

Abstract
Every 23 seconds a person sustains a traumatic brain injury in the United States leaving many patients with substantial cognitive impairment and epilepsy. Injury-induced alterations in the hippocampus underpin many of these disturbances of neurological function. Abnormalities in the dentate gyrus are likely to play a major role in the observed pathophysiology because this subregion functions as a filter impeding excessive or aberrant activity from propagating further into the circuit and following experimental brain injury, the dentate gyrus becomes more excitable. Although alteration in excitation or inhibition could mediate this effect in the dentate gyrus, we show a key role played by an impairment of GABAAergic inhibition. The efficacy of GABAA-mediated inhibition depends on a low [Cl−]i that is maintained by neuronal K-Cl co-transporter 2 (KCC2). Using fluid percussion injury (FPI) in the mouse, we demonstrate significant reductions in KCC2 protein and mRNA expression in the dentate gyrus that causes a depolarizing shift in GABAA reversal potential, due to impaired chloride clearance, resulting in reduced inhibitory efficiency. This study elucidates a novel mechanism underlying diminished dentate gyrus inhibitory efficacy and provides an innovative target for the development of potential therapeutics to restore the severe pathological consequences of traumatic brain injury.
Introduction
Traumatic brain injury is the leading cause of death in children and young adults in the United States and often results in neurological dysfunction i.e., cognitive impairment and epilepsy (BIAUSA, 2004). Disturbance in hippocampal function (Scoville and Milner, 1957; Zola-Morgan et al., 1986) plays a leading role in these pathologies since this structure is implicated in higher cognitive function (Zola-Morgan et al., 1986; Cave and Squire, 1991; Rempel-Clower et al., 1996) and is a frequent focus of seizure generation (Annegers et al., 1998; Asikainen et al., 1999). Despite the importance of the hippocampus, little is known about injury-induced regional alterations in neuronal circuitry underlying hippocampal dysfunction. Under normal conditions, robust GABAAergic synaptic inhibition in the dentate gyrus is thought to endow this hippocampal sub region with the ability to function as a low pass filter, impeding excessive or aberrant activity from propagating into the more seizure prone hippocampus (Heinemann et al., 1992; Sloviter, 1994; Cohen et al., 2003). Following fluid percussion injury (FPI), the dentate gyrus becomes more excitable (Lowenstein et al., 1992), which is likely due to a breakdown in GABAAergic inhibition (Reeves et al., 1997; Toth et al., 1997; Witgen et al., 2005). As a result, a decrease in dentate gyrus GABAAergic efficacy may contribute to both cognitive deficits and epilepsy. The efficacy of GABAA-mediated postsynaptic inhibition depends on the maintenance of low intracellular chloride [Cl−]i. Dentate gyrus neurons regulate intracellular [Cl−]i via a neuronal K-Cl co-transporter (KCC2) driven by the transmembrane potassium gradient for ionic transport (Kaila, 1994; Payne, 1997; Rivera et al., 1999). When Cl− extrusion is disrupted due to decreased KCC2 expression, [Cl−]i increases inside the neuron, decreasing the driving force for GABAA-mediated inhibitory currents resulting in significant disinhibition (Nabekura et al., 2002; Jin et al., 2005; Zhu et al., 2005).
Little is known about the mechanisms underlying diminished GABAAergic inhibition in dentate gyrus following TBI. Here we investigated whether KCC2 expression was altered following FPI in mice. A reduction in KCC2 expression would diminish Cl− extrusion, thereby impairing cellular ability to maintain low intracellular Cl− concentration following traumatic brain injury, resulting in decreased inhibitory synaptic efficacy.
Methods
Generation of TBI Animals
Experiments were performed on 5 to 7 week old, 20 to 25 gram C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME). All experimental procedures and protocols for animal studies were approved by the Children’s Hospital of Philadelphia Institution for Animal Care and Use Committee in accordance with international guidelines on the ethical use of animals. Experiments were designed to minimize the number of animals required. Furthermore, those used were cared for, handled and medicated to minimize suffering (National Research Council, National Academy Press, Washington, DC, 1996).
FPI was conducted as previously described in Witgen et. al., (Witgen et al., 2005). Briefly, on day one, a craniectomy was performed over the right parietal and subsequently visually inspected to ensure that no dural breeching had occurred. On day two, a 20 ms pulse (ranging between 1.4 to 2.1 atm) of saline was delivered to the brain resulting in a mild to moderate concussive injury. Sham animals underwent all FPI procedures with the single exception of delivery of the fluid pulse. All experiments were undertaken in animals that were allowed to recover for 7 days after sham surgery or FPI.
Sub regional dissection of Hippocampus
Sagital hippocampal sections were oriented on the dissecting surface such that the pyramidal cell layer, hippocampal fissure, and supra- and infra-granular blades of the dentate gyrus were clearly visible. CA1 and dentate gyrus were then micro dissected and subsequently placed into lysis buffer containing protease inhibitors (Roche, Indianapolis, IN). Because the amount of material from the micro-dissected hippocampus was < 50μg, we collected micro-dissected tissue from 5 animals and homogenized them to generate one sample for analysis. This process was repeated 2 more times thereby employing a total of 15 animals per group (FPI and sham, respectively) to have 3 samples for subsequent quantitative analysis. Regionally dissected dentate gyrus and CA1 were pooled from five sham and FPI animals respectively. The pooled tissue from five animals (sham or injured) was considered one sample. Thus, for statistical comparison n = 3 represents 3 independent pooled samples obtained from 15 animals (i.e., 15 injured and 15 sham respectively) per group.
Western blotting
Fifty μg of protein were run on a 10% gel using standard protocols. Samples were immunoblotted with a 1:1000 rabbit polyclonal antibody against KCC2 (Upstate, Lake Placid, NY), NKCC1 (Chemicon, Temecula, CA), VGAT, GAT1, EAAC1 (Alpha Diagnostics International San Antonio, TX) or, Beta-actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA) detected by chemiluminescence (Pierce, Rockford, IL) and analyzed using UN-SCAN-IT gel-scanning software (Silk Scientific, Orem, UT).
Immunostaining
Tissue preparation
Brain tissue was prepared by perfusion of the animals with 4% PFA in PBS. Brains were removed and postfixed in 4% PFA overnight. The following day, coronal sections (50 μm thickness) were cut on a Leica vibratome. Sections were collected and stored in cryoprotectant at 4°C until used. Prior to beginning the staining protocol, sections to be used were washed 4x15 min in 0.1M TRIS pH 7.6 to remove any trace cryoprotectant.
Staining protocol
Sections were treated in the following succession: 3% triton-X for 30 minutes; wash 3x15 min PBS; 3% hydrogen peroxide/30% methanol in PBS 1 hour; wash 3x15 min PBS; block for 60 minutes in 10% serum/PBS; apply the primary antibody (rabbit anti KCC2, 1μg/ml, Upstate, Lake Placid, NY or rabbit anti NKCC1, 3μg/ml, Chemicon, Temecula, CA) overnight at 4°C. The following morning, sections were washed 3x15 min in PBS, and the secondary antibody (FITC conjugated fluorescent) applied for 60 minutes. Sections were washed 3x10 min in PBS and coverslips subsequently mounted.
In additional sections, primary antibody was omitted as a negative control to assess nonspecific binding. Immuno labeling was analyzed using a Leitz Diaplan microscope equipped for epifluorescent illumination. Images were collected using a Leica DC 300 FX digital camera.
Real time RT-PCR
Microdissection was done in an identical fashion as described above, except tissue was dissected in ice cold RNAlater (Qiagen, Valencia, CA). RNA was isolated using an RNeasy Lipid tissue isolation kit (Qiagen). Approximately 50 ηg of RNA template, per reaction, was used to run a one step RT-PCR with Applied Biosystems Assays on Demand (GenBank Accession # NM_020333). All reactions were run on the ABI PRISM 7500 sequence detection system (Applied Biosystems, Foster city, CA). All samples were run in triplicate including standard curves, NTC (no template control), naïve, and normalized to the beta-actin endogenous reference gene. All normalized samples were then calibrated to naïve values.
Electrophysiology
Brain slices were prepared as previously reported (Witgen et al., 2005). Perforated patch voltage and current clamp recordings were conducted at room temperature from visually identified dentate gyrus neurons using infrared differential interference contrast video microscopy (Stuart et al., 1993). Cells were voltage clamped at −60 mV, and signals were recorded and amplified with Axopatch 200 (Axon instruments, Foster city, CA), filtered at 2 kHz, digitized at 5 kHz, and stored on a PC microcomputer for off line analysis. Electrodes were pulled to a resistance between 6 and 8 M when filled with an internal solution composed of (in mM): 100 KCl, 10 Hepes, pH 7.4 (KOH), on a two stage puller (P-30, Sutter instruments. Novato, CA). Gramicidin (Sigma, St. Louis) was dissolved in dimethylsulfoxide (DMSO, Sigma) (1–2mg/ml) then diluted in the pipette filling solution (1:1000). Progress of membrane patch perforation was evaluated by following the decrease in access resistance and recordings were begun once the access resistance had stabilized (17 – 45 m ). Current-voltage relationships were generated using a protocol that initially stepped the membrane voltage from a holding potential of –60 mV to −110mV for 100 msec and then ramped the voltage from −110 to + 40 mV at a rate of 107 mV/sec in the absence and presence of agonist (GABA, 100 μM, applied 125 ms, 40–60 PSI using a Picospritzer II, General Valve Corporation). For low K+ experiments, KCl concentration was reduced from 3 to 1 mM while the concentration of NaCl was increased to maintain tonicity.
Chloride fluorescence
The fluorescent chloride indicator 6-methoxy-N-ethylquinolinium iodide (MEQ, 100 μM) was included in the recording whole cell pipette in order to load DG neurons in slices derived from injured and sham animals. The internal whole cell solution for these experiments was composed of (in mM) Kgluconate, 145; MgCl2, 2; BAPTA, 0.1; KCl, 2.5; NaCl, 2.5; HEPES, 10; GTP–Tris, 0.5; Mg-ATP, 2; pH 7.4 (KOH). MEQ was excited by single wavelength illumination from an argon lamp band pass (340 – 480 nm) filtered and emitted fluorescence images (filtered at 435 – 485 nm) were captured with a CCD camera (Hamamatsu C4742–95, Orca, Japan), sampled at 1Hz and stored on a PC microcomputer. Images were subsequently analyzed offline with Image Pro software (Media Cybernetics, Silver Spring, MD). For the analysis of fluorescence changes in each region of interest (i.e., patched neuron), the index, - F/F, was used to estimate relative change of [Cl−]I. Background fluorescence (F) was the averaged fluorescent intensity obtained for two seconds before the stimulation, and F was the change from the value of F to fluorescent intensity excited at a given time. Absolute Cl− concentration was not determined in the present study.
Reagents and statistical tests
Reagents were purchased from the following vendors: all salts, Gramicidin, tetrodotoxin (TTX) and GABA from Sigma (St. Louis, MO). CGP 55845 from Tocris (Ellisville, MO) and MEQ from Invitrogen (Carlsbad, CA). All drugs were made as stock solutions and then diluted to their final concentration in bathing or recording medium. Two-tailed unpaired Student’s t-tests were performed to determine statistical significance at the p < 0.05 confidence level when comparing different treatment groups. All data are presented as group means ± standard deviation.
Results
KCC2 expression in dentate gyrus following FPI
Initial examination into proteins important in maintaining presynaptic and vesicular GABA content demonstrated no injury-induced alterations (Fig. 1A). Therefore to test whether increased dentate gyrus excitability (Lowenstein et al., 1992; Witgen et al., 2005) in slices from brain injured animals may be due to altered intracellular chloride concentration, KCC2 protein expression was assessed in regionally dissected dentate gyrus and area CA1 using Western blot analysis. Expression of KCC2 was robust in both regions isolated from sham animals (Fig. 1B1 & 2). Interestingly, KCC2 protein expression was unaltered in area CA1 but significantly reduced in the dentate gyrus dissected from animals 7 days following traumatic brain injury (Fig. 1B1 & 2). The quantity of KCC2 protein in the DG of injured animals was only 56% (a decrease in the pixel quantity from 26.4 ± 3.4 to 14.9 ± 6.2) of that observed in the dentate gyrus dissected from sham animals (Fig. 1B2, p < 0.05, n = 3, see methods for details). Injured dentate gyrus might respond to the loss of KCC2 by reverting back to a prior developmental stage by increasing the expression of different chloride transporter NKCC1 (Plotkin et al., 1997) present during early development. However, Western blot analysis revealed virtually no NKCC1 protein expression in regionally dissected dentate gyrus tissue (Fig. 1C, sham = 7%, FPI = 9%, of fetal control tissue) from injured or sham animals.
Figure 1. KCC2 protein and mRNA expression is significantly reduced in dentate gyrus seven days following FPI.
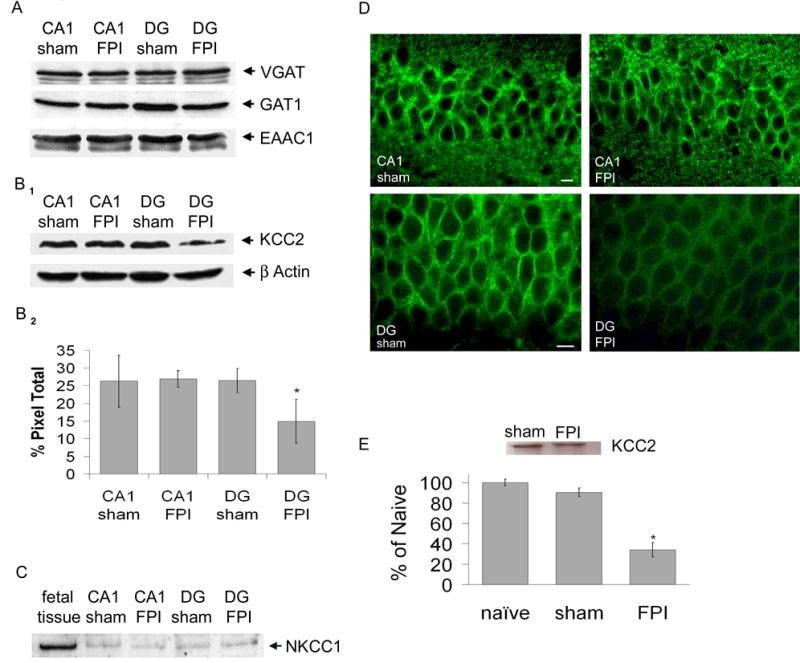
A) Representative Western blot showing no difference in expression of VGAT (55 kDa), GAT1 (67 kDa) and EAAC1 (69 kDa) within isolated CA1 and dentate gyrus (DG) from ipsilateral (injured) hippocampus of both sham and FPI animals (B1) Representative Western blot showing expression of KCC2 (142 kDa) within isolated CA1 and dentate gyrus from ipsilateral (injured) hippocampus of both sham and FPI animals. Control for protein loading is shown for β-actin (45kDa). (B2) Densitometry histogram demonstrating significant reduced expression of KCC2 in the dentate gyrus of injured animals. (C) Representative western blot showing expression of NKCC1 (145kDa) within isolated CA1 and DG from ipsilateral (injured) hippocampus of both sham and FPI animals compared to fetal brain tissue. (D) KCC2 immunohistochemistry in area CA1 (top panel) and dentate gyrus (lower panel) 7 days following FPI. Scale bar is 10 μm. (E) Inset: Representative phosphoimage of total mRNA isolated from DG tissue from FPI and sham mice. Histogram: Real time RT-PCR analysis of KCC2 mRNA expression in dentate gyrus 7 days following FPI compared to sham and naive.
Figure 2. Brain injury induces a depolarized shift in GABA reversal potential in dentate gyrus.

(A) A representative IV curve demonstrating EGABA to be −73 mV recorded in a dentate gyrus neuron from a sham animal. As a control the perforated patch was ruptured, thereby entering into the whole cell configuration and EGABA shifts to approximately 0. Insets are current recordings in response to the displayed voltage ramp protocol in the absence (left) and presence (right) of focally applied agonist (GABA). (B) Representative current-voltage curve shows a significant shift in EGABA in a dentate gyrus neuron recorded in a slice from an FPI animal to −51 mV. When the superfusing aCSF is changed to low K+ (1 mM, 2–3 min) aCSF the curve shifts toward EGABA observed in sham animals. Inset: negative shift in EGABA in 3 DG neurons recorded in slices from injured animals when aCSF is switched to low K+. (C) Histogram of the hyperpolarizing response to focally applied GABA (100μM) recorded in current clamp mode demonstrating a significant decrease in the amplitude of the response recorded in dentate gyrus from injured animals.
In addition to Western blot analysis, we used immunocytochemistry to inquire whether injury also altered the regional distribution of KCC2. Since action potential independent activity arises from synapses close to dentate gyrus somata (Soltesz et al., 1995), KCC2 immunoreactivity was detected as densely packed perisomatic puncta present in the dentate gyrus cell layer (Fig. 1D, lower sham panel). As predicted, less immunoreactivity and intense perisomatic dentate gyrus staining is evident in hippocampal slices derived from injured animals compared to that observed in slices generated from sham animals (Fig. 1D, lower FPI panel). Furthermore, in accordance with the Western blot analysis, KCC2 immunoreactivity was unaltered in area CA1 (Fig 1D upper panels).
To determine whether the reduction in KCC2 protein expression in the dentate gyrus derived from injured animals was due to a reduction in the amount of mRNA, real-time RT PCR was performed. Samples were normalized to the beta-actin endogenous reference gene, expressed as percent of naïve and run in a similar fashion to Western blots (n = 3 i.e., three independent groups of pooled tissue, each from 5 FPI and 5 sham animals from a total of 15 sham and injured animals, respectively). KCC2 dentate gyrus mRNA expression from injured animals was reduced 62.21 ± 7.16% compared to sham values (Fig. 1E, p < 0.05).
Brain injury decreases inhibitory efficacy
The functional consequence of reduced KCC2 expression in injured animals was examined by conducting gramicidin perforated patch clamp recordings in DG neurons in brain slices from sham and FPI animals. In the perforated patch recording mode the antibiotic gramicidin is added to the pipette solution thus forming pores in the plasma membrane at the tip of the electrode enabling the flow of monovalent ions. This recording configuration allows for electrical continuity between the pipette and the neuron without disruption of the intracellular milieu and importantly maintains the intracellular chloride concentration. For all electrophysiological experiments (voltage and current clamp) all neurons were held at −60 mV. Current-voltage relationships were created using a voltage ramp command and subsequently plotted by subtracting currents recorded in control (Fig. 2A & B control insets) artificial cerebrospinal fluid (aCSF) from currents recorded in the presence of focally applied agonist (GABA 100μM, Fig. 2A & B GABA insets). The GABA equilibrium potential (EGABA) is the value where there is no net ionic movement because the electrical gradient (set and controlled by the voltage clamp) is equal and opposite the concentration gradient. Furthermore the equilibrium potential is often referred to as the reversal potential since it is the potential around which the current reverses direction. EGABA values of naïve and sham animals were not significantly different and thus pooled. EGABA values of FPI animals were significantly shifted to more depolarized potentials. Mean EGABA values were −72 ± 11.4 mV (n = 7) in sham slices (Fig. 2A) and −51 ± 10.7 mV (n = 16) in FPI slices (Fig. 2B). To ensure that the recordings were done in an intact perforated patch configuration, following the experiment, the patch was ruptured to enter the whole cell configuration allowing for dialysis and consequent steady-state equilibrium of the intracellular milieu with the ionic contents present in the electrode. Entering into the whole-cell recording configuration caused EGABA to shift towards zero (−2 ± 4 mV, n = 18, Fig. 2A whole cell trace) due to high pipette chloride concentration dialysis with the cell resulting in near symmetrical chloride concentrations in the cell and aCSF. EGABA recorded in injured area CA1 pyramidal neurons was −75 ± 13.7 (n = 4), which was not significantly different from area CA1 values in slices from sham animals and is similar to previously reported control values (Sun et al., 2001). This data is consistent with the Western blot (Fig. 1B) and immunocytochemistry (Fig 1D) that shows no alterations in KCC2 in area CA1 and demonstrates dentate gyrus specificity. Furthermore, in additional experiments conducted in current clamp, all of the dentate gyrus neurons from naïve or sham animals responded to focally applied GABA (100μM) with a robust hyperpolarizing response (−14 ± 0.7 mV, n = 8, Fig. 2C). However, in dentate gyrus neurons recorded in slices from injured animals, focally applied GABA resulted in significantly smaller hyperpolarizing responses (22% of sham values, −3 ± 1.2 mV, n = 17, Fig. 2C) consistent with an increase in intracellular Cl− concentration and the recorded shift in EGABA.
Since the fidelity of the KCC2 transporter relies on the electrochemical gradient for both potassium and chloride, we sought to examine whether altering the driving force of potassium by external ionic substitution would restore a (negative) shift in EGABA recorded in the dentate gyrus from injured animals. After recording EGABA in dentate gyrus neurons in slices from injured animals in normal aCSF ([K+]o = 3mM), the superfusing external was switched to low [K+]o (1mM, 2–3 min) aCSF. The subsequently recorded EGABA was −66 ± 2.3 (n = 6), exhibiting a mean negative shift of −15 mV compared to the value recorded in normal aCSF (Fig. 2B and inset). Furthermore, EGABA recorded in DG neurons from injured animals superfused with low K+ external was significantly more negative compared to values obtained in normal external, (n = 6, −51 ± 4.0 mV, n = 16 p < 0.05). These data suggest that the efficacy of GABA-mediated inhibition can be partially restored by reducing [K+]o.
Altered chloride clearance after injury
To further examine the functional consequences of reduced KCC2 expression in dentate gyrus neurons the fluorescent chloride indicator (MEQ) was loaded into cells via a whole cell patch pipette to examine Cl− clearance. Quantification of chloride clearance was accomplished by measuring the time for GABA-induced (bath application 30μM, 10 sec) fluorescence intensity levels to return to pre-GABA baseline levels. This decay time was significantly longer in dentate gyrus neurons recorded in slices from injured animals compared to those recorded from sham animals (Fig. 3A, B & C; sham = 7.9 ± 6.4sec n = 7; FPI=> 60 sec n = 16, p < 0.05). The rate of rise of the Cl− transient was not significantly different in neurons recorded in slices from the two populations (3 ± 5.8; 2 ± 1.9 sec, n = 8, 16, for slices from sham and FPI animals respectively, p > 0.05).
Figure 3. Intracellular chloride homeostasis is disrupted seven days post FPI.
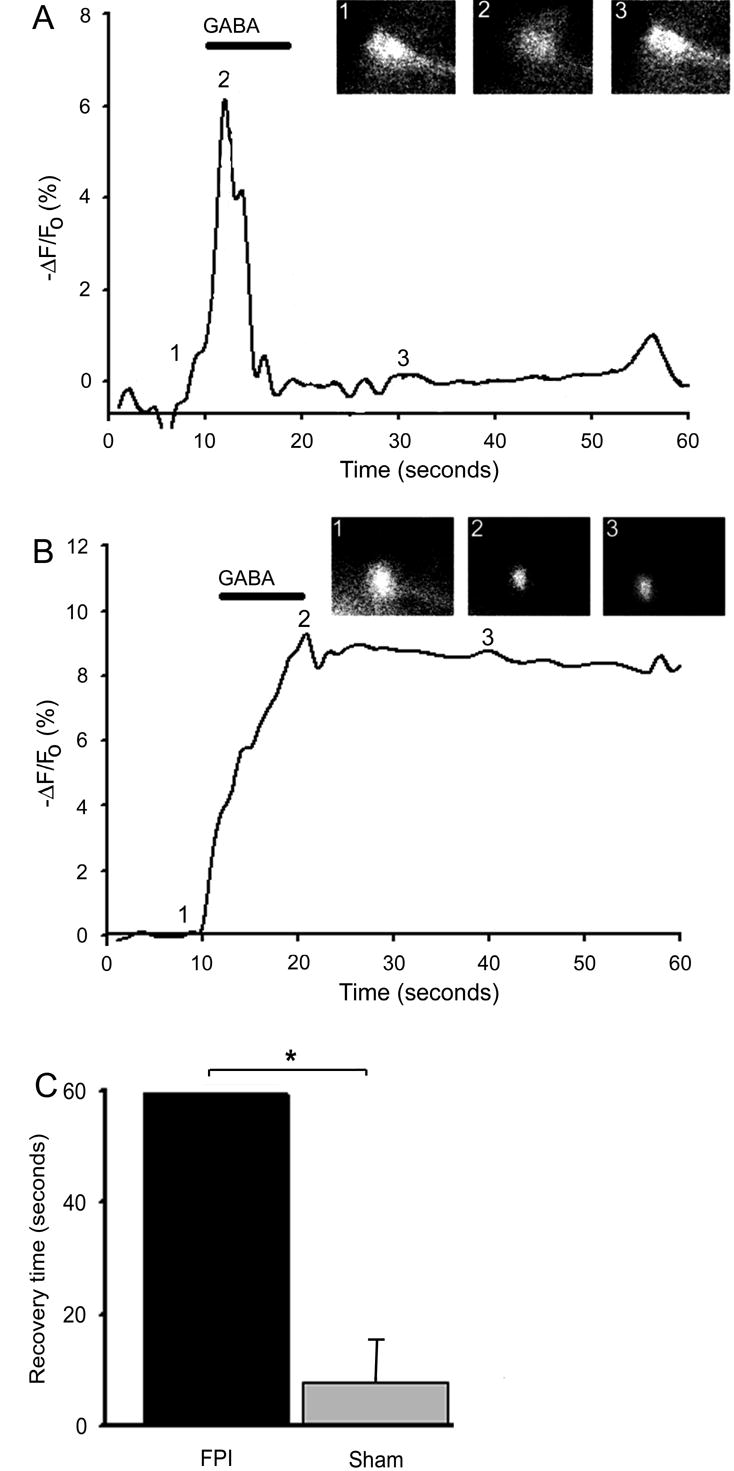
(A) In a dentate granule neuron from a sham animal, GABA (bath application 30μM, 10 sec) caused an influx of chloride, measured by MEQ fluorescence, which was quickly cleared. Inset: MEQ fluorescent image of same dentate gyrus neuron at: 1) baseline 2) immediately after GABA application 3) 20 seconds after GABA application. (B) In a dentate granule neuron from an FPI animal, GABA application caused an influx of chloride, however clearance was significantly protracted to > 60 seconds. inset: image of same dentate granule neuron at: 1) baseline 2) immediately after GABA application 3) 30 seconds after GABA application. (C) Histogram of group data summarizing the time for fluorescence intensity to return to baseline following application of GABA.
Discussion
The principal finding of the present study is that a mild to moderate episode of concussive brain injury leads to a significant reduction in the abundance and function of the primary neuronal chloride transporter KCC2 (Payne, 1997) in dentate gyrus neurons. This decrease (Fig. 1) results in reduced intracellular chloride extrusion (Fig. 3) reducing transmembrane chloride gradient shifting EGABA more positive (Fig. 2) thereby, diminishing the ability of GABA to cause neuronal membrane hyperpolarization.
Using the Nernst equation to estimate the putative change in [Cl−]i due to the injury-induced (44%) reduction in KCC2 protein expression, yields a value of approximately 8mM for [Cl−]i present in dentate gyrus neurons from sham animals, whereas 18mM for [Cl−]i present in dentate gyrus neurons recorded in slices from injured animals. These values correlate well with those reported by Zhu et al (2005) derived from cortical neurons cultured from KCC2 knockout embryos ([Cl−]i = 7 and 25mM for neurons from wild type and KCC2 knockout respectively). Furthermore, these data suggest that a relative minor increase in [Cl−]i (from 8 to 18 mM) results in significant diminished dentate gyrus inhibitory efficacy.
We examined alterations present at one week post-FPI because it is within the clinically relevant “therapeutic time window” (Grady and Lam, 1995). Low intracellular chloride is essential for dynamic GABAA-mediated synaptic inhibitory efficacy (Kaila, 1994; Payne, 1997; Rivera et al., 1999), thus the consequence of reduced KCC2 expression in this hippocampal subregion is twofold. First, robust GABAAergic inhibition is thought to give rise to the gatekeeper function, quelling synchronous or excessive neuronal activity from propagating from the entorhinal cortex into the seizure prone hippocampus (Buzsaki et al., 1983; Heinemann et al., 1992; Lothman et al., 1992; Sloviter, 1994; Cohen et al., 2003). Second, normal dentate gyrus function is thought to be important in anterograde memory formation (Eldridge et al., 2005). Therefore, a significant decrease in inhibitory efficacy would lead to dysfunctional dentate gyrus activity compromising gatekeeper and cognitive fidelity.
Injury-induced reduction in KCC2 expression decreases the transmembrane chloride gradient similar to the condition present during development. However, brain injury does not revert dentate gyrus neurons to a prior developmental stage since expression of the developmental chloride accumulator NKCC1 (Plotkin et al., 1997) was not detected in tissue derived from either sham or injured animals (Fig. 1C).
In conclusion, the dentate gyrus KCC2 expression is significantly reduced seven days following FPI. The significant decrease in KCC2 expression deteriorates cellular ability to maintain the transmembrane chloride gradient essential for robust GABAAergic inhibitory efficacy. Diminished inhibitory efficiency causes dysfunction of the dentate gyrus potentially contributing to anterograde cognitive deficits and seizures following human brain injury. Presently, few if any potential curative targets exist for TBI patients. Our data highlights KCC2 as an attractive candidate site for the development of therapeutic intervention to assuage the serious ramifications of head trauma.
Acknowledgments
We thank Drs: D. Weinreich, D. Henshall, M.B Robinson, R.G. Kalb and P. Haydon for constructive suggestions on a previous draft of the manuscript. This work was supported by NIH/NINDS RO1 NS45975 (A.S.C.).
References
- Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Engl J Med. 1998;338:20–24. doi: 10.1056/NEJM199801013380104. [DOI] [PubMed] [Google Scholar]
- Asikainen I, Kaste M, Sarna S. Early and late posttraumatic seizures in traumatic brain injury rehabilitation patients: brain injury factors causing late seizures and influence of seizures on long-term outcome. Epilepsia. 1999;40:584–589. doi: 10.1111/j.1528-1157.1999.tb05560.x. [DOI] [PubMed] [Google Scholar]
- BIAUSA. TBI Statistics. 2004. [Google Scholar]
- Buzsaki G, Leung LW, Vanderwolf CH. Cellular bases of hippocampal EEG in the behaving rat. Brain Res. 1983;287:139–171. doi: 10.1016/0165-0173(83)90037-1. [DOI] [PubMed] [Google Scholar]
- Cave CB, Squire LR. Equivalent impairment of spatial and nonspatial memory following damage to the human hippocampus. Hippocampus. 1991;1:329–340. doi: 10.1002/hipo.450010323. [DOI] [PubMed] [Google Scholar]
- Cohen AS, Lin DD, Quirk GL, Coulter DA. Dentate granule cell GABA(A) receptors in epileptic hippocampus: enhanced synaptic efficacy and altered pharmacology. Eur J Neurosci. 2003;17:1607–1616. doi: 10.1046/j.1460-9568.2003.02597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge LL, Engel SA, Zeineh MM, Bookheimer SY, Knowlton BJ. A dissociation of encoding and retrieval processes in the human hippocampus. J Neurosci. 2005;25:3280–3286. doi: 10.1523/JNEUROSCI.3420-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady MS, Lam AM. Management of acute head injury. In: Lam AM, editor. Anesthetic Management of Head Injury. NY: McGraw-Hill; 1995. pp. 87–100. [Google Scholar]
- Heinemann U, Beck H, Dreier JP, Ficker E, Stabel J, Zhang CL. The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res Suppl. 1992;7:273–280. [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Impaired Cl- extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex. J Neurophysiol. 2005;93:2117–2126. doi: 10.1152/jn.00728.2004. [DOI] [PubMed] [Google Scholar]
- Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Stringer JL, Bertram EH. The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res Suppl. 1992;7:301–313. [PubMed] [Google Scholar]
- Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12:4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Akaike N. Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci. 2002;22:4412–4417. doi: 10.1523/JNEUROSCI.22-11-04412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JA. Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am J Physiol. 1997;273:C1516–1525. doi: 10.1152/ajpcell.1997.273.5.C1516. [DOI] [PubMed] [Google Scholar]
- Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA’s excitatory role in immature brain. J Neurobiol. 1997;33:781–795. doi: 10.1002/(sici)1097-4695(19971120)33:6<781::aid-neu6>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Reeves TM, Lyeth BG, Phillips LL, Hamm RJ, Povlishock JT. The effects of traumatic brain injury on inhibition in the hippocampus and dentate gyrus. Brain Res. 1997;757:119–132. doi: 10.1016/s0006-8993(97)00170-4. [DOI] [PubMed] [Google Scholar]
- Rempel-Clower NL, Zola SM, Squire LR, Amaral DG. Three cases of enduring memory impairment after bilateral damage limited to the hippocampal formation. J Neurosci. 1996;16:5233–5255. doi: 10.1523/JNEUROSCI.16-16-05233.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Scoville WB, Milner B. Loss of recent memory after bilateral hippocampal lesions. J Neurochem. 1957;20:11–21. doi: 10.1136/jnnp.20.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS. The functional organization of the hippocampal dentate gyrus and its relevance to the pathogenesis of temporal lobe epilepsy. Ann Neurol. 1994;35:640–654. doi: 10.1002/ana.410350604. [DOI] [PubMed] [Google Scholar]
- Soltesz I, Smetters DK, Mody I. Tonic inhibition originates from synapses close to the soma. Neuron. 1995;14:1273–1283. doi: 10.1016/0896-6273(95)90274-0. [DOI] [PubMed] [Google Scholar]
- Stuart GJ, Dodt HU, Sakmann B. Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflugers Arch. 1993;423:511–518. doi: 10.1007/BF00374949. [DOI] [PubMed] [Google Scholar]
- Sun MK, Zhao WQ, Nelson TJ, Alkon DL. Theta rhythm of hippocampal CA1 neuron activity: gating by GABAergic synaptic depolarization. J Neurophysiol. 2001;85:269–279. doi: 10.1152/jn.2001.85.1.269. [DOI] [PubMed] [Google Scholar]
- Toth Z, Hollrigel GS, Gorcs T, Soltesz I. Instantaneous perturbation of dentate interneuronal networks by a pressure wave-transient delivered to the neocortex. J Neurosci. 1997;17:8106–8117. doi: 10.1523/JNEUROSCI.17-21-08106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witgen BM, Lifshitz J, Smith ML, Schwarzbach E, Liang SL, Grady MS, Cohen AS. Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: A systems, network and cellular evaluation. Neuroscience. 2005;133:1–15. doi: 10.1016/j.neuroscience.2005.01.052. [DOI] [PubMed] [Google Scholar]
- Zhu L, Lovinger D, Delpire E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J Neurophysiol. 2005;93:1557–1568. doi: 10.1152/jn.00616.2004. [DOI] [PubMed] [Google Scholar]
- Zola-Morgan S, Squire LR, Amaral DG. Human amnesia and the medial temporal region: enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J Neurosci. 1986;6:2950–2967. doi: 10.1523/JNEUROSCI.06-10-02950.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]