

Abstract
The electron transport chain enzyme complex I may play a role in Parkinson's disease (PD) pathogenesis. Association studies considering whether or not complex I-relevant gene polymorphisms contribute to PD risk are discordant. We evaluated four complex I-relevant gene polymorphisms alternatively reported to associate and not associate with PD (tRNAGln T4336C, ND1 T4216C, ND2 G5460A, and the NDUFV2 exon 2 C182T transition). Our study included 111 PD subjects and 106 controls in central Virginia. Individuals with at least one copy of the NDUFV2 182T allele were more likely to report a PD family history than non-carriers, but aside from this no positive associations were found. Indeed, the tRNAGln 4336C variant occurred more frequently in controls. We also observed that individuals in both groups often carried more than one of the assayed polymorphisms, and for the first time show bigenomic polymorphic variation (between nuclear and mtDNA complex I subunit genes) commonly occurs within individuals. In an exploratory sub-analysis, more control than case women had an ND1 4216C, NDUFV2 homozygous 182C compound genotype. Complex I compound genotype variation commonly occurs and may explain why particular complex I gene polymorphisms associate with PD in some populations but not others.
Keywords: Complex I, Electron transport chain, Mitochondria, Mitochondrial DNA, Parkinson's disease, Polymorphisms
1. Introduction
Parkinson's disease (PD) subjects show reduced NADH: ubiquinone oxidoreductase (complex I) activity and complex I inhibition causes both animal and human parkinsonism [1-6]. These findings suggest that complex I may play a pathogenically important role in PD. Complex I, an electron transport chain (ETC) enzyme, contains over 40 protein subunits encoded on an equal number of genes [7]. Seven of these genes reside on mitochondrial DNA (mtDNA) and the rest are nuclear genes. Association studies of polymorphisms in several complex I genes or genes related to complex I subunit production suggest complex I variation might increase or decrease one's risk of developing PD [8-15]. However, positive association study results are counter-balanced by negative studies [14,16-20]. The importance of complex I or complex I-related gene polymorphisms in PD remains controversial.
Past positive studies report polymorphism associations between complex I subunit genes on both mtDNA (T4216C in ND1, G5460A in ND2) [10,12-14] and nuclear DNA (a C→T transition at exon 2 nucleotide 182 of NDUFV2) [11]. Increased frequency of a T4336C polymorphism (in the mtDNA tRNAGln gene) may also occur in PD [8,9]. This is a synthetic gene required for translation of mtDNA-encoded ETC structural genes, and therefore can be considered a complex I-relevant gene. Negative association studies for each of these polymorphisms in PD also exist [16-20]. Because of our interest in how complex I dysfunction arises in and may contribute to PD, we attempted to replicate studies reporting a positive association between PD and complex I subunit or complex I-related gene polymorphisms.
2. Methods
2.1. Subjects
All parts of our study were conducted in accordance with the Declaration of Helsinki. Genotyped subjects provided signed consent; the University of Virginia institutional review board approved the protocol. PD subjects were recruited from the University of Virginia Movement Disorders Clinic. PD sub-specialty neurologists made each diagnosis and patients met UK Parkinson's Disease Society Brain Bank clinical diagnostic criteria [21]. We recruited a control cohort from the same geographical region. Subjects from both PD and control groups were overwhelmingly of white European descent. As a prior study of the T4216C polymorphism in PD reported a sex-specific effect [12], we took care to sex-match our cohorts. Since PD more commonly affects men than women [22,23], we were forced to limit inclusion of PD spouses in order to avoid female over-representation in the control group.
In order to reduce the chances of including Mendelian PD in our analysis, as well as limit the number of controls below the age of peak PD presentation, individuals less than 40 years of age were excluded. At the time of enrollment, subjects in both groups were asked to report any family members with PD.
2.2. Procedures
Blood samples were collected using standard vacutainer tubes containing EDTA as anticoagulant. We used a QIAamp DNA minikit (Qiagen, Valencia, CA) to prepare genomic DNA from these samples. Taq/Pwo polymerase (Roche Diagnostics, Indianapolis, IN) and a GeneAmp9600 thermocycler were used for polymerase chain reaction (PCR) generation of amplicons containing the polymorphisms of interest (T4336C in tRNAGln, T4216C in ND1, G5460A in ND2, and the C→T transition in exon 2 of NDUFV2 that causes an alanine to valine substitution at amino acid 29). Table 1 presents the primer sequences used for these analyses. PCR products were purified using a QIAquick kit (Qiagen). The polymorphisms of interest were interrogated with the restriction enzymes shown in Table 1, and the final digestion products electrophoresed within agarose gels containing ethidium bromide.
Table 1.
Oligonucleotide primers and restriction enzymes used
| Polymorphism genotyped | Primers | Restriction enzyme |
|---|---|---|
| ND1 T4216C | 5′–GCCGTTTACTCAATCCTCTG–3′ | NlaIII |
| 5′–GTGTGATAGGTGGCACGGAG–3′ | ||
| tRNAGln T4336C | 5′–GCCGTTTACTCAATCCTCTG–3′ | NlaIII |
| 5′–GTGTGATAGGTGGCACGGAG–3′ | ||
| ND2 G5460A | 5′–TCCCCACCATCATAGCCA–3′ | HphI |
| 5′–GGGTTTTGCAGTCCTTAG–3′ | ||
| NDUFV2 C182T | 5′–GATGGATAGGGTAGAATACCATATTCCTTA–3′ | MaeIII |
| 5′–ATAAAGCTCCTCCAGCTCCATTTTGCGTA–3′ |
The 4216C and 4336C polymorphisms both create NlaIII restriction sites, and because both polymorphisms reside within one PCR product spanning mtDNA nucleotides 3540–4390 (as numbered by the Cambridge sequence) [24], the 4216 and 4336 positions were genotyped simultaneously. A 4% agarose concentration (E-Gel, Invitrogen, Carlsbad, CA) facilitated clear interpretation of the NlaIII digestion products.
The 5460A polymorphism opens an HphI site. The NDUFV2 polymorphism corresponds to nucleotide 182 of the NDUFV2 GenBank exon 2 sequence (as listed in gi:3123713). The downstream primer used in the reaction that amplifies NDUFV2 exon 2 nucleotides 2–211 actually contains a mismatch near its 3′ end, so that when thymine rather than cytosine occurs at nucleotide 182 the mismatched primer generates a unique MaeIII restriction site [11]. Interpretation of HphI and MaeIII digestion products was obvious using a 2% agarose concentration.
To ensure fidelity of our approach, several amplicons shown by restriction fragment length analysis to contain polymorphic changes were corroborated by dideoxynucleotide sequencing using an Applied Biosystems (model 377; Foster City, CA) sequencer. All samples found on restriction analysis to contain the 4336C variant were sequence-corroborated.
PD and control subject group mean ages were compared by two-way Student's t-test with α set at 0.05. For our family history analysis, we anticipated a positive PD family history would prove a rare event, and therefore used the binomial exact test. All other statistical analyses, including exploratory sub-analyses of genotype interaction, were done by Pearson's χ2. Statistical calculations were performed using S-Plus (Insightful Corporation, Seattle, WA).
3. Results
We studied 111 PD subjects (68 males, 43 females) and 106 sex-matched controls (64 males, 42 females). Ages ranged from 40 to 91 years. There was a slight age mismatch between the groups, as the mean age of the controls was 4.2 years younger than that of the PD group. Table 2 provides demographic data on genotyped subjects. Although we excluded PD subjects with clear Mendelian inheritance patterns, 13.5% of PD subjects reported a positive family history of PD, which was greater than the 7.5% of controls reporting a positive PD family history (p = 0.028). This difference was primarily female-driven, as the number of female cases with a positive family history of PD exceeded the number of control females (p = 0.004).
Table 2.
Group demographics
| PD | Control | |
|---|---|---|
| Total subjects | 111 | 106 |
| Male:Female | 68:43 (1.6:1.0) | 64:42 (1.5:1.0) |
| Positive family history | 15 (13.5%) | 8 (7.5%)* |
| Males with a positive family history | 8 (11.8%) | 6 (9.4%) |
| Females with a positive family history | 7 (16.3%) | 2 (4.9%)** |
| Age of subjects (mean±S.E.M.) | 65.0±0.9 | 60.8±1.2*** |
p=0.028 by binomial exact test.
p=0.004 by binomial exact test.
p<0.01 by two-way Student's t-test.
Surprisingly, the tRNAGln 4336C polymorphism was over-represented in our controls (p = 0.039). Otherwise, polymorphism frequencies for ND2 5460A, NDUFV2 182T, and total subject 4216C were similar between PD cases and controls (Table 3). ND2 G5460A and tRNAGln T4336C polymorphisms were very rare in either group, precluding further analyses of these polymorphisms. NDUFV2 182T and ND1 4216C polymorphisms were relatively more common. Specifically, 34.6% of all genotyped individuals carried at least one NDUFV2 182T allele; our NDUFV2 genotype distributions exhibited Hardy–Weinberg equilibrium. We found 19.8% possessed the ND1 4216C variant. This enabled us to further stratify the NDUFV2 C182T and ND1 T4216C data by sex. Our sex-specific analysis suggested a trend in which more control women than PD women carried the ND1 4216C variant (p = 0.060) (Table 3).
Table 3.
Selected complex I subunit gene and complex I-related gene polymorphism frequencies in central Virginia
| PD | Control | ||
|---|---|---|---|
| tRNAGln 4336C | 0/111 (0%) | 4/106 (3.8%)* | |
| ND2 5460A | 2/111 (1.8%) | 3/106 (2.8%) | |
| ND1 4216C | Total | 19/111 (17.1%) | 24/106 (22.6%) |
| Males | 13/68 (19.1%) | 11/64 (17.2%) | |
| Females | 6/43 (14.0%) | 13/42 (31.0%)** | |
| NDUFV2 | Total with Ala/Val or Val/Val | 36/111 (32.4%) | 39/106 (36.8%) |
| Male Ala/Val or Val/Val | 23/68 (33.8%) | 25/64 (39.1%) | |
| Female with Ala/Val or Val/Val | 13/43 (30.2%) | 14/42 (33.3%) | |
| Total Ala/Val | 32/111 (28.8%) | 36/106 (34.0%) | |
| Total Val/Val | 4/111 (3.6%) | 3/106 (2.8%) | |
| Male Ala/Val | 21/68 (30.9%) | 25/64 (39.0%) | |
| Female Ala/Val | 11/43 (23.3%) | 11/42 (26.2%) | |
| Male Val/Val | 2/68 (2.9%) | 0/64 (0%) | |
| Female Val/Val | 2/43 (4.7%) | 3/42 (7.1%) | |
| NDUFV2+ND1 4216C: | Val Total | 6/111 (5.9%) | 9/106 (8.5%) |
| Males | 4/68 (5.9%) | 6/64 (9.4%) | |
| Females | 2/43 (4.7%) | 3/42 (7.1%) |
p=0.039 by χ2.
p= 0.060 by χ2.
We also considered whether carriage of the NDUFV2 182T (36 of 111 individuals) and/or ND1 4216C (19 of 111 individuals) polymorphisms contributed to age of PD onset. We plotted PD subject age at the time of phlebotomy (a rough surrogate for age of disease onset) according to polymorphism genotype. This analysis did not support the possibility that either polymorphism modifies age of PD onset, either alone or in combination (Fig. 1).
Fig. 1.
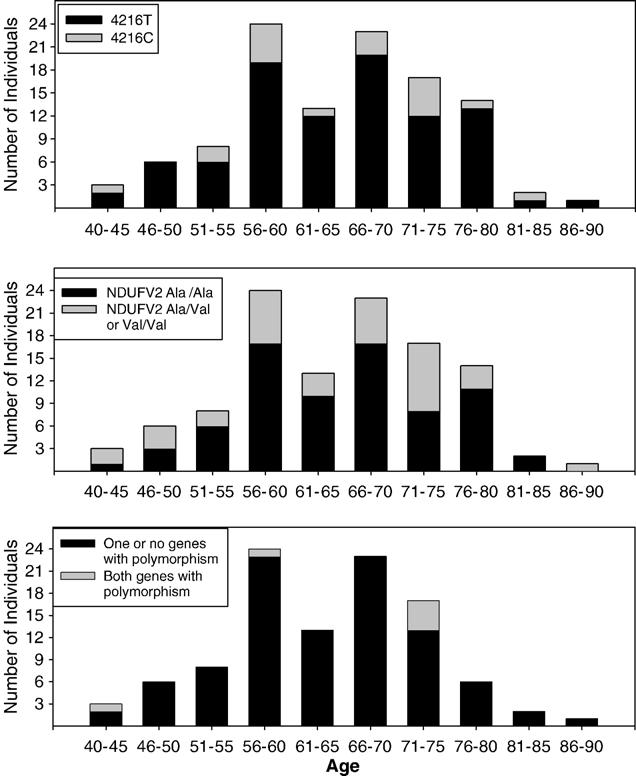
Distribution (by age at the time of genotyping) of PD subjects with ND1 4216C (top), either one or two NDUFV2 182T (Val) alleles (middle), or both ND1 4216C and NDUFV2 182T polymorphisms (bottom).
We evaluated the impact of NDUFV2 182T or ND1 4216C polymorphisms on PD family history reporting across cases and controls (Table 4). Possessing the ND1 4216C variant was not associated with greater likelihood of reporting a family history of PD. Carriers of at least one NDUFV2 182T allele were more likely to report a positive PD family history than those with two copies of NDUFV2 182C (p = 0.019). Other smaller sub-group analyses evaluating the effects of NDUFV2 genotype on family history reporting were significant as well, and for this reason in Table 4 we included a breakdown of family history reporting by genotype and sex. However, as the numbers in these subgroup analyses were small, in Table 4 we only report significance for the key aggregate NDUFV2 C182T data.
Table 4.
ND1 T4216C and NDUFV2 C182T distribution in those with a positive PD family history
| PD subjects |
Control subjects |
All subjects |
||||
|---|---|---|---|---|---|---|
| Polymorphism present | Polymorphism not present | Polymorphism present | Polymorphism not present | Polymorphism present | Polymorphism not present | |
| ND1 T4216C | ||||||
| All | 4/19 (27%) | 11/92 (12%) | 2/24 (8%) | 6/82 (7%) | 6/43 (14%) | 17/174 (10%) |
| Males | 2/13 (15%) | 6/55 (11%) | 2/11 (18%) | 4/53 (8%) | 4/24 (17%) | 10/108 (9%) |
| Females | 2/6 (33%) | 5/37 (14%) | 0/13 (0%) | 2/29 (7%) | 2/19 (10%) | 7/66 (11%) |
| NDUFV2 C182T (polymorphism present = either heterozygous or homozygous for 182T) | ||||||
| All | 7/36 (19%) | 8/75 (10%) | 6/39 (15%) | 2/67 (3%) | 13/75 (17%)* | 10/142 (7%) |
| Males | 3/23 (13%) | 5/45 (6%) | 5/25 (20%) | 1/39 (3%) | 8/48 (17%) | 6/84 (7%) |
| Females | 4/13 (31%) | 3/30 (10%) | 1/14 (7%) | 1/28 (4%) | 5/27 (19%) | 4/58 (7%) |
p=0.019 by χ2.
Six PD and nine control subjects possessed both ND1 4216C and NDUFV2 182T polymorphisms (Table 3). These 15 subjects comprised 7% of the overall study population. The presence of bigenomic-derived ETC heterogeneity within individuals seems conceptually predictable and not surprising. Nonetheless, to our knowledge these data for the first time actually demonstrate this phenomenon. We further evaluated the degree to which just these two polymorphisms contribute to complex I heterogeneity in central Virginia. The ND1 T4216C and NDUFV2 C182T polymorphisms allowed us to define and demonstrate six different genetically-determined complex I subgroups (Table 5).
Table 5.
Genotype breakdown of 217 individuals assessed for the mtDNA ND1 T4216C and nuclear NDUFV2 C182T complex I subunit gene polymorphisms (182C = Ala; 182T = Val)
| ND1 4216T | ND1 4216C | |
|---|---|---|
| NDUFV2 Ala/Ala | 114 (53%) | 28 (13%) |
| NDUFV2 Ala/Val | 55 (25%) | 13 (6%) |
| NDUFV2 Val/Val | 5 (2%) | 2 (1%) |
Although the numbers were small, the proportion of individuals carrying concomitant ND1 4216C, NDUFV2 182T polymorphisms did not vary between PD and control groups (Table 3). To further consider whether interactive effects exist between these two variants in an exploratory sub-analysis, we sorted the ND1 4216C subjects by NDUFV2 status, and also the NDUFV2 182T carriers by ND1 status (Table 6). This analysis found the previously suggested trend towards ND1 4216C over-representation in the female control group (Table 3) primarily arose from an excess of 4216C women that were also NDUFV2 182C homozygous (the Ala/Ala genotype). This exploratory finding was significant (p = 0.047), and implies a potential protective effect for the ND1 4216C and NDUFV2 Ala/Ala compound genotype in women. However, there was no evidence that an ND1 4216C and NDUFV2 Ala/Ala combination protected men from PD. Indeed, there was a non-significant trend towards over-representation of the ND1 4216C and NDUFV2 Ala/Ala compound genotype in PD men.
Table 6.
NDUFV2 C182T against “fixed” ND1 T4216C background, and ND1 T4216C against “fixed” NDUFV2 C182T background
| PD | Control | |
|---|---|---|
| A) ND1 4216C when NDUFV2 is Ala/Ala | ||
| Total | 13/75 (17.3%) | 15/67 (22.4%) |
| Males | 9/45 (20.0%) | 5/39 (12.8%) |
| Females | 4/30 (13.3%) | 10/28 (35.8%)* |
| B) ND1 4216C when NDUFV2 is Ala/Val or Val/Val | ||
| Total | 6/36 (16.7%) | 9/39 (23.1%) |
| Males | 4/23 (17.4%) | 6/25 (24.0%) |
| Females | 2/13 (15.4%) | 3/14 (21.4%) |
| C) NDUFV2 Ala/Val or Val/Val when ND1 is 4216C | ||
| Total | 6/19 (31.5%) | 9/24 (37.5%) |
| Males | 4/13 (30.8%) | 6/11 (45.5%) |
| Females | 2/6 (33.3%) | 3/13 (23.1%) |
| D) NDUFV2 Ala/Val or Val/Val when ND1 is 4216T | ||
| Total | 30/92 (32.6%) | 30/82 (36.6%) |
| Males | 19/55 (34.5%) | 19/53 (35.8%) |
| Females | 11/37 (29.7%) | 11/29 (37.9%) |
p=0.047 by χ2.
4. Discussion
This study joins several others finding sporadic PD subjects more commonly report positive PD family histories than control subjects [23,25-32]. Perhaps PD itself increases ones awareness of PD or parkinsonism in others, or control status reduces awareness, rendering in either case a secondary artifact [33]. Environmental overlap between family members requires consideration, as toxic exposure can cause parkinsonism [3]. Genetic factors might also explain this finding.
Despite controversy regarding the accuracy of PD family history reporting [33-35], our family history data agree reasonably well with recent “standard for comparison” PD family history data derived using a rigorous, secondarily verified ascertainment method in Olmsted county [33]. Furthermore, we believe geographic congruence between our PD and control groups reduces the chance that environmental factors account for our increased PD subject reporting of PD family histories. We therefore feel unable to summarily dismiss the difference we found between PD and control family history reporting as artifact.
Prior studies of tRNAGln 4336C in PD reported either positive or absent associations [8,9,16,17,19,20].None found, as we did, an over-representation of tRNAGln 4336C in control subjects. While particular mtDNA signatures may reduce PD risk [15], the rarity of tRNAGln 4336C in our study precludes us from postulating this polymorphism protects against PD in some populations. The small sample size further renders the statistical finding suspect. Nevertheless, we do feel our data adequately argue that the tRNAGln 4336C variant does not meaningfully contribute to the central Virginia PD burden.
A previous PD association study from Germany reported an excess of the ND1 5460A polymorphism in PD [10].We failed to replicate this finding in our central Virginia PD cohort. In the German controls, the 5460A frequency (5/77; 6.5%) more than doubled the central Virginia 5460A control rate of 2.8%. Although this difference is not statistically significant, it does suggest potential baseline genetic dissimilarities may exist between the populations, which perhaps could account for the discrepant results between these two studies. Regardless, we do feel our data adequately argue the ND2 5460A variant does not meaningfully contribute to the central Virginia PD burden.
Our finding that ND1 4216C may occur more commonly in central Virginia female control subjects than in female PD subjects does not dispute Kirchner et al., who reported a sex-dependent disparity of this polymorphism existed between western Washington state PD and control cohorts [12]. That study showed a trend towards more 4216C female (21%) than male controls (16%). The authors further noted a non-significant tendency towards over-representation of 4216C in male but not female PD subjects, perhaps consistent with the trend we observed in PD 4216C males carrying the NDUFV2 Ala/Ala genotype.
In their 1998 Japanese PD case–control study, Hattori et al. reported NDUFV2 Val/Val frequencies greater than those we found in central Virginia PD (23.8% vs. 3.6%) and control (11.5% vs. 2.8%) subjects [11]. While NDUFV2 Val/Val frequencies are equivalent between PD and control subjects in central Virginia, the frequency of the NDUFV2 Val/Val genotype in Japanese PD subjects exceeded that of the Japanese controls. Interestingly, NDUFV2 Ala/Val did not appear to confer PD risk in the Hattori et al. study, as the heterozygous state was significantly greater in the control than in the PD group (42.5% control heterozygotes, 30.2% PD heterozygotes). The absolute percentage of control NDUFV2 heterozygotes in our study was also greater than that of our PD subjects, but this was not a statistically significant difference. Our central Virginia populations further showed a tight Hardy–Weinberg equilibrium distribution, whereas the Japanese cohort did not.
While our NDUFV2 genotype data do not indicate NDUFV2 Val/Val contributes to the PD burden of central Virginia, two findings in our study nevertheless suggest the NDUFV2 C182T polymorphism might still influence PD risk in at least some instances. First, individuals with the 182T allele were more likely to report a positive family history than those who were 182C homozygous. Second, in our control group we found an excess of homozygous NDUFV2 182C women with the ND1 4216C polymorphism. Taken together, these two observations could imply NDUFV2 182C homozygosity lessens the risk of PD in certain sub-populations, such as women with the ND1 4216C polymorphism. If so, the absence of an NDUFV2 182C allele would abrogate or reduce any protective effect that NDUFV2 182C confers to that subgroup.
Large association studies of 500–1000 subjects are typically required to identify polymorphism-disease associations worthy of further consideration. Our study was neither suited nor intended to identify new associations, but sought only to evaluate certain polymorphisms previously claimed by others to affect PD risk. The size of our study is similar to or even larger than other published PD association studies [9-14]. Nevertheless, we acknowledge that some of our subgroups contain small numbers, and we therefore consider the results of our exploratory subgroup analyses preliminary. We performed multiple statistical comparisons and obtained limited significant findings. None were exceedingly robust and most would not have survived correction for multiple comparisons, raising the possibility of type II error. As discussed above, we suspect over-representation of the tRNAGln 4336C in controls may indeed represent type II error. On the other hand, our two other genotype-related findings (that the NDUFV2 Ala/Ala genotype might reduce the risk of PD in women with ND1 4216C and perhaps in general reduce the chance of reporting a PD family history) corroborate each other.
To our knowledge, our data for the first time explicitly show different bigenomic ETC polymorphism combinations co-exist within individuals and actually contribute to ETC enzyme heterogeneity. While perhaps obvious to the point of overlooking, our data represent the variability that was found from just one mtDNA complex I polymorphism and one nuclear DNA complex I polymorphism. Considering there are perhaps 46 complex I genes encoding complex I subunits [7], and that at least some of these genes (certainly the mtDNA ND genes) can have multiple polymorphisms [36], the potential for inter-individual variation is enormous.
Since it indeed occurs, the question of whether bigenomic polymorphic variability represents an incidental or phenotypically relevant phenomenon requires consideration. Although the small size of our data set renders any insight into this question preliminary, our finding that NDUFV2 182C homozygosity might reduce PD risk in women that also carry the ND1 4216C polymorphism suggests compound genotypes might prove potentially relevant. If correct, bigenomic ND gene heterogeneity between individuals could help explain discrepancies between complex I or complex I-related polymorphism association studies performed in ethnically or geographically distinct PD populations.
Acknowledgments
This work was supported by the National Institutes of Health (AG00800, R01 AG022407) and the American Parkinson's Disease Association (Cotzias Award).
References
- 1.Schapira AHV, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;i:1289. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 2.Parker WD, Boyson SJ, Parks JK. Electron transport chain abnormalities in idiopathic Parkinson's disease. Ann Neurol. 1989;26:719–23. doi: 10.1002/ana.410260606. [DOI] [PubMed] [Google Scholar]
- 3.Langston JW, Ballard PA, Tetrud JW, Irwin I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–80. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 4.Nicklas WJ, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenylpyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Life Sci. 1985;36:2503–8. doi: 10.1016/0024-3205(85)90146-8. [DOI] [PubMed] [Google Scholar]
- 5.Burns RS, Chiueh CC, Markey SP, Ebert MH, Jacobowitz DM, Kopin IJ. A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci U S A. 1983;80:4546–50. doi: 10.1073/pnas.80.14.4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–6. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 7.Carroll J, Fearnley IM, Shannon RJ, Hirst J, Walker JE. Analysis of the subunit composition of complex I from bovine heart mitochondria. Mol Cell Proteomics. 2003;2:117–26. doi: 10.1074/mcp.M300014-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Shoffner JM, Brown MD, Torroni A, Lott MT, Cabell MF, Mirra SS, et al. Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics. 1993;17:171–84. doi: 10.1006/geno.1993.1299. [DOI] [PubMed] [Google Scholar]
- 9.Egensperger R, Kosel S, Schnopp NM, Mehraein P, Graeber MB. Association of the mitochondrial tRNA(A4336G) mutation with Alzheimer's and Parkinson's diseases. Neuropathol Appl Neurobiol. 1997;23:315–21. [PubMed] [Google Scholar]
- 10.Kosel S, Lucking CB, Egensperger R, Mehraein P, Graeber MB. Mitochondrial NADH dehydrogenase and CYP2D6 genotypes in Lewy-body parkinsonism. J Neurosci Res. 1996;44:174–83. doi: 10.1002/(SICI)1097-4547(19960415)44:2<174::AID-JNR10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 11.Hattori N, Yoshino H, Tanaka M, Suzuki H, Mizuno Y. Genotype in the 24-kDa subunit gene (NDUFV2) of mitochondrial complex I and susceptibility to Parkinson disease. Genomics. 1998;49:52–8. doi: 10.1006/geno.1997.5192. [DOI] [PubMed] [Google Scholar]
- 12.Kirchner SC, Hallagan SE, Farin FM, Dilley J, Costa-Mallen P, Smith-Weller T, et al. Mitochondrial ND1 sequence analysis and association of the T4216C mutation with Parkinson's disease. Neurotoxicology. 2000;21:441–5. [PubMed] [Google Scholar]
- 13.Ross OA, McCormack R, Maxwell LD, Duguid RA, Quinn DJ, Barnett YA, et al. mt4216C variant in linkage with the mtDNA TJ cluster may confer a susceptibility to mitochondrial dysfunction resulting in an increased risk of Parkinson's disease in the Irish. Exp Gerontol. 2003;38:397–405. doi: 10.1016/s0531-5565(02)00266-8. [DOI] [PubMed] [Google Scholar]
- 14.Otaegui D, Paisan C, Saenz A, Marti I, Ribate M, Marti-Masso JF, et al. Mitochondrial polymporphisms in Parkinson's Disease. Neurosci Lett. 2004;370:171–4. doi: 10.1016/j.neulet.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 15.van der Walt JM, Nicodemus KK, Martin ER, Scott WK, Nance MA, Watts RL, et al. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am J Hum Genet. 2003;72:804–11. doi: 10.1086/373937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayr-Wohlfart U, Rodel G, Hennesberg A. Mitochondrial tRNA (Gln) and tRNA (Thr) gene variants in Parkinson's disease. Eur J Med Res. 1997;2:111–3. [PubMed] [Google Scholar]
- 17.Garcia-Lozano JR, Mir P, Alberca R, Aguilera I, Gil Neciga E, Fernandez-Lopez O, et al. Mitochondrial DNA A4336G mutation in Alzheimer's and Parkinson's diseases. Eur Neurol. 2002;48:34–6. doi: 10.1159/000064955. [DOI] [PubMed] [Google Scholar]
- 18.Tan EK, Chai A, Zhao Y, Lum SY, Fook-Chong SM, Teoh ML, et al. Mitochondrial complex I polymorphism and cigarette smoking in Parkinson's disease. Neurology. 2002;59:1288–9. doi: 10.1212/01.wnl.0000031809.71668.a1. [DOI] [PubMed] [Google Scholar]
- 19.Bandmann O, Sweeney MG, Daniel SE, Marsden CD, Wood NW. Mitochondrial DNA polymorphisms in pathologically proven Parkinson's disease. J Neurol. 1997;244:262–5. doi: 10.1007/s004150050082. [DOI] [PubMed] [Google Scholar]
- 20.Simon DK, Mayeux R, Marder K, Kowall NW, Beal MF, Johns DR. Mitochondrial DNA mutations in complex I and tRNA genes in Parkinson's disease. Neurology. 2000;54:703–9. doi: 10.1212/wnl.54.3.703. [DOI] [PubMed] [Google Scholar]
- 21.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:185–8. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanner CM, Goldman SM. Epidemiology of Parkinson's disease. Neurol Clin. 1996;14:317–35. doi: 10.1016/S0733-8619(05)70259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swerdlow RH, Parker WD, Jr, Currie LJ, Bennett JP, Harrison MB, Trugman JM, et al. Gender ratio differences between Parkinson's disease patients and their affected relatives. Parkinsonism Relat Disord. 2001;7:129–33. doi: 10.1016/s1353-8020(00)00029-8. [DOI] [PubMed] [Google Scholar]
- 24.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–65. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 25.Martin WE, Young WI, Anderson VE. Parkinson's disease: a genetic study. Brain. 1973;96:495–506. doi: 10.1093/brain/96.3.495. [DOI] [PubMed] [Google Scholar]
- 26.Payami H, Larsen K, Bernard S, Nutt J. Increased risk of Parkinson's disease in parents and siblings of patients. Ann Neurol. 1994;36:659–61. doi: 10.1002/ana.410360417. [DOI] [PubMed] [Google Scholar]
- 27.Bonifati V, Fabrizio E, Vanacore N, De Mari M, Meco G. Familial Parkinson's disease: a clinical genetic analysis. Can J Neurol Sci. 1995;22:272–9. doi: 10.1017/s0317167100039469. [DOI] [PubMed] [Google Scholar]
- 28.Marder K, Tang MX, Mejia H, Alfaro B, Cote L, Louis E, et al. Risk of Parkinson's disease among first-degree relatives: a community-based study. Neurology. 1996;47:155–60. doi: 10.1212/wnl.47.1.155. [DOI] [PubMed] [Google Scholar]
- 29.Uitti RJ, Shinotoh H, Hayward M, Schulzer M, Mak E, Calne DB. “Familial Parkinson's disease”: a case–control study of families. Can J Neurol Sci. 1997;24:127–32. doi: 10.1017/s0317167100021454. [DOI] [PubMed] [Google Scholar]
- 30.Elbaz A, Grigoletto F, Baldereschi M, Breteler MM, Manubens-Bertran JM, Lopez-Pousa S, et al. Familial aggregation of Parkinson's disease: a population based case–control study in Europe. Neurology. 1999;52:1876–82. doi: 10.1212/wnl.52.9.1876. [DOI] [PubMed] [Google Scholar]
- 31.Taylor CA, Saint-Hilaire MH, Cupples LA, Thomas CA, Burchard AE, Feldman RG, et al. Environmental, medical, and family history risk factors for Parkinson's disease: a New England-based case control study. Am J Med Genet. 1999;88:742–9. [PubMed] [Google Scholar]
- 32.Autere JM, Moilanen JS, Myllyla VV, Majamaa K. Familial aggregation of Parkinson's disease in a Finnish population. J Neurol Neurosurg Psychiatry. 2000;69:107–9. doi: 10.1136/jnnp.69.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elbaz A, McDonnell SK, Maraganore DM, Strain KJ, Schaid DJ, Bower JH, et al. Validity of family history data on PD: evidence for a family information bias. Neurology. 2003;61:11–7. doi: 10.1212/01.wnl.0000068007.58423.c2. [DOI] [PubMed] [Google Scholar]
- 34.Marder K, Levy G, Louis ED, Mejia-Santana H, Cote L, Andrews H, et al. Accuracy of family history data on Parkinson disease. Neurology. 2003;61:18–23. doi: 10.1212/01.wnl.0000074784.35961.c0. [DOI] [PubMed] [Google Scholar]
- 35.Tanner CM. PD or not PD? That is the question. Neurology. 2003;61:5–6. doi: 10.1212/wnl.61.1.5. [DOI] [PubMed] [Google Scholar]
- 36.Brandon MC, Lott MT, Nguyen KC, Spolim S, Navathe SB, Baldi P, et al. MITOMAP: a human mitochondrial genome database-2004 update. Nucleic Acids Res. 2005:33. doi: 10.1093/nar/gki079. [Database Issue: D611–13] [DOI] [PMC free article] [PubMed] [Google Scholar]