

Abstract
Many peroxidase inhibitors have been used in horseradish peroxidase (HRP) mediated immunostaining and in situ hybridization to quench background peroxidase activity. However, the efficacy of these inhibitors has been controversial, partially due to the lack of a quantitative study. Tyramide signal amplification (TSA) is much more sensitive than other HRP-mediated methods but its super-sensitivity also demands effective inhibition of background peroxidase activity. In searching for an effective peroxidase inhibitor, we have systematically evaluated the efficacy of several peroxidase inhibitors by quantifying the fluorescence intensity in cultured fibroblasts and tissue sections treated with the inhibitors. For cultured cells, 0.05 mM of phenylhydrazine and 1 unit/ml of glucose oxidase gave only moderate inhibition of HRP activity while 1mM of sodium azide (NaN3), 3% of hydrogen peroxide (H2O2), NaN3/H2O2 combined and 0.02 N hydrochloric acid (HCl) provided more complete inhibition. However, the inhibitory effect of NaN3/H2O2 is reversible upon removal of the inhibitors and followed by incubation and wash to mimic antibody interactions. Similar results were obtained from rat skin wound tissues that have strong endogenous peroxidase activity. Our results recommend the use of HCl and caution the use of phenylhydrazine, glucose oxidase, NaN3 and H2O2 as potent peroxidase inhibitors.
Keywords: phenylhydrazine, glucose oxidase, hydrochloric acid, hydrogen peroxide, azide
Introduction
Horseradish peroxidase (HRP) mediated detection methods have been widely used in cytochemistry and histochemistry to detect proteins and polynucleotides in situ. It is necessary to quench the endogenous peroxidase in the sample for clean background staining. For decades, a variety of HRP inhibitors have been used to quench endogenous peroxidase activity in cell and tissue samples. However, controversial claims on the efficacy of these inhibitors have been reported. For example, phenylhydrazine was recommended for use to inhibit HRP (Straus 1972). However, it was found that the inhibition of HRP by phenylhydrazine was incomplete (Andrew et al. 1987). Additionally glucose oxidase was found to inhibit endogenous peroxidase activity better than sodium azide (NaN3) and hydrogen peroxide (H2O2) (Andrew et al. 1987). Nevertheless, NaN3 and H2O2 have been the two most widely used peroxidase inhibitors. The cause of this discrepancy remains unknown. One of the possible causes may be because the scoring of inhibition efficacy was mostly done by visual judgment, which is less sensitive and only qualitative at best.
Tyramide signal amplification (TSA) is an HRP-mediated method that was developed in the late 1990s to detect proteins and polynucleotides in situ (Van Gijlswijk et al. 1996; Hopman et al. 1997; van de Corput et al. 1998). TSA is much more sensitive than the conventional HRP-mediated methods and becomes a powerful tool to detect low abundant targets in situ. However, its super-sensitivity also demands that unwanted background peroxidase activity be effectively quenched. In our earlier study of mRNA localization, we used two rounds of TSA sequentially to detect two mRNA species in the same cells. This requires effective quenching of the antibody-conjugated HRP used in the first round of TSA. Previously H2O2 and HCl were used to inhibit exogenous HRP for TSA (Zaidi et al. 2000; Van Gijlswijk et al. 1996; Hopman et al. 1997). However, under our experimental conditions, widely used peroxidase inhibitors NaN3 and H2O2 did not effectively inhibit the HRP activity. In searching for an effective peroxidase inhibitor, we started systematically testing several previously used peroxidase inhibitors using quantitative fluorescence intensity measurement.
Materials and Methods
Reagents
Diethyl-pyrocarbonate (DEPC), E. coli tRNA, heparin, phenylhydrazine, glucose oxidase and hydrogen peroxide were from Sigma (St. Louis, MO). Bovine serum albumin (BSA, protease-and nuclease-free), digoxigenin (DIG)-11-dUTP, sheep anti-fluorescein (peroxidase-conjugated) and RNase inhibitor were from Roche (Indianapolis, IN). Goat anti-mouse IgG (HRP conjugated) was from Molecular Probes (Eugene, OR) and mouse anti-DIG antibody was from Jackson ImmunoResearch Lab (Westgrove, PA). Minimum essential medium Eagle, fetal bovine serum (FBS) and trypsin/EDTA were from Mediatech (Herndon, VA). Pathogen-free fertile chicken eggs were purchased from Charles River SPAFAS (Franklin, CT). TSA reagents were purchased from Perkin Elmer (Boston, MA). Other general chemicals were from Sigma (St. Louis, MO) and Fisher (Pittsburgh, PA).
Cell and tissue preparation
Chicken embryo fibroblasts (CEFs) were isolated from 12-day embryos and cultured as described previously (Liu et al. 2002). These cells were plated onto 0.5% gelatin coated cover glasses for 24 h before fixation for fluorescence in situ hybridization (FISH) and TSA. Rat skin tissue samples were prepared from wounded female adult CD1 rats (~200 g, Charles River Laboratory, Wilmington, MA). After the animals were anesthetized with 4% isofluane, wounds were created by making punch biopsies (4 mm diameter) in the flanks of the rats. Four days after wounding, the animals were euthanized. Wound tissues were harvested and embedded in OCT (Electron Microscopy Sciences, Washington, PA), followed by quick freezing on dry ice. Cryostat sections (10 μm) from the rat tissue were placed on Superfrost Plus slides (Fisher, Pittsburgh, PA) and fixed with 4% paraformaldehyde in PBS (1 mM KH2PO4, 10 mM Na2HPO4, 137 mM NaCl, 2.7 mM KCl, pH7.4). All procedures for animal care and handling were approved by Albany Medical College Institutional Animal Care and Use Committee.
Fluorescence in Situ Hybridization
Cells grown on glass cover slips were fixed with 4% formaldehyde in PBS for 20 min then washed with PBS. Cells were permeabilized with 0.5% Triton X-100 in PBS for 5 min then washed with PBS/5mM MgCl2. The cells were pre-hybridized with Hybridization Buffer (HYB) (50% formamide, 5 x SSC, 50 μg/ml Heparin, 100 μg/ml E. coli tRNA, 0.1% Tween 20, and adjusted to pH 6.5–6.8 with 1 N HCl) for at least an hour at 70 °C in a moisture chamber. Digoxiginin (DIG) labeled RNA probe for beta-actin and fluorescein-labeled RNA probe for actin related protein 2 (Arp2) were prepared as previously described (Mingle et al. 2005). These two probes were used either alone or in combination at concentration of 0.5 ng/μl each for hybridization at 70 °C overnight in a moisture chamber. After hybridization, the cover slips were washed extensively with HYB and PBT (0.1% Tween 20 in PBS) at 70 °C, then blocked at room temperature (RT) with Blocking Buffer (0.5% blocking reagent of Perkin-Elmer, 1% BSA and 4 % FBS in PNT (0.3% Triton X-100 in PBS)). The cover slips were incubated with 1 μg/ml of mouse anti-DIG for o1 h at RT, washed with PNT then incubated with 1–2 μg/ml of HRP conjugated goat anti-mouse for 1 h. After three times of 10 min washes with PNT, the cover slips were treated either with PBS or each of the tested peroxidase inhibitors.
Treatment with peroxidase inhibitors
For cultured cells, after hybridization and antibody bindings, the cover slips were treated for 20 min at RT with either PBS (no treatment control) or tested reagents. The samples were then washed 3x10 min with PBS followed by TSA. To test the reversibility of the inhibition, after removal of the tested reagents, some samples were subjected to two rounds of 1-h incubation in Blocking Buffer and 3x10 min wash with PBS to mimic the normal antibody binding process employed in FISH. TSA was then used to evaluate the HRP activity.
For rat skin tissues, the fixed cryostat sections were permeabilized with 0.5% Triton X-100 in PBS for 10 min, treated with PBS or tested reagents at RT for 20 min, followed by 3x5 min washes with PBS. These tissue section samples were blocked with Blocking Buffer for 30 min then subjected to two rounds of 1-h incubation in Blocking Buffer and 3x10 min wash with PBS to mimic the normal antibody binding process. TSA was then used to evaluate the HRP activity.
Tyramide Signal Amplification and immunofluorescence staining
After the aforementioned removal of tested reagents and/or mock antibody binding process, the samples were incubated with tetramethylrhodamine-tyramide at a 1:100 dilution (TSA-PLUS system, Perkin-Elmer) at RT for 15 min for cell samples to detect the remaining HRP activity or with fluorescein-tyramide at a 1:100 dilution at RT for 30 min for tissue sections to detect the remaining endogenous peroxidase activity. For detection of both beta-actin and Arp2 mRNAs in the same cells, tetramethylrhodamine-tyramide was used for the TSA reaction for beta-actin mRNA. After treatment with PBS, or 1 mM of NaN3 combined with 3% of H2O2 or 0.02 N HCl at RT for 20 min, followed by incubation with sheep anti-fluorescien antibody (HRP-conjugated), the samples were processed for second TSA with fluorescein-tyramide. For immunofluorescence staining, after the first round of TSA for detecting beta-actin mRNA and the subsequent treatment with HRP inhibitors, the samples were blocked with Blocking Buffer for 30 min then incubated with mouse antibodies against beta-actin, eukaryotic translation elongation factor 1A (eEF-1A), or vinculin for 1 h. Cy2-conjugated goat anti-mouse antibodies were used as secondary antibody. All the samples were stained with 10 ng/ml of DAPI for 5 min then washed with PBS before mounted to glass slides with Mounting Medium (25% glycerol, 10% polyvinyl alcohol, 2.2% n-propyl gallate, 2% ethanol and 0.1 M Tris-HCl, pH7.5).
Fluorescence Microscopy and fluorescence pixel intensity quantification
All the cell samples were viewed and imaged with a BX60 Olympus microscope equipped with a cooled CCD camera (SensiCam from Cooke) and Slidebook software (Intelligent Imaging Innovation, Inc. Denver, CO). To ensure equal treatment of all the samples from an experiment, positive controls (no inhibitor treated, with the brightest fluorescence signal) were first tested for optimal exposure time of imaging. Once the exposure time was determined, all the images from an experiment were acquired with the same parameters. To minimize the influence of fluorescence variation within a cover slip, two randomly selected fields were imaged from each of five defined regions of a cover slip (designated as center, north, south, east and west). Each image generally contains from 1 to 5 cells. With the Slidebook program, the cell area was selected (highlighted) on the basis of threshold fluorescence intensity using the Mask function. For some cells which were partially imaged, they were de-selected manually to ensure only the area of entire cells was selected. The fluorescence signal in the selected cells was quantified by the software and the signal was expressed as units of average pixel intensity. For background fluorescence control, a region that was outside the cells in the same image was separately masked and the fluorescence was quantified.
For the tissue samples, they were viewed and imaged with a BX61 Olympus equipped with a cooled CCD camera (SensiCam from Cooke) and IP-Lab software (Scanalytics, Inc., Fairfax, VA). Images were acquired with identical parameters. Four images were taken from the wound bed of each tissue section sample and out-of-focus fluorescence was removed by deconvolution. All the pixels in each image were selected and quantified using the functions of segmentation and quantification. An area outside the tissue sample on the same slide was also imaged and quantified as background fluorescence. The average pixel intensity of wound bed tissue was subtracted by that of the background to produce a net average fluorescence pixel intensity. All the values of net average fluorescence pixel intensity from each experiment were normalized as percentage of that of the corresponding PBS-only control. Student’s t test was used for statistical evaluation.
Co-localization assay
Multiple channel fluorescence images for beta-actin and Arp2 mRNAs were acquired as described above. Positive mRNA signal was separated from the background by using the mask function of Slidebook software with identical threshold for both red (beta-actin mRNA) and green (Arp2 mRNA) channels of all the samples. The number of pixels of positive signal in each channel was calculated. The number of pixels with both positive red and green fluorescence signals were further selected by the mask function then calculated. The co-localized pixel number was expressed as the percentage of positive pixel number of red channel (beta-actin mRNA) that overlaps with the positive signal of the green channel.
Results
In this study several peroxidase inhibitors were tested for their efficacy. These include phenylhydrazine, glucose oxidase, sodium azide (NaN3), hydrogen peroxide (H2O2) and hydrochloric acid (HCl). We first asked how effective each of these inhibitors was in quenching exogenous HRP for TSA mediated multiple mRNA detection. Since beta-actin is a relatively stably expressed housekeeping gene, its transcripts were chosen as detection targets to minimize the difference between the cells. After hybridization for beta-actin mRNA using DIG-labeled RNA probe, exogenous HRP molecules were immobilized through the use of mouse anti-DIG antibody and goat anti-mouse antibody (HRP conjugated). After treatment with peroxidase inhibitors, the HRP activity was detected by TSA. If the HRP activity is inhibited, there will be a diminished fluorescence signal in the samples compared to the buffer-treated positive control. As shown by typical images in Fig. 1 and the quantitative summary in Fig. 2, after 20 min of treatment with 0.05 mM of phenylhydrazine or 10 mM of glucose with 1unit/ml of glucose oxidase, there was only a moderate reduction of fluorescence signal in the cells (about 40% reduction of net fluorescence readout compared to the positive control). Treatment with 1 mM of NaN3 or 3% of H2O2 or 1 mM of NaN3 combined with 3% of H2O2 gave a more significant inhibition of the HRP (about 60% reduction in net fluorescence readout as compared to control). Treatment with 0.02 N of HCl gave the most potent inhibition of HRP among the tested inhibitors (about 80% reduction in net fluorescence readout).
Fig. 1. Inhibition of conjugated HRP in cultured cells by peroxidase inhibitors.

Fixed fibroblasts with equally immobilized exogenous HRP were treated with peroxidase inhibitors at RT for 20 min followed by TSA. Representative cells are shown in: a control treated with PBS; b treated with 0.05 mM of phenylhydrazine; c treated with 10 mM of glucose and 1 unit/ml of glucose oxidase; d treated with 1 mM of NaN3; e treated with 3% of H2O2; f treated with 1 mM of NaN3 combined with 3% of H2O2; and g treated with 0.02 N of HCl. Beta-actin mRNA is shown in red. Dotted lines indicate cell border. Scale bar = 10 μm.
Fig. 2. Quantitative results of exogenous HRP inhibition.
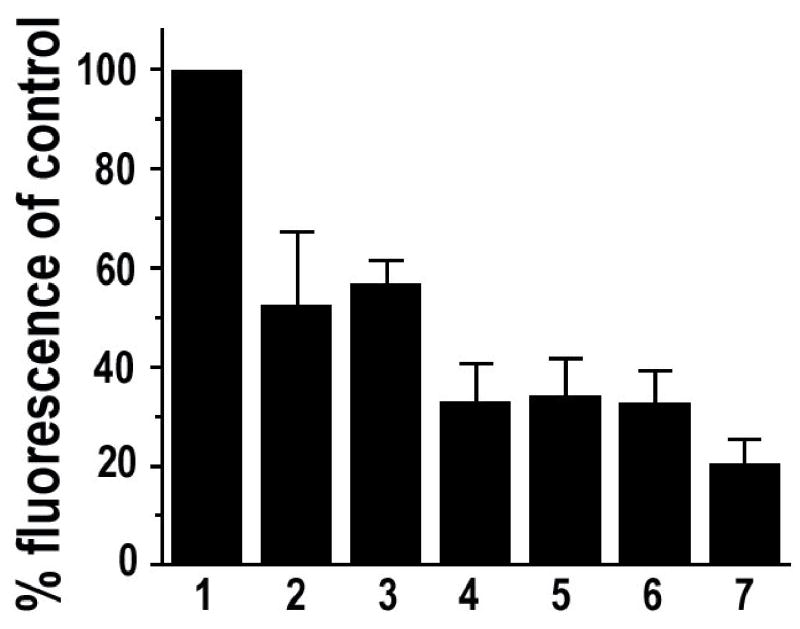
Each column represents normalized data for samples treated with: 1 PBS; 2 0.05 mM of phenylhydrazine; 3 10 mM of glucose and 1 unit/ml of glucose oxidase; 4 1 mM of NaN3; 5 3% of H2O2; 6 1 mM of NaN3 combined with 3% of H2O2; 7 0.02 N of HCl. Compared to the PBS control, all the other treatments resulted in significant inhibition of the HRP (P< 0.01). Error bar = SEM. Data are from at least three independent experiments and represent measurements of 80–190 cells for each treatment group.
Andrew and Jasani (1987) reported that a potent inhibition of peroxidase in tissue samples was observed after treating the samples with 10 mM of glucose and 1 unit/ml of glucose oxidase for 60 min. The moderate effect of this inhibitor under our conditions as shown in Figs. 1 and 2 may be due to the shorter incubation time (20 min). To test this possibility, we also treated the samples for 60 min using this inhibitor. As shown in Fig. 3, treatment of the samples with this inhibitor for 60 min only slightly increased the inhibition of HRP over the treatment for 20 min.
Fig. 3. Comprehensive analyses of HRP inhibitors.
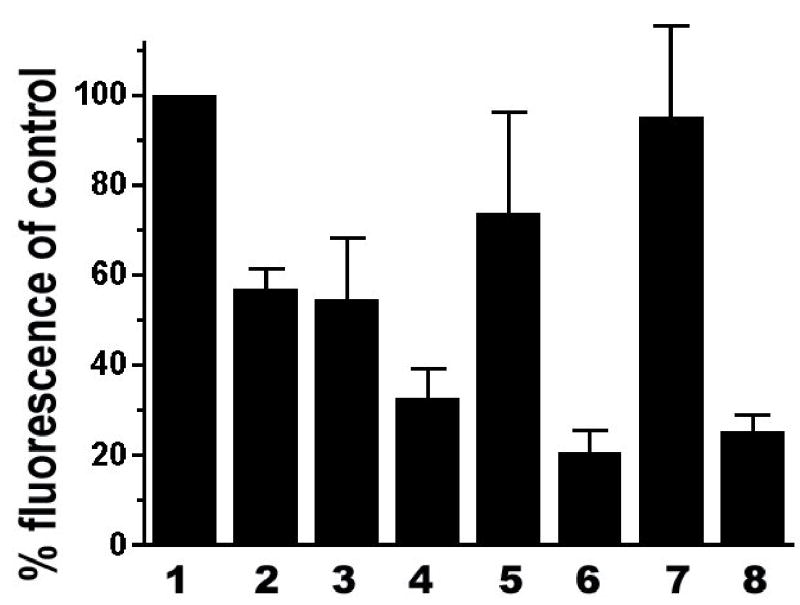
Cell samples were treated with the inhibitors then the effects were quantified the same way as in Fig. 2. 1 PBS control; 2 treated with 10 mM of glucose and 1 unit/ml of glucose oxidase for 20 min; 3 treated with 10 mM of glucose and 1 unit/ml of glucose oxidase for 60 min; 4 treated with 1 mM of NaN3 and 3% of H2O2 for 20 min; 5 treated with 1 mM of NaN3 and 3% of H2O2 for 20 min then washed/incubated in the absence of the inhibitor for 3 hours. There is a significant difference in the fluorescence signal between 4 and 5 (P < 0.05); 6 Treated with 0.02 N of HCl for 20 min; 7 re-fixation with 4% formaldehyde then treated with PBS for 20 min; 8 re-fixation with 4% of formaldehyde then treated with 0.02 N of HCl for 20 min. Error bar = SEM. Data are from at least three independent experiments and represent measurements of 80–190 cells for each treatment group.
The above data indicate that the combination of 1 mM of NaN3 and 3% of H2O2 gave a significant inhibition of the HRP. These inhibitors are commonly used at the early steps of histochemical and cytochemical processes (mostly after fixation or permeabilization but before in situ hybridization and antibody incubation). Given the discrepancy about the efficacy of these two inhibitors, we asked whether their inhibitory effect could be reversed after a process mimicking antibody incubation and wash upon removal of the inhibitors. After treatment with the NaN3 and H2O2 for 20 min, the cell samples were washed then divided into two sets. One set was processed immediately for fluorescence signal development and the other set was incubated and washed for a total of 3 h to mimic the process of first and secondary antibody binding before fluorescence signal development. The incubation and wash process led to partial but significant recovery of the HRP activity as indicated by the increased fluorescence signal in the cells (comparing columns 4 and 5 in Fig. 3).
As shown in Figs. 1 and 2, treatment with 0.02 N of HCl led to about 80% reduction of fluorescence signal in the cells compared to the control, suggesting that HCl is a potent inhibitor of HRP. However, because the binding of antibody to antigen can be weakened significantly at a low pH, which is a commonly used method to dissociate antibody from antigen (Arvieux et al. 1988), the observed reduction of fluorescence signal could have been a result of loss of antibody-conjugated HRP after HCl treatment. To test how effectively 0.02 N of HCl can inhibit the HRP under our experimental conditions, after binding of HRP-conjugated antibody to the samples, we immobilized the conjugated HRP by re-fixing the sample with 4% formaldehyde, quenched the residual formaldehyde activity in the sample with 0.1 M glycine then treated the sample with either PBS or 0.02 N of HCl for 20 min. As shown in Fig. 3, while immobilization had little effect on the HRP activity, after treatment with 0.02 N of HCl the immobilized HRP was effectively inhibited, suggesting that the diminished fluorescence signal was due to the inhibition of HRP instead of wash-off of the antibody-conjugated HRP.
To test whether HCl is superior than NaN3/H2O2 in quenching of HRP for multiple mRNA detection in situ, we used NaN3/H2O2 and HCl as HRP inhibitors in sequential TSA for beta-actin and Arp2 mRNAs in cultured CEFs. After the first round of TSA for detecting beta-actin mRNA, PBS, 1 mM of NaN3 combined with 3% of H2O2 or 0.02 N HCl was used to treat the samples before the second round of TSA for detecting Arp2 mRNA. As presented in Fig. 4a, samples treated with PBS show significant yellow fluorescence, an indication of overlapping red and green fluorescence. Very few distinct red fluorescence spots were observed. Samples treated with NaN3/H2O2 showed less yellow and more distinct red and green fluorescence (Fig. 4b), suggesting significant quenching of the HRP. The samples treated with HCl showed the least yellow fluorescence and the most distinct red and green fluorescence (Fig. 4c). However, since visual evaluation tends to be insensitive and subjective, we quantified the HRP-quenching effect of NaN3/H2O2 versus HCl using a co-localization assay. As summarized in Fig. 5, although the percentage of the red pixels (beta-actin mRNA) that are overlapped with green pixels in samples treated with NaN3/H2O2 is significantly lower (37% ± 6.4) than that in samples treated with PBS control (88.3% ± 2.3), this number is still significantly higher than that in samples treated with HCl (11.4% ± 2.4), suggesting an incomplete inhibition of the HRP from the first round of TSA by NaN3/H2O2. To further evaluate how much of the observed overlapping green fluorescence was from the un-quenched HRP of the first round of TSA, other sets of samples were designed to detect only beta-actin mRNA but were processed as if for both beta-actin and Arp2 mRNAs using sequential TSA. In this setting, the green fluorescence can only be derived from the residual HRP of the first round of TSA. As illustrated in Figs. 4e-g and 5, about 22% ( ± 4) of the co-localization observed in samples treated with NaN3/H2O2 was due to the incomplete quenching of the HRP. In contrast to NaN3/H2O2, HCl quenched the HRP very effectively (close to zero co-localization, Fig. 5, column 6).
Fig. 4. Cells treated with HCl show less overlap of red fluorescence (beta-actin mRNA) with green fluorescence than those treated with NaN3/H2O2.
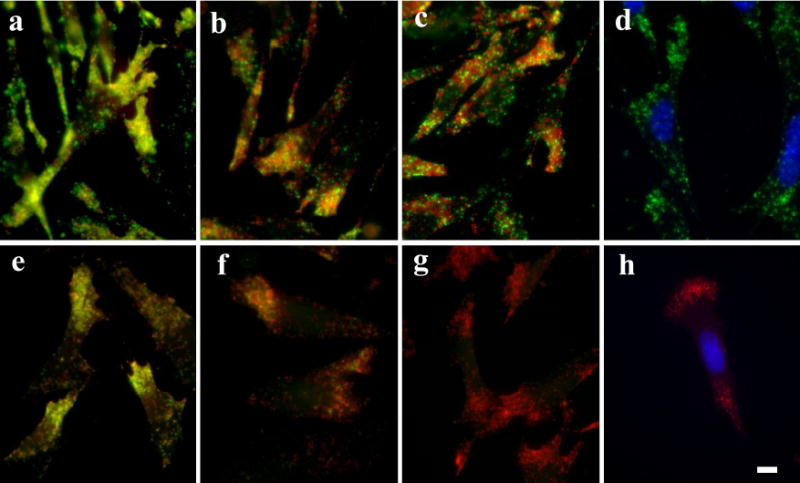
ac Merged images of cells detected for both beta-actin mRNA (red) and Arp2 mRNA (green). e-g Merged images of cells detected for only beta-actin mRNA (red) but processed for second TSA (green) to detect the residual HRP activity of the first TSA. Between the first and second TSA, samples were treated with PBS (a, e), 1 mM NaN3 combined with 3% H2O2 (b, f), or 0.02 N HCl (c, g). Images d and h are results of separately processed FISH-TSA for Arp2 mRNA (d) or beta-actin mRNA (h) without any HRP quenching treatment. Compared to d and h, there was no deleterious effect on the fluorecein probe and the immobilized tetramethylehodamine-tyramide in HCl treated samples (c, g). Blue: DAPI staining for nucleus. Scale bar = 10 μm.
Fig. 5. Incomplete quench of HRP by NaN3/H2O2 contributes to higher co-localization index of beta-actin mRNA with Arp2 mRNA.
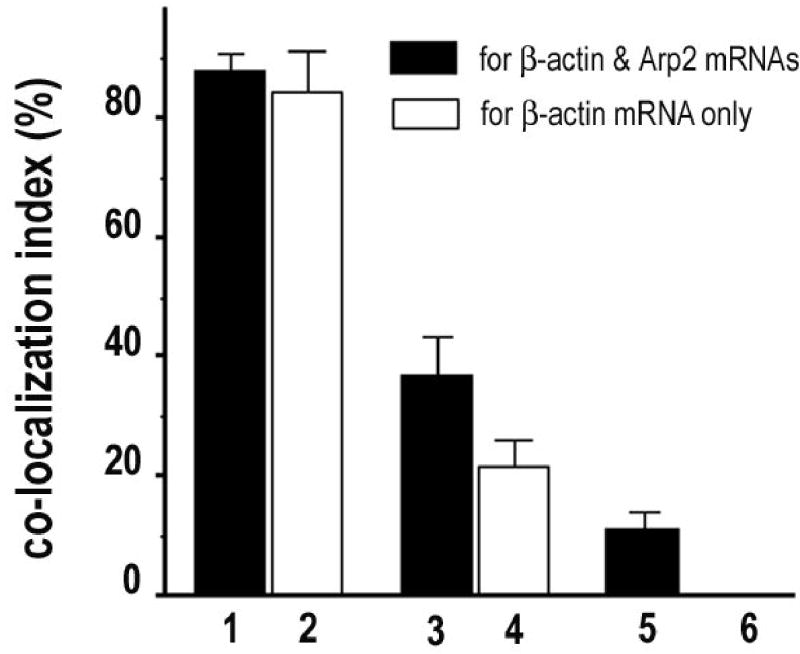
Results of co-localization assay. 1, 3, and 5 (black columns) are samples for both beta-actin and Arp2 mRNAs. 2, 4 and 6 (white columns) are samples for only beta-actin mRNA but processed with the second TSA for residual HRP activity of the first TSA. Between the two TSA, cell samples were treated with PBS (1–2); NaN3/H2O2 (3–4), or HCl (5–6). Significant difference between each treatment group (P < 0.01). Error bar = SEM.
To evaluate the efficacy of the inhibitors in quenching endogenous peroxidase, we extended our tests to tissue samples. Skin wounds at early healing stages contain many inflammatory cells such as neutrophils and macrophages that have relatively strong endogenous peroxidase activity (Martin et al. 2005). We chose day-four rat skin wound tissue for its relatively better structural integrity than day-two wound tissue and stronger endogenous peroxidase activity than day-seven wound tissue. Fixed cryostat tissue sections were treated with the tested peroxidase inhibitors. After removal of the inhibitors, incubations and washes were followed to mimic antibody binding and washing. We did not test phenylhydrazine and glucose oxidase because of their weak inhibitory results in the cell samples. As shown in Figs. 6 and 7, azide fared poorly on the tissue samples with only 11.2% ( ± 20.8) of reduction of fluorescence compared to PBS control. H2O2 fared better than azide on tissue sample (50.1%, ± 15.1 reduction of fluorescence) while HCl fared the best with 67.6% ( ± 5.2) of reduction of fluorescence.
Fig. 6. Quenching endogenous peroxidase in rat skin wound tissue.

Representative fluorescence images of the rat skin wound tissue treated with peroxidase inhibitors. The samples were treated with a PBS (control); b 3% H2O2; c 1 mM NaN3; and d 0.02 N HCl. Green fluorescence indicates endogenous peroxidase activity. Blue fluorescence indicates DAPI staining of cell nucleuses.
Fig. 7. Quantitative analysis of quenching endogenous peroxidase in rat skin wound tissue.

Percentage of net fluorescence intensity in rat skin wound tissue samples treated with: 1 PBS control; 2 3% H2O2; 3 1 mM NaN3 and 4 0.02 N HCl. Error bar = SEM. Data are from three independent experiments and represent measurements of 12 fields of wound bed for each treatment group.
One concern about the use of HCl as an HRP inhibitor is whether it will damage the antigenicity of targets. To address this issue systematically, we evaluated the protein signals for beta-actin, eEF-1A and vinculin in cell samples after detection of beta-actin mRNA and treatment with or without HCl before immunostaining. Visually, there is no significant difference in morphology and protein fluorescence signal between PBS and HCl treated samples (Fig. 8). The conclusion about the intact antigenicity after the HCl treatment is confirmed by quantitative data (Fig. 9): there is no significant difference in protein fluorescence intensity between the samples treated with PBS or 0.02 N HCl.
Fig. 8. No detectable deleterious effects on cell morphology and antigenicity in CEFs treated with HCl.

After detection of beta-actin mRNA (red), the cells were treated with either PBS (a-c) or 0.02 N HCl for 20 min before processed for immunostaining (green) for beta-actin (a, d), eEF-1A (b, e) or vinculin (c, f). Scale bar = 10 μm.
Fig. 9. No detectable deleterious effect on antigenicity in CEFs treated with HCl.

Protein fluorescence intensity of HCl treated cells was quantified and normalized to that of corresponding PBS treated cells. 1 beta-actin, 2 eEF-1A, and 3 vinculin. n = 10. Error bar = SEM.
Discussion
Unlike previous qualitative tests that relied on visual estimation of the effects, in this study we have quantitatively evaluated several peroxidase inhibitors in cell and tissue samples in the context of TSA-mediated detection. In cultured cells with equally immobilized exogenous HRP, we quantified the fluorescence intensity after peroxidase inhibitor treatment, which is very sensitive and more objective. To the best of our best knowledge, this is the first quantitative study of these inhibitors in the context of cytochemistry and histochemistry. Our results indicate that phenylhydrazine and glucose oxidase are poor inhibitors of HRP. NaN3 or H2O2 alone or in combination can produce reasonable inhibition but their effect can be reversed, at least partially, upon removal of the inhibitors and 3 h of incubations and washes to mimic antibody binding. In contrast to the above inhibitors, HCl provided the most potent inhibitory effect which appeared irreversible.
The mechanisms for the observed reversible effects of phenylhydrazine and azide in inhibiting peroxidase can be at least partially explained by earlier biochemical studies. Experimental data derived from using purified enzyme in solution revealed that complete inhibition of HRP by phenylhydrazine was blocked by the accumulation of an unknown product (Ator et al. 1987). The inhibitory effect of azide on HRP is based on the production of azidyl from the interaction of azide and HRP. The azidyl modifies a methionine residue near the heme of HRP (Brill et al. 1967), or the heme group itself (Ortiz de Montellano et al. 1988). However, the complete modification of HRP by azide, like that by phenylhydrazine (Ator et al. 1987), was blocked by the accumulation of an unidentified product that could be removed by gel filtration (Ortiz de Montellano et al. 1988). This may at least partially explain why the inhibitory effects of these inhibitors are reversible. The mechanism of the partial reversal for H2O2 mediated inhibition of peroxidase is less clear. Nevertheless, inhibition of peroxidase by H2O2 was also partially reversible under our experimental conditions.
In contrast to the apparent reversible inhibition of peroxidase by azide and H2O2, HCl appeared to be an irreversible inhibitor of peroxidase. It was known that acid treatment could cleave off heme proteins (Lewis 1954). At pH 2.4 in HCl, HRP can be effectively split into heme and apoprotein, and such acid treatment was used to prepare apoprotein of HRP for biochemical studies (Modesto et al. 1971; Mauk et al. 1974). The loss of the small molecule heme is probably the cause of irreversible inhibition of peroxidase by HCl. Our evaluation also indicates that treatment of samples with 0.02 N HCl did not cause any noticeable deleterious effect to cell morphology and antigenicity, making HCl superior to other inhibitors in quenching peroxidases for clean background and multiple mRNA/protein detection. The importance of irreversible and complete inhibition of HRP is also clearly demonstrated by our co-localization assay. Overlapping fluorescence has been used as a criterion for co-localization. From our samples treated with NaN3 and H2O2 (Figs. 4 and 5), one may conclude that beta-actin mRNA and Arp2 mRNA co-localize due to the relatively significant co-localization index (37 ± 6.4%). This could be misleading since the index was significantly lower if the HRP was effectively quenched by HCl. The 11.4% ( ± 2.4) of co-localization of beta-actin mRNA with Arp2 mRNA in HCl treated samples may come from random distribution of these two mRNAs at the same spots. The effective quenching of HRP with HCl leads to the likely conclusion that although both beta-actin and Arp2 mRNAs are relatively concentrated at the lamellae of some CEFs, these two messages are not precisely co-localized.
Despite their incomplete inhibition of peroxidase, NaN3 and H2O2 are currently the two most used inhibitors for endogenous peroxidase in cytochemistry and histochemistry. We checked 100 research articles from Histochemistry and Cell Biology, published between Volume 121 (issue 2) 2004 and volume 123 (issue 1) 2005. Of the 100 articles, 52 articles used an HRP-mediated detection method. Twelve of the 52 articles did not indicate whether an inhibitor was used to quench endogenous peroxidase. In 40 of the 52 articles, azide and H2O2, either alone or in combination, were used as the inhibitors to quench endogenous peroxidase. No phenylhydrazine, glucose oxidase or HCl was used. The popular use of azide and H2O2 may be due to that (1) most of the biochemical studies of the inhibitors decades ago were not done in the context of cytochemistry and histochemistry so most of the researchers in this field may not be fully aware of these results; (2) less sensitive methods were used to detect abundant targets therefore endogenous peroxidase was not a big problem; and (3) lack of systematical quantitative evaluation of the inhibitors. With the increasingly popular use of TSA as a sensitive method to detect targets in situ and the increasing detection capability of digital cameras for imaging, effective quenching of peroxidase becomes more important than ever. We believe that our studies address this issue timely and provide a useful guide for the selection of peroxidase inhibitors. Our results recommend the use of HCl and caution the use of phenylhydrazine, glucose oxidase, NaN3 and H2O2, as potent peroxidase inhibitors for cytochemistry and histochemistry.
Acknowledgments
Dr Livingston Van De Water and Ms. Jessica Germond for help in tissue preparation. This research was supported by NIH grants R01-GM70560 to GL, R25-GM062460 to SA and T32-HL07194 to LAM.
References
- Andrew SM, Jasani B. An improved method for the inhibition of endogenous peroxidase non-deleterious to lymphocyte surface markers. Application to immunoperoxidase studies on eosinophil-rich tissue preparations. Histochem J. 1987;19:426–430. doi: 10.1007/BF01675753. [DOI] [PubMed] [Google Scholar]
- Arvieux J, Williams AF. Immunoaffinity chromatography. In: Catty D, editor. Antibodies: a practical approach. I. IRL Press; Oxford: 1988. pp. 113–136. [Google Scholar]
- Ator MA, Ortiz de Montellano PR. Protein control of prosthetic heme reactivity. Reaction of substrates with the heme edge of horseradish peroxidase. J Biol Chem. 1987;262:1542–1551. [PubMed] [Google Scholar]
- Brill AS, Weinryb I. Reactions of horseradish peroxidase with azide. Evidence for a methionine residue at the active site. Biochemistry. 1967;6:3528–3535. doi: 10.1021/bi00863a026. [DOI] [PubMed] [Google Scholar]
- Hopman AH, Claessen S, Speel EJ. Multi-colour brightfield in situ hybridisation on tissue sections. Histochem Cell Biol. 1997;108:291–298. doi: 10.1007/s004180050168. [DOI] [PubMed] [Google Scholar]
- Lewis UJ. Acid cleavage of heme proteins. J Biol Chem. 1954;206:109–120. [PubMed] [Google Scholar]
- Liu G, Grant WM, Persky D, Latham VM, Jr, Singer RH, Condeelis J. Interactions of elongation factor 1 alpha with F-actin and beta-actin mRNA: implications for anchoring mRNA in cell protrusions. Mol Biol Cell. 2002;13:579–592. doi: 10.1091/mbc.01-03-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P, Leibovich SJ. Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol. 2005;15:599–607. doi: 10.1016/j.tcb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Mauk MR, Girotti AW. The protoporphyrin-apoperoxidase complex. Photooxidation studies. Biochemistry. 1974;13:1757–1763. doi: 10.1021/bi00705a031. [DOI] [PubMed] [Google Scholar]
- Mingle LA, Okuhama NN, Shi J, Singer RH, Condeelis J, Liu G. Localization of all seven messenger RNAs for the actin-polymerization nucleator Arp2/3 complex in the protrusions of fibroblasts. J Cell Sci. 2005;118:2425–2433. doi: 10.1242/jcs.02371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modesto RR, Pesce AJ. The reaction of 4,4'-difluoro-3,3'-dinitro-diphenyl sulfone with gamma-globulin and horseradish peroxidase. Biochim Biophys Acta. 1971;229:384–395. doi: 10.1016/0005-2795(71)90197-8. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano PR, David SK, Ator MA, Tew D. Mechanism-based inactivation of horseradish peroxidase by sodium azide. Formation of meso-azidoprotoporphyrin IX. Biochemistry. 1988;27:5470–5476. doi: 10.1021/bi00415a013. [DOI] [PubMed] [Google Scholar]
- Straus W. Phenylhydrazine as inhibitor of horseradish peroxidase for use in immunoperoxidase procedures. Journal of Histochem Cytochem. 1972;20:949–951. doi: 10.1177/20.11.949. [DOI] [PubMed] [Google Scholar]
- van de Corput MP, Dirks RW, van Gijlswijk RP, van Binnendijk E, Hattinger CM, de Paus RA, Landegent JE, Raap AK. Sensitive mRNA detection by fluorescence in situ hybridization using horseradish peroxidase-labeled oligodeoxynucleotides and tyramide signal amplification. Journal of Histochem Cytochem. 1998;46:1249–1259. doi: 10.1177/002215549804601105. [DOI] [PubMed] [Google Scholar]
- Van Gijlswijk RPM, Viegant R, Larsan R, Raap AK. Horseradish peroxidase-labeled oligonucleotides and fluorescence tyramide for rapid detection of chromosome-specific repeat sequences. Cytogenet Cell Genet. 1996;75:258–262. doi: 10.1159/000134496. [DOI] [PubMed] [Google Scholar]
- Zaidi AU, Enomoto H, Milbrandt J, Roth KA. Dual fluorescent in situ hybridization and immunohistochemical detection with tyramide signal amplification. J Histochem Cytochem. 2000;48:1369–1375. doi: 10.1177/002215540004801007. [DOI] [PubMed] [Google Scholar]