human homolog of PKC-3 (Tabuse et al., 1998), can phosphorylate PAR-2 in vitro. A commercial preparation of PKCζ could phosphorylate Escherichia coli-produced MBP:PAR-2, but not MBP alone (Figure 2B). Conversion of the serines in the seven PKC sites to alanines (PAR-27S/A) reduced phosphorylation by PKCζ (Figure 2B), consistent with at least some of the PKC sites being targeted for phosphorylation. We conclude that PAR-2 is a substrate for PKCζ in vitro, and thus a possible substrate for PKC-3 in vivo.
To investigate whether PAR-2 is phosphorylated in vivo, we examined the mobility of GFP:PAR-2(178–412) after immunoprecipitation from embryonic extracts. GFP:PAR-2(178–412) migrated as a doublet (Figure 2C). The slower migrating band accounted for the majority (69%) of the protein and was sensitive to alkaline phosphatase treatment, consistent with representing a phosphorylated isoform (Figure 2C). GFP:PAR-2(178– 412)7S/A extracted from wild-type embryos, and GFP: PAR-2(178–412) extracted from pkc-3(RNAi) embryos, exhibited reduced levels of phosphorylation (43.7% and 48.8%) (Figure 2C). We conclude that PAR-2 is phosphorylated in vivo and that this phosphorylation is at least partially dependent on PKC-3 and the PKC sites.
To test the function of the PKC sites in vivo, we examined the effect of the 7S/A mutations on the localization of GFP:PAR-2(178–412) and full-length GFP:PAR-2. We found that, unlike wild-type fusions, GFP:PAR-2(178–412)7S/A and GFP:PAR-27S/A localized to the cortex at high levels even before pronuclear formation (data not shown), and remained on both the anterior and posterior cortex through mitosis (Figure 2D). Some preference for the posterior cortex could still be observed in many embryos, consistent with the observation that the 7S/A mutations do not completely eliminate phosphorylation (Figure 2C). In contrast to all transgenic lines in this study, which could be propagated stably for 20 generations or more, the two lines expressing GFP:PAR-27S/A could not be maintained, suggesting that ectopic PAR-2 is toxic. We conclude that the PKC sites are essential to exclude PAR-2 from the anterior cortex.
To test whether phosphorylation of the PKC sites might be sufficient for cortical exclusion, we mutated all seven alanines to the negatively charged amino acid glutamate. GFP:PAR-27S/E accumulated on the posterior cortex, but at lower levels compared to wild-type (Figure 2D), consistent with phosphorylation interfering with cortical localization. The residual accumulation of GFP:PAR-27S/E on the posterior cortex suggests that GFP:PAR-27S/E remains sensitive to PKC-3 and that additional PKC sites exist, as also implied by the analysis of GFP:PAR-27S/A described above. Consistent with this view, in pkc-3(RNAi) zygotes, GFP:PAR-27S/E accumulated weakly on both the anterior and posterior cortex (7/9 zygotes; data not shown). Furthermore, in par-2(lw32) embryos where PKC-3 levels are uniform throughout the cortex, GFP:PAR-27S/E was cytoplasmic, whereas GFP:PAR-2(178–412)7S/A remained cortical (Figure 2D). The observation that GFP:PAR-27S/E behaves differently from GFP:PAR-27S/A and GFP:PAR-2(178–412)7S/A strongly suggests that phosphorylation regulates PAR-2's ability to associate with the cortex.
To investigate whether PKC-3 is sufficient to exclude PAR-2 from the cortex, we examined the distribution of GFP:PAR-2 when expressed with or without PKC-3 in HEK293 cells. For these experiments, we used a deleted version of PKC-3 lacking the domain (amino acids 1–161) required for interaction with PAR-6 (Drier et al., 2002; Betschinger et al., 2003). PKC-3(162–597) is predicted to be constitutively active (Drier et al., 2002) and, as expected, was cytoplasmic when expressed in HEK293 cells (Figure 2E). In contrast, GFP:PAR-2 was primarily cortical when expressed alone in HEK293 cells. Coexpression with PKC-3(162–597), however, caused GFP:PAR-2 to partially relocalize to the cytoplasm (Figure 2E). This change was dependent on the PKC sites in PAR-2, as GFP:PAR-27S/A remained mostly cortical in HEK293 cells whether or not PKC-3 was present (Figure 2E). We conclude that PKC-3 interferes with PAR-2 localization to the cortex, and that this inhibition depends on PKC sites in the PAR-2 localization domain.
The RING Domain Is Required In Vivo
To determine which domains are required for PAR-2 activity in vivo, we examined the localization and activity of GFP:PAR-2 fusions expressed in zygotes lacking endogenous PAR-2. GFP:PAR-2 transgenes were crossed into strains carrying the par-2 loss-of-function mutation lw32 and examined for their ability to (1) rescue the maternal effect lethality of par-2(lw32)) (2) rescue the polarity defects of par-2(lw32) embryos, and (3) localize to the posterior cortex in par-2(lw32) zygotes.
We found that transgenes lacking, or with mutations in, the ATP binding domain rescued the maternal effect lethality of par-2(lw32) as efficiently as the wild-type transgene (Figure 3A). par-2(lw32) zygotes expressing GFP:PAR-2, GFP:PAR-2C-termΔ, or GFP:PAR-2G433A, K435A divided unequally (data not shown), like par-2(+) zygotes and unlike par-2(lw32) zygotes, which divide symmetrically (Kemphues et al., 1988). Both GFP:PAR-2G433A, K435A and GFP:PAR-2C-termΔ localized to the posterior cortex with wild-type dynamics (Figure 4). The only defect we noted for GFP:PAR-2C-termΔ and GFP:PAR-2G433A, K435A was that both were found at reduced levels at cell-cell contacts in somatic blastomeres in eight-cell and older embryos compared to wild-type (data not shown). The significance of this localization is not known. We conclude that the ATP binding domain is not required for PAR-2's polarity function in the zygote, although it may play a nonessential role in later blastomeres.
Figure 3.
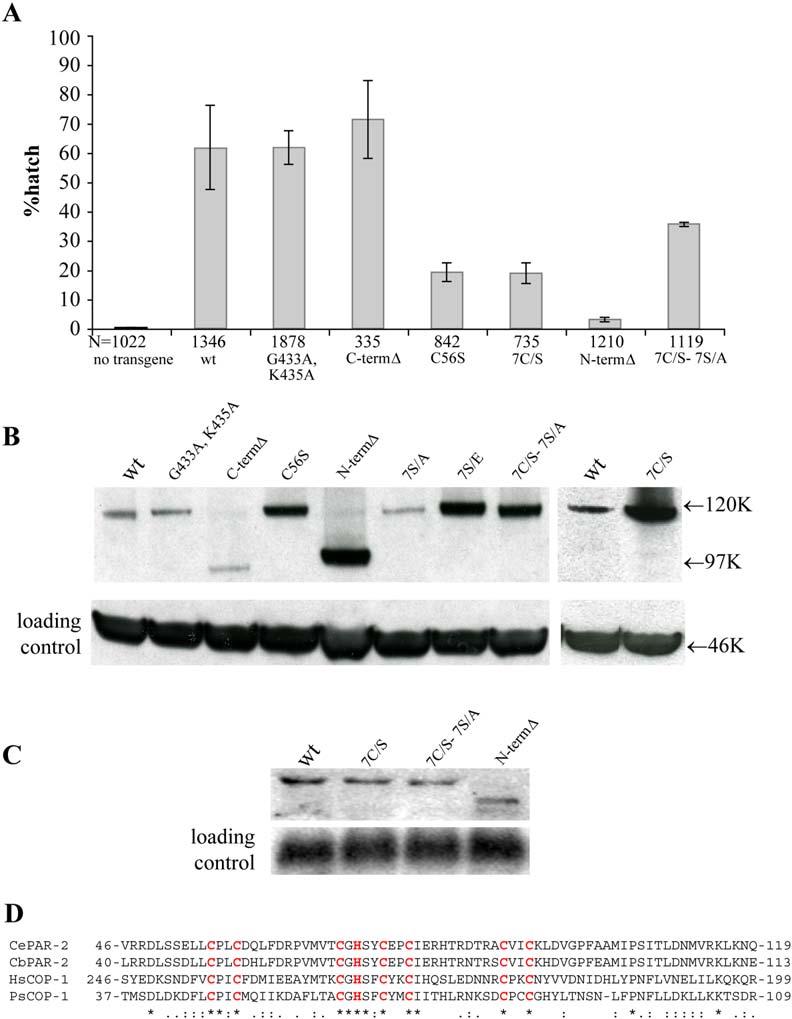
- Graph depicting the percent of par-2(lw32) embryos that completed embryogenesis (% hatching rate) in the presence of the indicated GFP:PAR-2 transgenes. Five independent experiments were performed for each line, and two independent lines were examined for each transgene, except for GFP:PAR-2N-termΔ and GFP:PAR-2C-termΔ, for which only one line was characterized. Error bars represent standard deviation of the values obtained for each experiment. Total number of embryos scored (N) is indicated below each bar. G433A and K435A are point mutations in the ATP binding consensus, C-termΔ is a C-terminal deletion (216 amino acids) that removes the ATP binding consensus, C56S is a mutation in the first cysteine in the RING, 7C/S is a RING mutant with all seven cysteines mutated, N-termΔ is an N-terminal deletion (177 amino acids) that removes the RING domain (73 amino acids; [D]) plus flanking amino acids, and 7C/S-7S/A is a RING/PKC “double” mutant where all seven cysteines in the RING are mutated to serines and all seven serines in the consensus PKC sites are mutated to alanines.
- Western blot of anti-GFP immunoprecipitated embryonic extracts probed with anti-GFP antibody and run on 4%–12% SDS polyacrylamide gel. The left and right panels are from two different blots. Anti-tubulin against total extract was used as loading control.
- Northern analysis comparing levels of GFP:PAR-2 RNA in four transgenic lines. GFP:PAR-27C/S, GFP:PAR-27C/S-7S/A, and GFP:PAR-2N-termΔ accumulate higher levels of protein compared to wild-type (see [B]), yet do not express more RNA.
- Alignment of the RING domain of C. elegans PAR-2, C. briggsae PAR-2, human COP1, and Pisum sativum COP1 (Bianchi et al., 2003; Raghuvanshi et al., 2001). Conserved Cys and His residues are highlighted.
Figure 4.
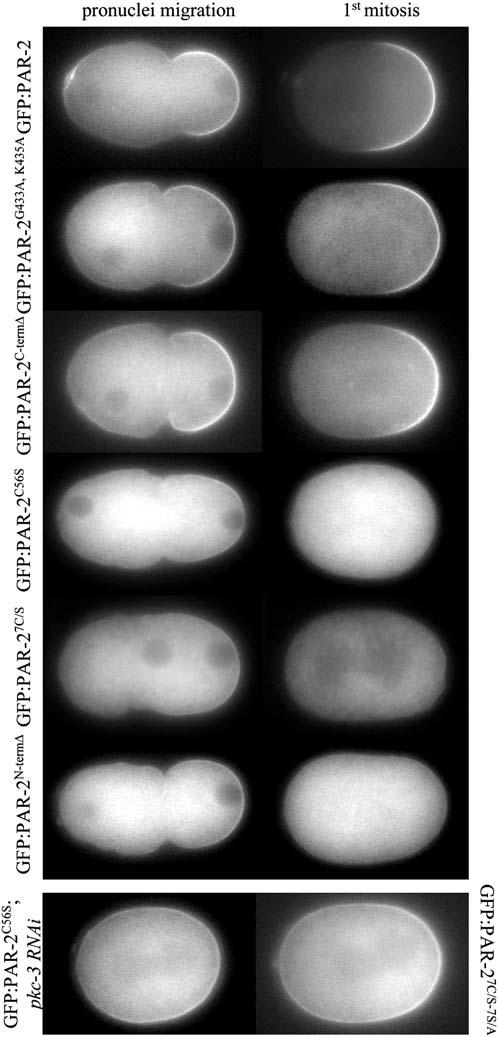
The RING Domain Is Required to Maintain GFP:PAR-2 on the Posterior Cortex in the Absence of Endogenous PAR-2
Fluorescence micrographs of par-2(lw32) zygotes expressing the indicated GFP:PAR-2 fusions. The zygotes are shown during pronu-clear migration (cortical flow) and during mitosis (after cortical flow). Movie S2 shows dynamics of GFP:PAR-2N-termΔ in par-2(lw32). The two bottom panels are of embryos in mitosis.
In contrast to mutations in the ATP binding site, mutations in the RING domain disrupted the activity and localization of GFP:PAR-2 fusions. GFP:PAR-2C56S, GFP:PAR-27C/S, and GFP:PAR-2N-termΔ rescued the embryonic lethality and polarity defects of par-2(lw32) zygotes less efficiently than the wild-type transgene (Figure 3A). Time-lapse microscopy revealed that all three fusions localized weakly to the posterior cortex during pronuclear migration, and disappeared completely from the cortex after pronuclear meeting (Figure 4; Movie S2). By the time of mitosis, the RING mutant fusions were primarily cytoplasmic and appeared to accumulate there at higher levels than wild-type (see below). The dynamics of GFP:PAR-2C56S, GFP:PAR-27C/S, and GFP:PAR-2N-termΔ suggested that these fusions might be displaced from the posterior cortex by the anterior PAR complex, which returns to the posterior after pronuclear meeting in par-2(lw32) zygotes (Cuenca et al., 2003). Consistent with this hypothesis, RFP:PAR-6 was delocalized in par-2(lw32) zygotes expressing GFP:PAR-2C56S (2/2 zygotes in mitosis and 5/5 in two- and four-cell embryos; data not shown). Furthermore, removal of PKC-3 by RNAi restored the cortical localization of GFP:PAR-2C56S (Figure 4). A GFP:PAR-2 fusion with mutations in both the seven cysteines of the RING and the seven PKC sites (GFP: PAR-27C/S-7S/A) also localized efficiently throughout the cortex with some preference for the posterior (Figure 4). Remarkably, GFP:PAR-27C/S-7S/A rescued the embryonic lethality of par-2(lw32) zygotes more efficiently than GFP:PAR-2C56S, GFP:PAR-27C/S, or GFP:PAR-2N-termΔ (Figure 3A). This observation suggests that the RING is required primarily to maintain PAR-2 at the cortex and resist exclusion by PKC-3.
If the RING stabilizes cortical localization, we might expect GFP:PAR-27C/S to be less efficient at localizing to the cortex even in the presence of endogenous PAR-2. To explore this possibility, we compared the cortical to cytoplasmic ratio of GFP:PAR-2, GFP:PAR-27C/S, and GFP:PAR-27C/S-7S/A in wild-type and par-1(RNAi) embryos. In par-1(RNAi) zygotes, GFP:PAR-6 initially localizes to the anterior cortex as in wild-type but returns to the posterior during mitosis, whereas GFP:PAR-2 remains posterior (Cuenca et al., 2003). We found that GFP:PAR-27C/S localized to the posterior cortex with lower efficiency in par-1(RNAi) zygotes compared to wild-type. In contrast, GFP:PAR-2 and GFP:PAR-27C/S-7S/A localized to the posterior cortex with similar efficiency in wild-type and par-1(RNAi) zygotes (Figure 5). We conclude that the RING is required to counteract cortical exclusion and maintain high levels of PAR-2 at the cortex.
Figure 5.
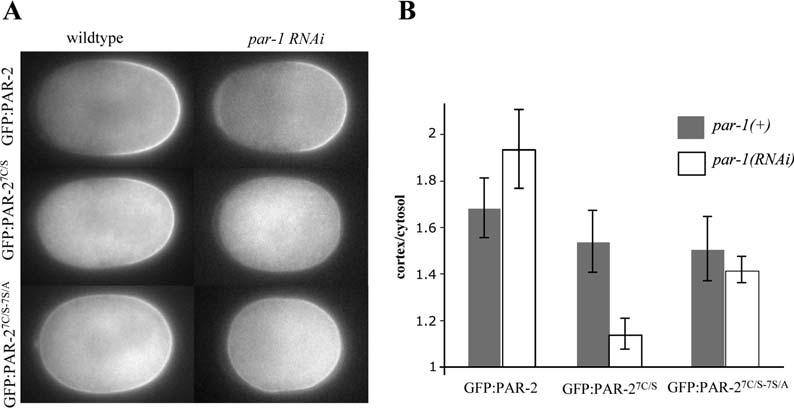
- Fluorescence micrographs of wild-type and par-1(RNAi) zygotes in mitosis expressing the indicated GFP:PAR-2 fusions. At this stage, in wild-type zygotes, GFP:PAR-6 is restricted to the anterior cortex, whereas, in par-1(RNAi) zygotes, GFP:PAR-6 is both on the anterior and posterior cortex (12/12 wild-type zygotes, and 6/6 par-1(RNAi) zygotes examined by time-lapse fluorescent microscopy; Cuenca et al., 2003; A. Cuenca and G.S., unpublished data).
- Comparison of cortex/cytosol GFP intensity ratios (Experimental Procedures) for the genotypes shown in (A). Error bars represent standard deviations derived from six zygotes analyzed for each genotype.
The RING Finger Downregulates PAR-2 Levels
An important control when comparing the in vivo activity of transgenes is to verify that the transgenic proteins are expressed at similar levels. Immunoblots of immunoprecipitated extracts from the various transgenic lines revealed that all fusions were expressed at levels equivalent or higher than wild-type GFP:PAR-2 (Figure 3B). Remarkably, high levels of protein accumulation were observed in all fusions where the RING finger was mutated, suggesting that the RING finger normally negatively regulates PAR-2 levels. Elevated PAR-2 levels were also observed in GFP:PAR-27S/E (Figure 3B), which has a wild-type RING finger but does not localize to the cortex efficiently (Figure 2D). This observation suggests a potential link between cortical localization and RING activity. Northern analyses confirmed that the differences observed in protein levels were not due to differences in RNA levels (Figure 3C), consistent with the fact that all transgenes were driven by the same promoter. We conclude that the RING domain accelerates PAR-2 turnover.
Overexpression of PAR-2 Interferes with Cortical Localization of PAR-3
PAR-2 is required to keep PAR-3/PAR-6/PKC-3 excluded from the posterior cortex during mitosis (Cuenca et al., 2003; Munro et al., 2004). To investigate the requirements for this activity, we developed an ectopic expression assay for PAR-2 in 12- to 30-cell stage embryos. At these stages, in somatic blastomeres, PAR-2 and PAR-1 localize to basolateral cortices at sites of cell-to-cell contacts, and PAR-3, PAR-6, and PKC-3 localize in a complementary pattern to contact-free, apical cortices (Nance et al., 2003). We used the heat shock promoter to drive zygotic expression of PAR-2:GFP fusions in somatic blastomeres. To avoid cortical exclusion by PKC-3, we used the PAR-27S/A mutant. As expected, upon heat shock, PAR-27S/A:GFP was found throughout the cortex of most embryonic blastomeres in 12-cell and older embryos (Figure 6). Remarkably, immunostaining against PAR-3 revealed that endogenous PAR-3 was no longer on apical cortices, except in the cells where PAR-2 failed to be expressed (Figure 6). To test whether the RING is required for this activity, we repeated this experiment with PAR-27C/S-7S/A:GFP. We found that PAR-27C/S-7S/A:GFP was as efficient as PAR-27S/A:GFP in excluding PAR-3 from the apical cortex (Figure 6). This result is consistent with our earlier finding that GFP:PAR-27C/S-7S/A can partially rescue the embryonic lethality of par-2(lw32) mutants, suggesting that the RING is not essential for PAR-2's polarity function when PAR-2 is no longer an efficient target for PKC-3. We conclude that cortical PAR-2 can exclude PAR-3 from the cortex and that this activity does not require the RING.
Figure 6.
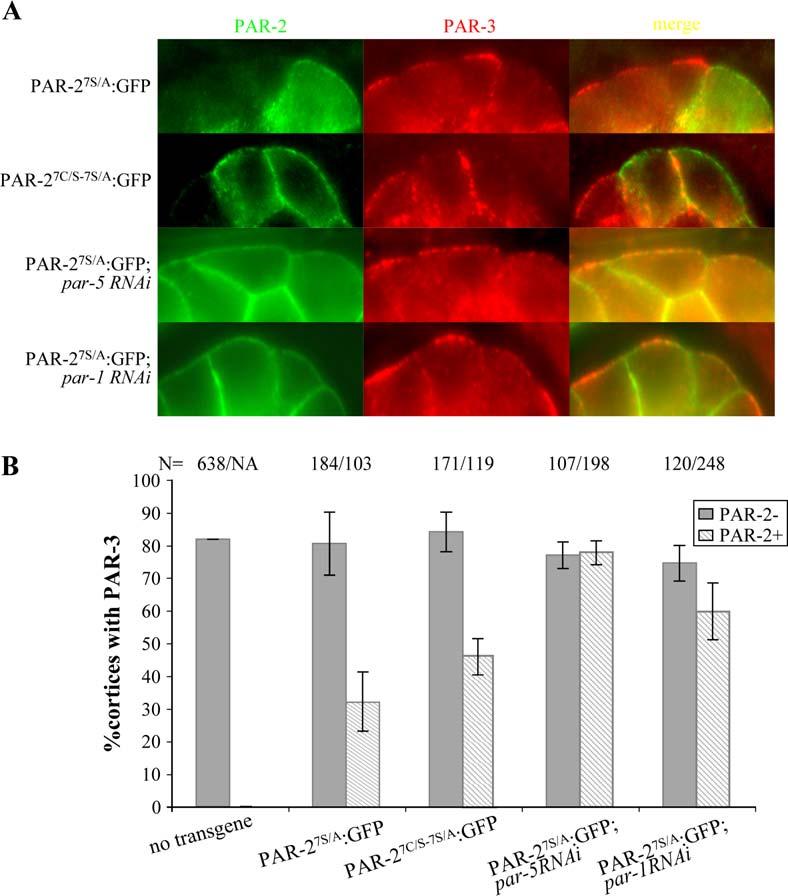
- Somatic blastomeres (apical surfaces are up) expressing the indicated PAR-2:GFP fusion and immunostained for endogenous PAR-3. Note that PAR-2:GFP is sufficient to exclude PAR-3 from apical but not lateral cortices.
- Graphs depicting the percent of PAR-3-positive apical cortices in PAR-2-negative (dark gray bars) and PAR-2-positive cells (hatched bars). Numbers (N) of cortices examined are indicated above each bar. Error bars represent standard deviations computed from two independent experiments for each condition.
In wild-type zygotes, PAR-2 is required to recruit PAR-1 to the posterior cortex, and RNAi experiments have suggested that PAR-1 functions with PAR-2 to exclude anterior PARs (Cuenca et al., 2003). Consistent with this possibility, we found that endogenous PAR-1 was recruited to apical cortices positive for PAR-27S/A:GFP (data not shown). To test whether PAR-1 is required to exclude PAR-3 in the heat shock assay, we inactivated par-1 by RNAi before inducing PAR-27S/A:GFP by heat shock. We found that in par-1(RNAi) embryos, PAR-27S/A:GFP was no longer sufficient to displace PAR-3, and could now be found on apical cortices with PAR-3 (Figure 6). The 14-3-3 protein PAR-5 has also been implicated in the mechanisms that prevent overlap between anterior and posterior PARs (Morton et al., 2002; Cuenca et al., 2003). We found that RNAi of par-5 also blocked PAR-2's ability to exclude PAR-3 (Figure 6). We conclude that cortical PAR-2 excludes PAR-3 from apical cortices in a manner that depends on both par-1 and par-5.
Discussion
We have used transgenic assays to dissect the function of PAR-2 during polarization of the C. elegans zygote. Our observations indicate that (1) cortical localization of PAR-2 is antagonized by PKC-3, (2) the RING domain of PAR-2 is required to overcome cortical exclusion by PKC-3, and (3) cortical PAR-2 excludes anterior PARs from the cortex with the help of PAR-1 and PAR-5.
Cortical Exclusion of PAR-2 by PKC-3
Genetic analyses have shown that par-3, par-6, and pkc-3 are required to restrict PAR-2 to the posterior cortex (Kemphues, 2000). We have identified a 235 amino acid domain in PAR-2, containing seven consensus PKC phosphorylation sites, that is sufficient to localize GFP to the posterior cortex in par-2(+) zygotes. Several lines of evidence suggest that PAR-2 is a substrate for PKC-3, and that phosphorylation by PKC-3 prevents PAR-2 from associating with the anterior cortex. First, PAR-2 can be phosphorylated by human PKCζ in vitro, and this phosphorylation depends in part on consensus PKC sites in the PAR-2 localization domain. Second, a GFP:PAR-2 fusion is phosphorylated in vivo and this phosphorylation depends in part on the PKC sites and on pkc-3. Third, alanine substitutions in the PKC sites that reduce phosphorylation in vitro and in vivo cause GFP:PAR-2 to invade the anterior cortex in vivo. Fourth, glutamic acid substitutions predicted to mimic phosphorylation interfere with GFP:PAR-2's ability to localize to the cortex. Fifth, when expressed in HEK293 cells, PAR-2 is enriched on the cortex, but becomes more cytoplasmic when PKC-3 is also expressed in these cells.
The observation that PKC-3 can affect PAR-2localization in human HEK293 cells suggests that cortical exclusion involves a conserved mechanism. Indeed, PKC-3 homologs have also been implicated in cortical exclusion in two other organisms. In Drosophila, aPKC is enriched with PAR-3 and PAR-6 on the apical cortex of neuroblasts, where it directs the localization of cell fate determinants to the basal cell cortex. Recent evidence suggests that aPKC functions by phosphorylating and inactivating Lethal (2) giant larvae protein (Lgl) specifically on the apical cortex (Betschinger et al., 2003, 2005). Studies in cultured cells indicate that phosphorylation by aPKC releases Lgl from the actin cytoskeleton (Betschinger et al., 2005). In mammalian epithelial cells, aPKC phosphorylates PAR-1b on threonine 595, causing PAR-1 to dissociate from the cortex (Hurov et al., 2004; Suzuki et al., 2004). Interestingly, threonine 595 is conserved in all PAR-1 homologs, and recent studies from our lab suggest that C. elegans PAR-1 may also be a target of PKC-3 (A. Cuenca, J. Levy, and G.S., unpublished data). Together, these studies suggest that phosphorylation by aPKC/PKC-3 may be a common mechanism to exclude proteins from cortices rich in the PAR-3/PAR-6/PKC-3 complex.
Fighting Back: The RING Domain of PAR-2
We have found that the RING domain of PAR-2 is essential to overcome exclusion by PKC-3 and establish a stable PAR-2 domain in the posterior cortex. GFP:PAR-2 fusions lacking the RING localize transiently to the posterior cortex during cortical flow, presumably because PKC-3 levels drop during that time, but are unable to remain on the cortex during mitosis. Because par-2 is required to maintain anterior PARs out of the posterior cortex during mitosis (Cuenca et al., 2003), the localization defects of RING mutants could be due to (1) an inability to exclude anterior PARs after reaching the cortex or (2) an inability to fully overcome exclusion by PKC-3 and establish sufficient levels of PAR-2 on the cortex to exclude anterior PARs. We favor the latter possibility based on two observations. First, the localization and rescuing activity defects of the RING mutants can be suppressed by mutations in the PKC phosphorylation sites, suggesting that the RING is required primarily to counteract the effect of PKC-3. Second, the RING is not essential to exclude PAR-3 from the cortex, when PAR-2 is ectopically expressed in somatic blastomeres (see below). We propose that the primary function of the RING is to protect cortical PAR-2 from exclusion by PKC-3, perhaps by increasing PAR-2's affinity for the cortex or by making PAR-2 a poorer substrate for PKC-3.
The PAR-2 RING belongs to the C3HC4 class characteristic of single-subunit E3 ubiquitin ligases (Moore and Boyd, 2004). Consistent with functioning as an E3 ubiquitin ligase, PAR-2 interacts with E2 ubiquitin conjugation enzymes in the yeast two-hybrid assay (Gudgen et al., 2004) and can auto-ubiquitinate when combined with ubiquitin and E1 and E2 enzymes in vitro (L.B., unpublished data). E3 ubiquitin ligases capable of auto-ubiquitination are often unstable in vivo (Jackson et al., 2000). This is also the case for PAR-2; GFP:PAR-2 fusions with mutations in the RING domain accumulate up to 5-fold higher levels compared to wild-type, suggesting that the RING accelerates PAR-2 turnover. How this activity relates to the RING's ability to resist PKC-3 is not known. The two functions could depend on different enzymatic activities or could result from one common process. Biochemical investigations, including the identification of PAR-2 targets, will be essential to clarify the role of the RING.
Cortical Exclusion of PAR-3 by PAR-2, PAR-1, and PAR-5
Once on the posterior cortex, PAR-2 prevents the return of anterior PAR proteins (Cuenca et al., 2003). To analyze this aspect of PAR-2 function, we used a heat shock promoter to express PAR-27S/A:GFP in somatic blastomeres, where PAR-3, PAR-6, and PKC-3 are normally on apical cortices (Nance et al., 2003). The use of PAR-27S/A allowed us to focus specifically on the effect of cortical PAR-2 on the PAR-3/PAR-6/PKC-3 complex and avoid negative regulation by PKC-3. We found that PAR-27S/A:GFP interferes with PAR-3 localization to apical cortices. This activity was not dependent on the RING, consistent with the RING functioning primarily to overcome cortical exclusion by PKC-3. Exclusion of PAR-3 by PAR-2, however, was dependent on par-1 and par-5, suggesting that PAR-2 acts indirectly. As described in the Introduction, studies in Drosophila have implicated PAR-1 in the cortical exclusion of PAR-3. Phosphorylation by PAR-1 creates a PAR-5 (14-3-3) binding site in PAR-3, which disrupts the PAR-3/PAR-6/aPKC complex leading to cortical exclusion (Benton and St Johnston, 2003). That a similar mechanism may function in C. elegans is supported by the fact that (1) anterior PARs overlap with posterior PARs in par-5 mutant zygotes (Morton et al., 2002) and (2) GFP:PAR-6 invades the posterior cortex during mitosis in par-1(RNAi) zygotes (Cuenca et al., 2003). The latter observation, however, is at odds with results obtained with par-1(b274) mutants, where PAR-3 and GFP:PAR-6 remain excluded from the posterior cortex through mitosis (Etemad-Moghadam et al., 1995; A. Cuenca and G.S., unpublished data). Whether this discrepancy is due to par-1(b274) not being a null allele or to par-1(RNAi) disrupting the expression of another gene remains to be determined. In any case, our results suggest that the effect of PAR-2 on PAR-3 is indirect and involves other par genes.
A Working Model
The results described in this paper and in earlier studies (Kemphues, 2000; Cuenca et al., 2003; Munro et al., 2004) lead us to the following working model to describe polarization of the C. elegans zygote. Immediately before polarization, uniformly high levels of PKC-3 exclude most PAR-2 (and PAR-1) from the cortex. Upon maturation of the sperm asters, cortical flow enriches PKC-3 in the anterior, leaving only low levels in the posterior. This change in PKC-3 distribution allows PAR-2 to contact the posterior cortex, activate its RING activity, and establish a PAR-2 domain in the posterior. High concentration of PAR-2 in the posterior facilitates the loading of PAR-1 and possibly other factors, which in turn prevent anterior PARs from returning to the posterior. How PAR-2 recruits PAR-1 is not yet known. Munro et al. (2004) have observed that PAR-2 prevents the formation of posteriorly directed flows that would otherwise redistribute the PAR-3/PAR-6/PKC-3 complex throughout the cortex. Because PAR-1 likely is also excluded from the cortex by PKC-3 (A. Cuenca, J. Levy, and G.S., unpublished data), by blocking posteriorly directed flows, PAR-2 could effectively tip the balance in favor of PAR-1 (and possibly other factors) and against PKC-3. This model is consistent with the observation that a par-2 null allele can be partially suppressed by removing one copy of par-6, indicating that the primary role of PAR-2 is to oppose the activity of anterior PARs (Watts et al., 1996). Analysis of cortical dynamics in zygotes lacking different combinations of PAR proteins will be critical to explore this model and alternatives further.
In summary, our findings indicate that, by resisting cortical exclusion by PKC-3, the RING activity of PAR-2 potentiates a feedback loop that locks in high levels of PAR-2/PAR-1, and low levels of PAR-3/PAR-6/PKC-3, in the posterior cortex. Among the PAR proteins originally identified in C. elegans, PAR-2 is the only one without apparent homologs in other organisms. Our finding that the RING domain, but not the ATP binding domain, of PAR-2 is essential in vivo raises the possibility that RING proteins in other systems could fulfill the role of PAR-2. Interestingly, proteomic analysis of PAR-1 complexes from human cells identified the RING protein RNF41 (Brajenovic et al., 2004). It will be important to investigate whether RNF41 or other RING proteins contribute to stabilization of cell polarity in mammalian cells.
Experimental Procedures
Nematode Strains and Transgenics
Caenorhabditis elegans strains were derived from the wild-type Bristol strain N2 using standard procedures (Brenner, 1974), except that transgenic strains were kept at 24°C. par-2(lw32) is a nonsense mutation at amino acid position 234 (Levitan et al., 1994) that prevents the production of PAR-2 protein (Boyd et al., 1996).
PAR-2 transgenes were constructed in either pID3.01, a GATEWAY destination vector containing the pie-1 promoter, GFP, GATEWAY recombination sequences, and the pie-1 3′UTR, or pCD6.09AP, a GATEWAY destination vector containing the hsp16-42 promoter, GATEWAY recombination sequences, and the unc-54 3′UTR. The RFP:PAR-6 transgene was constructed in pKS1, a GATEWAY destination vector containing the pie-1 promoter, RFP, GATEWAY recombination sequences, and the pie-1 3′UTR (Sato et al., 2005). Mutations in par-2 were created in GATEWAY entry clones by the QuickChange site-directed and multisite-directed mutagenesis kit (Stratagene, La Jolla, CA) and confirmed by DNA sequencing.
All transgenes were introduced into worms by biolistic transformation (Praitis et al., 2001). Four independent lines or more were generated for each transgene. In all cases, lines with the same trans-gene exhibited the same GFP pattern. A single representative line was selected for further experiments, except for the rescue assay (Figure 3), where two independent lines were characterized for each transgene, except for GFP:PAR-2N-termΔ and GFP:PAR-2C-termΔ for which only one line was characterized. Transgenic lines were crossed to unc-45(e286ts)par-2(lw32)/sC1[dpy-1(s2171)let] males, balanced, and made homozygous for the transgene. For each rescue experiment, 10–20 Unc GFP+ hermaphrodites (par-2 mutant homozygotes containing the transgene) were cut open in egg salts (Edgar, 1995) to release eggs, which were placed on plates and scored 2 days later for viability. Data presented in Figure 3 were compiled from five independent experiments for each transgenic line. Strains used in this study are described in Table S1.
RNA interference was performed using the feeding method (Timmons and Fire, 1998). Bacteria were grown overnight on nematode nutritional growth medium plates containing 60 μg/ml ampicillin and 80 μg/ml isopropyl-β-D-thiogalactopyranoside. L4 hermaphrodites were allowed to feed for 24 hr at 25°C before examination.
Microscopy
Time-lapse microscopy was performed as described in Cuenca et al. (2003). Images were acquired using a Photometrics CoolSnap FX digital camera (Huntington Beach, CA) attached to a Zeiss Axioplan 2 (Thornwood, NY) equipped with Ludl shutters and a mercury lamp. In all pictures, anterior is to the left and posterior is to the right. HEK293 cell images in Figure 2E were collected on a Zeiss LSM510 confocal microscope.
Cortex/cytosol GFP intensity ratios were calculated using IPlab software (Scanalytics, BD Biosciences, Rockville, MD) as follows: For each embryo, a single cross-section image was used to create a fluorescence intensity histogram along the long axis of the embryo. The cortex/cytosol ratio was calculated by dividing the peak value at the cortex by the value at a 30-pixel distance toward the center of the embryo. Six embryos were scored for each genotype.
Kinase Assay
In vitro kinase assays were performed as described (Betschinger et al., 2003; Lin et al., 2000) with human PKCζ (Calbiochem, San Diego, CA) for 30 min.
HEK293 Culture and Transfection
HEK293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (FCS) on poly-L-lysine-coated coverslips and transfected using FuGENE6 (Roche, Indianapolis, IN) according to the manufacturer's protocol. GFP:PAR-2 (pYH2.91), GFP:PAR-27S/A (pYH2.92), and GST:PKC-3(162–597) (pYH2.93) were all driven by the CMV promoter. Transfected cells were fixed with 4% paraformaldehyde and stained with a mouse anti-GFP antibody (3E6, Molecular Probes, Carlsbad, CA; 1:100 dilution) and a rabbit anti-GST antibody (Z5, Santa Cruz Biotechnology, Santa Cruz, CA; 1:50 dilution).
Immunoprecipitations and Western Blots
C. elegans embryos of mixed stages were harvested from gravid adults by standard procedures (Epstein and Liu, 1995) and suspended in three volumes of 15 mM HEPES (pH 7.6), 10 mM KCl, 1.5 mM MgCl2, 0.1 mM EDTA, 0.5 mM EGTA, 44 mM sucrose, protease inhibitor (Roche). The embryo suspension was frozen in liquid nitrogen, ground with a mortar and pestle, and centrifuged at 4°C, 7,800 × g for 10 min. The supernatant was further centrifuged at 4°C, 14,000 rpm for 15 min. The final supernatant was frozen in liquid nitrogen and stored at −80°C.
Five microliters of polyclonal anti-GFP antibody (BD Biosciences, Mountain View, CA) or 60 μl of rabbit IgG (1 mg/ml, Sigma, St. Louis, MO) was coupled to protein A-agarose (Pierce, Rockford, IL). Fifty microliters of embryo extract from each transgenic line was preabsorbed to protein A-agarose coupled to rabbit IgG and incubated with protein A-agarose coupled with anti-GFP antibody to precipitate GFP fusion protein. After an overnight incubation, the beads were washed four times with 15 mM HEPES (pH 7.6), 10 mM KCl, 1.5 mM MgCl2, 0.1 mM EDTA, 0.5 mM EGTA, 44 mM sucrose, and protease inhibitor (Roche) added with 1/10 volume of 1 M NaCl, 15 mM HEPES (pH 7.6). Precipitates and inputs were run on a 4%–12% SDS polyacrylamide gel (Invitrogen, Carlsbad, CA). The GFP fusions were visualized by Western blotting using monoclonal anti-GFP antibody JL-8 (BD Biosciences; 1:1000). Anti-tubulin antibody E7 (Developmental Studies Hybridoma Bank, Iowa City, IA) was used as a loading control against inputs. Horseradish peroxidaseconjugated sheep anti-mouse antibody (1:10,000; Amersham Pharmacia, Piscataway, NJ) was used as secondary antibody. Protein bands were detected by enhanced chemiluminescence (Amersham Pharmacia).
Detection of phosphorylated PAR-2 isoforms was performed using the same methods except that phosphatase inhibitors (1 mM DTT, 1 mM sodium molybdate, 1 mM sodium orthovanadate, 1 mM sodium fluoride, 5 mM β-glycerol phosphate) were included during homogenization and immunoprecipitation. Twenty microliters of washed immunoprecipitates were incubated with ten units of calf intestinal alkaline phosphatase for 1.5 hr at 37°C. The samples were run on Nupage 7% Tris-acetate gel (Invitrogen) and detected using the SuperSignal West Pico kit (Pierce).
Northern Analysis
Total RNA was isolated from gravid hermaphrodites using TRIZOL reagent (Life Technologies, Gaithersburg, MD). Twenty micrograms of total RNA for each strain was hybridized with two random-primed probes: GFP to detect the transgenic RNAs, and RP21 (ribosomal protein) to control for loading.
Heat Shock and Immunofluorescence
Transgenic adult hermaphrodites were heat shocked for 1 hr at 34°C, and recovered for 1 hr at 20°C before immunostaining. This treatment leads to expression of the heat shock transgene in a subset of somatic blastomeres (Schubert et al., 2000). For RNAi experiments, transgenic L4 hermaphrodites were exposed to RNAi by feeding for 24 hr at 25°C before the heat shock procedure. Embryos were fixed on slides in −20°C methanol (15 min) and −20°C acetone (10 min), which preserves GFP fluorescence, and stained with mouse anti-PAR-3 as described (Guo and Kemphues, 1995; Nance et al., 2003). Sixteen- to 30-cell stage embryos with one or more outer blastomeres expressing PAR-2:GFP were analyzed by scoring all apical cortices for the presence of PAR-2:GFP and PAR-3. Standard deviations were computed using data from two independent experiments.
Supplementary Material
Acknowledgments
We thank E. Munro and K. Kemphues for critical reading of the manuscript, and the anonymous reviewers for many helpful comments. We also thank K. Kemphues, J. Priess, J. Nance, C. DeRenzo, K. Cheng, B. Grant, J. Betschinger, J.A. Knoblich, M. Lo, R. Lin, and the Caenorhabditis Genetics Center (funded by the NIH Center for Research Resources) for reagents, protocols, and strains. This work was supported by NIH RO1 grant M400-150-2298. G.S. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Supplemental Data
Supplemental Data include two movies and one table and are available at http://www.developmentalcell.com/cgi/content/full/10/2/199/DC1/.
References
- Benton R, Johnston D. Drosophila PAR-1 and 14-3-3 inhibit Bazooka/PAR-3 to establish complementary cortical domains in polarized cells. Cell. 2003;115:691–704. doi: 10.1016/s0092-8674(03)00938-3. [DOI] [PubMed] [Google Scholar]
- Betschinger J, Knoblich JA. Dare to be different: asymmetric cell division in Drosophila, C. elegans and vertebrates. Curr. Biol. 2004;14:R674–R685. doi: 10.1016/j.cub.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Betschinger J, Mechtler K, Knoblich JA. The Par complex directs asymmetric cell division by phosphorylating the cytoskeletal protein Lgl. Nature. 2003;422:326–330. doi: 10.1038/nature01486. [DOI] [PubMed] [Google Scholar]
- Betschinger J, Eisenhaber F, Knoblich JA. Phosphor-ylation-induced autoinhibition regulates the cytoskeletal protein Lethal (2) giant larvae. Curr. Biol. 2005;15:276–282. doi: 10.1016/j.cub.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Bianchi E, Denti S, Catena R, Rossetti G, Polo S, Gasparian S, Putignano S, Rogge L, Pardi R. Characterization of human constitutive photomorphogenesis protein 1, a RING finger ubiquitin ligase that interacts with Jun transcription factors and modulates their transcriptional activity. J. Biol. Chem. 2003;278:19682–19690. doi: 10.1074/jbc.M212681200. [DOI] [PubMed] [Google Scholar]
- Boyd L, Guo S, Levitan D, Stinchcomb DT, Kemphues KJ. PAR-2 is asymmetrically distributed and promotes association of P granules and PAR-1 with the cortex in C. elegans embryos. Development. 1996;122:3075–3084. doi: 10.1242/dev.122.10.3075. [DOI] [PubMed] [Google Scholar]
- Brajenovic M, Joberty G, Kuster B, Bouwmeester T, Drewes G. Comprehensive proteomic analysis of human Par protein complexes reveals an interconnected protein network. J. Biol. Chem. 2004;279:12804–12811. doi: 10.1074/jbc.M312171200. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeks RJ, Canman JC, Gabriel WN, Meyer N, Strome S, Goldstein B. C. elegans PAR proteins function by mobilizing and stabilizing asymmetrically localized protein complexes. Curr. Biol. 2004;14:851–862. doi: 10.1016/j.cub.2004.05.022. [DOI] [PubMed] [Google Scholar]
- Cuenca AA, Schetter A, Aceto D, Kemphues K, Seydoux G. Polarization of the C. elegans zygote proceeds via distinct establishment and maintenance phases. Development. 2003;130:1255–1265. doi: 10.1242/dev.00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drier EA, Tello MK, Cowan M, Wu P, Blace N, Sacktor TC, Yin JC. Memory enhancement and formation by atypical PKM activity in Drosophila melanogaster. Nat. Neurosci. 2002;5:316–324. doi: 10.1038/nn820. [DOI] [PubMed] [Google Scholar]
- Edgar LG. Blastomere culture and analysis. Methods Cell Biol. 1995;48:303–321. doi: 10.1016/s0091-679x(08)61393-x. [DOI] [PubMed] [Google Scholar]
- Epstein HF, Liu F. Proteins and protein assemblies. Methods Cell Biol. 1995;48:437–450. doi: 10.1016/s0091-679x(08)61398-9. [DOI] [PubMed] [Google Scholar]
- Etemad-Moghadam B, Guo S, Kemphues KJ. Asymmetrically distributed PAR-3 protein contributes to cell polarity and spindle alignment in early C. elegans embryos. Cell. 1995;83:743–752. doi: 10.1016/0092-8674(95)90187-6. [DOI] [PubMed] [Google Scholar]
- Gonczy P, Rose L. Asymmetric cell division and axis formation in the embryo. In: The C. elegans Research Community, editor. WormBook. 2005. 10.1895/wormbook.1.7.1 ( http://www.wormbook.org) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudgen M, Chandrasekaran A, Frazier T, Boyd L. Interactions within the ubiquitin pathway of Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 2004;325:479–486. doi: 10.1016/j.bbrc.2004.10.047. [DOI] [PubMed] [Google Scholar]
- Guo S, Kemphues KJ. par-1, a gene required for establishing polarity in C. elegans embryos, encodes a putative Ser/Thr kinase that is asymmetrically distributed. Cell. 1995;81:611–620. doi: 10.1016/0092-8674(95)90082-9. [DOI] [PubMed] [Google Scholar]
- Hurov JB, Watkins JL, Piwnica-Worms H. Atypical PKC phosphorylates PAR-1 kinases to regulate localization and activity. Curr. Biol. 2004;14:736–741. doi: 10.1016/j.cub.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Jackson PK, Eldridge AG, Freed E, Furstenthal L, Hsu JY, Kaiser BK, Reimann JD. The lore of the RINGs: substrate recognition and catalysis by ubiquitin ligases. Trends Cell Biol. 2000;10:429–439. doi: 10.1016/s0962-8924(00)01834-1. [DOI] [PubMed] [Google Scholar]
- Kemphues K. PARsing embryonic polarity. Cell. 2000;101:345–348. doi: 10.1016/s0092-8674(00)80844-2. [DOI] [PubMed] [Google Scholar]
- Kemphues KJ, Priess JR, Morton DG, Cheng NS. Identification of genes required for cytoplasmic localization in early C. elegans embryos. Cell. 1988;52:311–320. doi: 10.1016/s0092-8674(88)80024-2. [DOI] [PubMed] [Google Scholar]
- Levitan DJ, Boyd L, Mello CC, Kemphues KJ, Stinch-comb DT. par-2, a gene required for blastomere asymmetry in Caenorhabditis elegans, encodes zinc-finger and ATP-binding motifs. Proc. Natl. Acad. Sci. USA. 1994;91:6108–6112. doi: 10.1073/pnas.91.13.6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Edwards AS, Fawcett JP, Mbamalu G, Scott JD, Pawson T. A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat. Cell Biol. 2000;2:540–547. doi: 10.1038/35019582. [DOI] [PubMed] [Google Scholar]
- Macara IG. Parsing the polarity code. Nat. Rev. Mol. Cell Biol. 2004a;5:220–231. doi: 10.1038/nrm1332. [DOI] [PubMed] [Google Scholar]
- Macara IG. Par proteins: partners in polarization. Curr. Biol. 2004b;14:R160–R162. [PubMed] [Google Scholar]
- Moore R, Boyd L. Analysis of RING finger genes required for embryogenesis in C. elegans. Genesis. 2004;38:1–12. doi: 10.1002/gene.10243. [DOI] [PubMed] [Google Scholar]
- Morton DG, Shakes DC, Nugent S, Dichoso D, Wang W, Golden A, Kemphues KJ. The Caenorhabditis elegans par-5 gene encodes a 14-3-3 protein required for cellular asymmetry in the early embryo. Dev. Biol. 2002;241:47–58. doi: 10.1006/dbio.2001.0489. [DOI] [PubMed] [Google Scholar]
- Munro E, Nance J, Priess JR. Cortical flows powered by asymmetrical contraction transport PAR proteins to establish and maintain anterior-posterior polarity in the early C. elegans embryo. Dev. Cell. 2004;7:413–424. doi: 10.1016/j.devcel.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Nance J, Munro EM, Priess JR. C. elegans PAR-3 and PAR-6 are required for apicobasal asymmetries associated with cell adhesion and gastrulation. Development. 2003;130:5339–5350. doi: 10.1242/dev.00735. [DOI] [PubMed] [Google Scholar]
- Praitis V, Casey E, Collar D, Austin J. Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics. 2001;157:1217–1226. doi: 10.1093/genetics/157.3.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuvanshi S, Kelkar A, Khurana JP, Tyagi AK. Isolation and molecular characterization of the COP1 gene homolog from rice, Oryza sativa L. subsp. Indica var. Pusa Basmati 1. DNA Res. 2001;8:73–79. doi: 10.1093/dnares/8.2.73. [DOI] [PubMed] [Google Scholar]
- Sato M, Sato K, Fonarev P, Huang CJ, Liou W, Grant BD. Caenorhabditis elegans RME-6 is a novel regulator of RAB-5 at the clathrin-coated pit. Nat. Cell Biol. 2005;7:559–569. doi: 10.1038/ncb1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert CM, Lin R, de Vries CJ, Plasterk RH, Priess JR. MEX-5 and MEX-6 function to establish soma/germline asymmetry in early C. elegans embryos. Mol. Cell. 2000;5:671–682. doi: 10.1016/s1097-2765(00)80246-4. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Hirata M, Kamimura K, Maniwa R, Yamanaka T, Mizuno K, Kishikawa M, Hirose H, Amano Y, Izumi N, et al. aPKC acts upstream of PAR-1b in both the establishment and maintenance of mammalian epithelial polarity. Curr. Biol. 2004;14:1425–1435. doi: 10.1016/j.cub.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Tabuse Y, Izumi Y, Piano F, Kemphues KJ, Miwa J, Ohno S. Atypical protein kinase C cooperates with PAR-3 to establish embryonic polarity in Caenorhabditis elegans. Development. 1998;125:3607–3614. doi: 10.1242/dev.125.18.3607. [DOI] [PubMed] [Google Scholar]
- Timmons L, Fire A. Specific interference by ingested dsRNA. Nature. 1998;395:854. doi: 10.1038/27579. [DOI] [PubMed] [Google Scholar]
- Watts JL, Etemad-Moghadam B, Guo S, Boyd L, Draper BW, Mello CC, Priess JR, Kemphues KJ. par-6, a gene involved in the establishment of asymmetry in early C. elegans embryos, mediates the asymmetric localization of PAR-3. Development. 1996;122:3133–3140. doi: 10.1242/dev.122.10.3133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.