

Abstract
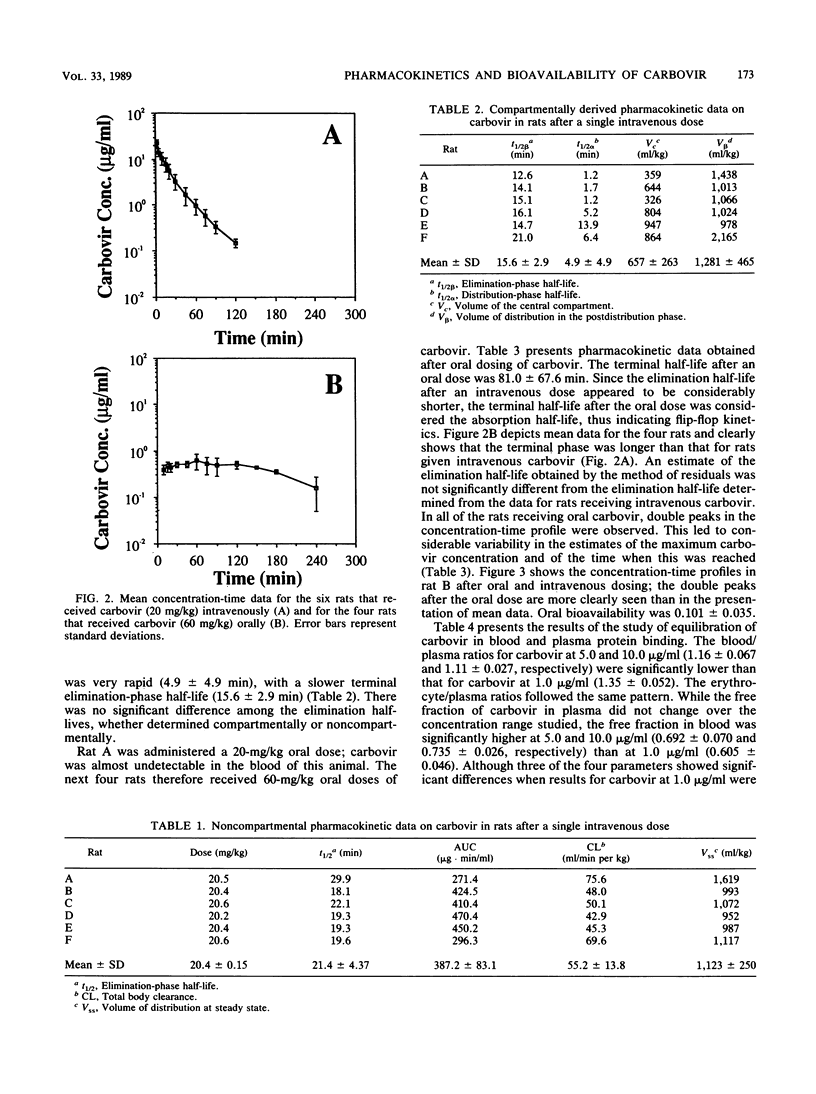
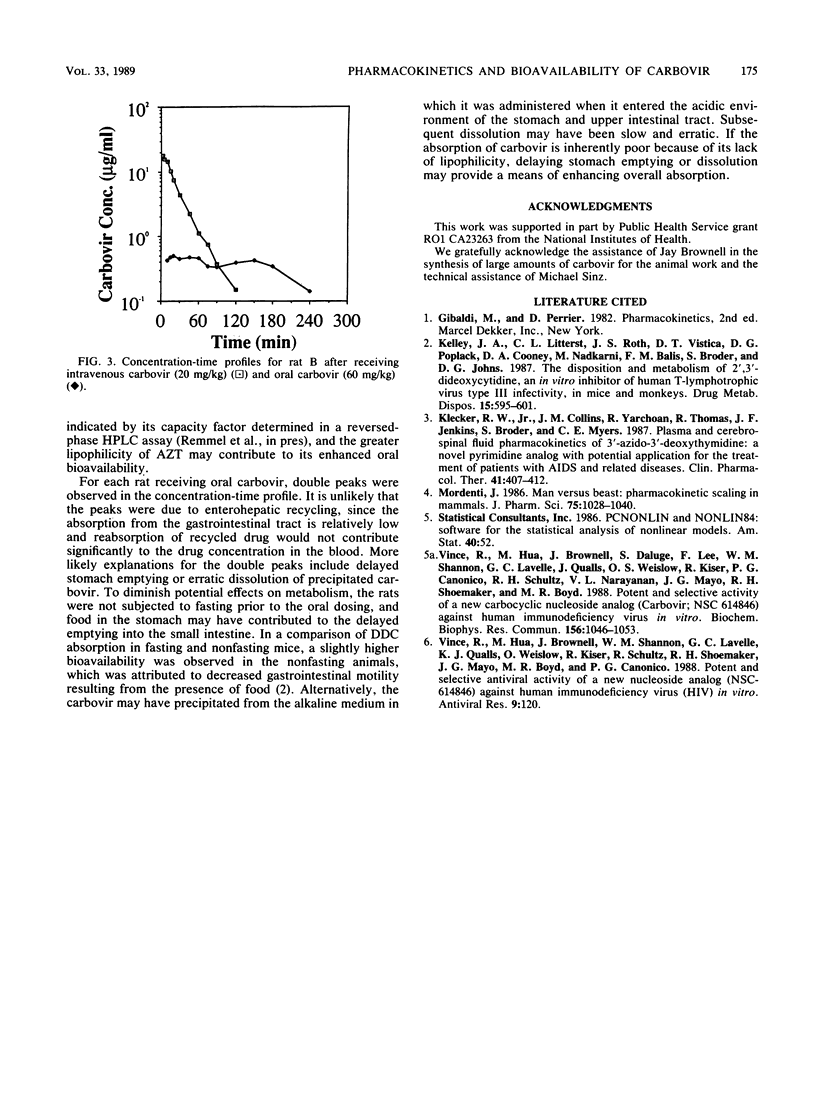
Carbovir is a novel carbocyclic nucleoside which has been shown to have potent in vitro activity against human immunodeficiency virus, the causative agent of acquired immunodeficiency syndrome. Sprague-Dawley male rats were used to investigate the pharmacokinetics and bioavailability of carbovir. Six rats received carbovir (20 mg/kg of body weight) through the jugular vein, and blood samples were collected through the femoral vein 2, 5, 10, 15, 20, 30, 45, 60, 75, 90, 120, 150, 180, and 240 min after the dose. Four of these rats also received a 60-mg/kg oral dose of carbovir, and a similar blood sampling schedule was followed. Whole-blood samples were prepared by solid-phase extraction, and the carbovir concentration in the samples was analyzed by reversed-phase high-pressure liquid chromatography. The profile of carbovir concentration in blood versus time after the intravenous dose was biexponential, with a very rapid distribution phase. Terminal elimination half-life was 21.4 +/- 4.37 min, and total body clearance was 55.2 +/- 13.8 ml/min per kg, which was within the range of the hepatic blood flow. The volume of distribution at steady state was 1,123 +/- 250 ml/kg. The blood/plasma ratio and the plasma protein binding of carbovir in rat blood were determined in vitro by ultrafiltration. The plasma protein binding of carbovir was only 20% and was not concentration dependent. However, the blood/plasma ratio decreased significantly as concentration increased, indicating saturable binding sites in erythrocytes. After the oral dose, the terminal half-life was 81.0 +/- 67.6 min, indicating that oral carbovir followed "flip-flop" kinetics, with absorption being much slower than elimination of the drug from the body. Oral bioavailability was 0.101 +/- 0.035. Double peaks were present in the concentration-time profile for each rat receiving the oral dose, indicating either a delay in stomach emptying of the drug or slow dissolution of precipitated carbovir in the stomach and upper small intestine.
Full text
PDF


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Kelley J. A., Litterst C. L., Roth J. S., Vistica D. T., Poplack D. G., Cooney D. A., Nadkarni M., Balis F. M., Broder S., Johns D. G. The disposition and metabolism of 2',3'-dideoxycytidine, an in vitro inhibitor of human T-lymphotrophic virus type III infectivity, in mice and monkeys. Drug Metab Dispos. 1987 Sep-Oct;15(5):595–601. [PubMed] [Google Scholar]
- Klecker R. W., Jr, Collins J. M., Yarchoan R., Thomas R., Jenkins J. F., Broder S., Myers C. E. Plasma and cerebrospinal fluid pharmacokinetics of 3'-azido-3'-deoxythymidine: a novel pyrimidine analog with potential application for the treatment of patients with AIDS and related diseases. Clin Pharmacol Ther. 1987 Apr;41(4):407–412. doi: 10.1038/clpt.1987.49. [DOI] [PubMed] [Google Scholar]
- Mordenti J. Man versus beast: pharmacokinetic scaling in mammals. J Pharm Sci. 1986 Nov;75(11):1028–1040. doi: 10.1002/jps.2600751104. [DOI] [PubMed] [Google Scholar]
- Vince R., Hua M., Brownell J., Daluge S., Lee F. C., Shannon W. M., Lavelle G. C., Qualls J., Weislow O. S., Kiser R. Potent and selective activity of a new carbocyclic nucleoside analog (carbovir: NSC 614846) against human immunodeficiency virus in vitro. Biochem Biophys Res Commun. 1988 Oct 31;156(2):1046–1053. doi: 10.1016/s0006-291x(88)80950-1. [DOI] [PubMed] [Google Scholar]