

Abstract
A newly identified gene, p51, is a functional and structural homologue of the p53 gene and thus a candidate tumor suppressor gene. To elucidate the role of the p51 gene in lung carcinogenesis, we determined the sequences of exon-intron boundaries and the 5′- and 3′-flanking regions of all the 15 coding exons and performed a mutation analysis, as well as detailed analysis for gene expression. A frameshift mutation was detected in 1 of 44 lung cancer cell lines, whereas no mutation was detected in 45 primary lung cancers. Thus, p51 mutation occurs only in a small subset of lung cancer. Expression of the p51 gene was detected in 23 of 43 cell lines by Northern blot analysis and 34 of 44 by reverse transcriptase-polymerase chain reaction (RT-PCR) analysis. Thus, p51 expression is low or absent in a subset of lung cancer. The ΔN isotype of p51 transcripts was dominantly expressed in several cell lines, particularly in cell lines with high levels of p51 expression. Because the ΔN isotype encodes a protein that transdominantly suppresses the transactivation function of the TA type of p51, it is possible that p51 protein is not functionally active, even in lung cancer cells with p51 mRNA expression, due to expression of dominant-negative p51 protein. These results suggested that the p51 gene is inactive in a considerable proportion of lung cancers. RT-PCR analysis also revealed the presence of a novel type of mRNA transcript, p51δ, which lacks exons 12 and 13 by alternative splicing. The δ isotype was expressed in 18 of 44 lung cancer cell lines and in diverse normal tissues. Further analysis on p51 expression in cancerous as well as noncancerous cells will provide us with valuable information for the understanding of multiple functions of the p53 family proteins in human carcinogenesis.
Keywords: p51 gene, p53 family proteins, mutation, alternative splicing, lung cancer
Introduction
The p53 tumor suppressor gene plays a key role in human carcinogenesis through regulating its target genes involved in cell cycle control and apoptosis in response to cellular damage [1]. Inactivation of the p53 gene appears to be the most common genetic alteration in human cancers and contributes to the development of over 50% of all human cancers [2]. Recently, two of the p53 structural homologues, p51 (also designated as p40, p63 and p73L) and p73, were cloned [3–8]. Because both p51 and p73 share critical functions with p53, including the induction of growth arrest and apoptosis, it is important to elucidate the functions of the newly identified p53 family of genes in more detail in association with human carcinogenesis.
To date, mutations of the p51 gene were detected only in 3 of 101 primary tumors and cancer cell lines [3]. Thus, it has been considered that p51 mutation occurs rarely in human cancers, as in the case of the p73 gene [9,10]. However, it is still possible that the p51 gene is frequently mutated in certain types of human cancers, particularly in cancer cells without p53 mutations. It is also possible that p51 is inactivated by mechanisms other than genetic alterations, including transcriptional silencing and expression of a dominant-negative form of the protein. To perform a detailed molecular analysis on the status of the p51 gene in human cancer cells, it is indispensable to determine the genomic structure of the p51 gene. For this reason, we first determined the genomic structure and designed intron-based primers for the mutation analysis of the p51 gene.
Yang et al. reported that there are at least 6 major types of p51 mRNA transcripts. Three of them (TA isotypes) encode proteins with the transactivation domain, DNA binding domain, and oligomerization domain, whereas the other three (ΔN isotypes) encode proteins without the acidic N-terminal domain, which is required for transactivation [7]. The ΔN isotypes of p51 proteins were predominantly expressed in a variety of epithelial cells and could act as dominant-negative agents toward the TA isotypes of p51 protein as well as p53 protein. Thus, it is also important to elucidate which types of p51 protein are predominantly expressed in human cancer cells.
In this study, we performed structural analysis of the p51 gene and its transcripts in human lung cancer cells for the following reasons. First, the p51 gene has been mapped to chromosome 3q28 [3], and comparative genomic hybridization (CGH) analysis indicated that this chromosomal region is frequently amplified in human lung cancers [11–16]. Second, a subset of lung cancers does not have mutated p53 genes, and responsible genes for the development of those cancers have not been identified yet. Third, a p51 mutation was previously detected in an undifferentiated squamous cell carcinoma of the lung [3]. We determined the exonintron structure of the p51 gene and intron sequences flanking all 15 coding exons and designed 15 sets of intron-based primers for the mutation analysis of the p51 gene. Then 44 cases of lung cancer cell lines and 45 cases of primary lung cancers were screened for mutations in the entire coding region as well as intronic splicing donor and acceptor sites of the p51 gene by polymerase chain reaction-single strand conformation polymorphism (PCR-SSCP) analysis. We also performed reverse transcriptase-polymerase chain reaction (RT-PCR) analysis to identify the isotypes of p51 transcripts that are predominantly expressed in lung cancer cells.
Materials and Methods
Samples
Forty-four lung cancer cell lines, including 33 non-small cell lung carcinomas (NSCLCs) and 11 small cell lung carcinomas (SCLCs) were used in this study. NSCLC cell lines were A427, A549, PC3, PC7, PC9, PC14, LCMS, H23, H441, H322, Ma1, Ma3, Ma10, Ma12, Ma17, Ma24, Ma26, Ma29, RERF-LCOK, VMRC-LCD, ABC1, H596, PC10, LC1-Sq, EBC1, H520, H157, H1155, Ma2, PC13, Lu65, Lu99A, and Ma25. SCLC cell lines were Lu24, Lu130, Lu134, Lu135, Lu139, H69, H82, N417, SBC5, H526, and H209 [17]. Detailed information on these cell lines can be obtained upon request. Primary lung cancers analyzed were obtained from patients with 15 SCLC and 30 NSCLC (15 adenocarcinomas and 15 squamous cell carcinomas). High-molecular-weight DNA was prepared from cell lines, tumors, and adjacent noncancerous tissues as described previously [18]. mRNAs of normal lung tissues were obtained from Clontech (NL1) and patients with lung cancer (NL2 and NL3). Poly (A)+ mRNA was prepared from cell lines and normal lung tissues by the Fast Track mRNA isolation kit (Invitrogen Corp).
Determination of the Exon-Intron Organization
Genomic DNA fragments covering the p51 gene locus were obtained by the following procedure. First, several sets of PCR primers based on the cDNA sequence were designed to construct a contig of genomic DNA fragments covering the p51 locus. One hundred nanograms of genomic DNA from human colon cancer cell lines, LoVo and HCT116, were used for PCR. PCR products were electrophoresed on 0.8% agarose gel, and their sizes were compared with those of the corresponding cDNA fragments. Amplified DNA fragments containing introns were sequenced to design intron primers. Next, human bacterial artificial chromosome (BAC) DNA Pools Release IV (Research Genetics) was screened by PCR with 3 sets of intron-based primers, which amplified exons 2, 6, and 15 of the p51 gene (Table 1). DNA from a BAC clone containing the p51 gene locus was isolated according to the supplier's protocol and was used to determine the sequences at the exon-intron boundaries and the 5′- and 3′-flanking regions of the exons of the p51 gene.
Table 1.
Primer Sequencesa for Amplification of Individual Exons of the p51 Gene.
| Exon | Sense Primer Sequence (5′–3′) | Antisense Primer Sequence (5′–3′) | Size of PCR Product (bp) |
| 2 | GGAAAGAAAGTTATTACCGATCCA | atattttgaccacccacatttacc | 163 |
| 3 | cctttccatgcctaactcact | gactgaagagaaagcctgtgc | 201 |
| 3′ | CAGAAGAAAGGACAGCAGCAT | tgaccgagaaccgcaaatacg | 173 |
| 4 | ggctaatattggggtttctgg | aactgggaagggggaaaaatc | 376 |
| 5 | catgcagctctaaaaagtgga | aggtgggtctcaaacaaaaat | 376 |
| 6 | ggctatgggatctgttcgtt | tcagtccatagaggtgttgtt | 318 |
| 7 | cttaaatagagggtagaactgag | cactttgccctctgctcgtg | 252 |
| 8 | gggaagtggaagtggtagatc | caccccattctccaacatctg | 286 |
| 9 | catcttccactccttattgc | actgtaaaagcccattcaaag | 380 |
| 10 | ggattgaccacacttctaacag | gtcactcatgcctcctaaaa | 248 |
| 11 | ccgtgctcaccatcatttcca | agctagcctgttcatccttca | 243 |
| 12 | tgatggcagtaaccctttttg | agtatccgccctccaccatcc | 256 |
| 13 | ggcatccaagggcaaaata | gggcatgacaaaaacaaat | 313 |
| 14 | ctccctgttttcattctccat | TAGCCTCTTACTTCTCCTTCC | 481 |
| 15 | ttttgtctggttcctctctgc | TGTCTTCAAGTCCTGTTTATG | 252 |
Upper case letters correspond to exons and lower case letters to introns.
Polymerase Chain Reaction-Single Strand Conformational Polymorphism Analysis
All samples were examined for mutations in the entire coding sequence of the p51 gene (exons 2–15 and exon 3′) by PCR-SSCP analysis. Each exon was amplified by PCR with primer pairs shown in Table 1. Fifty nanograms of genomic DNA templates were suspended in a total volume of 20-µL PCR buffer containing 10 mmol/L Tris-HCl (pH 9.0), 50 mmol/L KCl, 1.5 mmol/L MgCl2, 100 nmol/L of each primer, 200 µmol/L of each deoxynucleotide triphosphate, 1.5 µCi of α-[32P]dCTP (Amersham Pharmacia Biotech), and 0.5 U Taq DNA polymerase (Amersham Pharmacia Biotech). PCR conditions were as follows; 30 seconds at 95°C, 30 seconds at 55°C or 60°C, and 90 seconds at 72°C for 35 cycles, followed by 10 minutes at 72°C. SSCP analysis was performed in the low pH buffer system that showed improved separation of long mutant fragments of up to 800 base pairs (bp) [19]. PCR products were denatured and loaded on nondenaturing polyacrylamide gel containing 5% polyacrylamide (99:1 acrylamide to bisacrylamide) and TPE (30 mmol/L Tris, 20 mmol/L PIPES, and 1 mmol/L Na2 EDTA; pH6.8), electrophoresed in TPE buffer at 15°C, and exposed to Kodak XAR films for 24 to 48 hours at -80°C. PCR products showing different mobilities were purified and directly sequenced in both directions.
Sequence Analysis
PCR fragments were purified by using QIA quick-spin PCR purification kit (QIAGEN) and directly sequenced by using Thermo Sequence dye terminator cycle sequencing pre-mix kits (Amersham Pharmacia Biotech) and ABI 373S DNA Sequence System (Perkin-Elmer). DNA from BAC clone was sequenced by using the fmol DNA Sequencing System according to the supplier's protocol (Promega).
Southern and Northern Blot Analyses
Southern and Northern blot analyses were carried out under the stringent condition described previously [20]. Genomic DNA was digested with EcoRI and electrophoresed for Southern blot analysis. The cDNA fragment corresponding to nucleotide 170–781 of p51A cDNA (Accession No. AB016072 in Genbank) was used for hybridizations as a probe.
Reverse Transcriptase-Polymerase Chain Reaction Analysis
Randomly primed cDNAs were reverse-transcribed from 0.5 µg mRNAs by using SuperScript II Rnase H-Reverse Transcriptase (Gibco BRL) in a 20-µL mixture according to the supplier's protocol. One microliter of the cDNA conversion mixture was amplified by PCR in a 10-µL reaction mixture. PCR conditions were as follows: 30 seconds at 95°C, 30 seconds at 55°C or 60°C, and 90 seconds at 72°C for 35 cycles, followed by 10 minutes at 72°C. The following primers were used: TA, 5′-ATGTCCCAGAGCACACAG-3′ and 5′-AGCTCATGGTTGGGGCAC-3′; ΔN, 5′-CAGACTCAATTTAGTGAG-3′ and 5′-AGCTCATGGTTGGGGCAC-3′; α and β, 5′-GACTTATGAAATGCTGTTGAA-3′ and 5′-TCGCACAGCATCAATAACACG-3′; γ, 5′-GACTTATGAAATGCTGTTGAA-3′ and 5′-CGTCAGATTGTTTTGGAGTTT-3′. PCR products were electrophoresed on 2.0% agarose gel and stained with ethidium-bromide.
Results
Genomic Structure of the p51 Locus
It was previously shown that the p51 gene consisted of 16 exons and 15 introns [7]. Exon 1 is a noncoding exon, whereas all others are coding exons. Genomic structure of exons 2 to 15 was determined by the following methods. A remarkable conservation of exon-intron organizations between two of the p53 family genes, p53 and p73, permitted us to estimate intron positions of the p51 gene. We therefore designed PCR primers that would specifically anneal to regions in possible contiguous exons of the p51 gene. Genomic DNA fragments that contained introns 2, 5, 6, 7, 8, 9, and 15 were successfully amplified and sequenced to design sets of intron-based primers that specifically amplified exons 2, 6, 7, 8, 9, and 15. Then, these sets of primers were used to screen a BAC library. A BAC clone, 224P11, covering exons 2 to 15 of the p51 locus, was obtained and used to determine the sequences of the remaining flanking introns.
It was previously shown by the structural analysis of p51 cDNA that there are two different start codons and two different stop codons in the p51 gene [7,8]. The ATG start codons were located in exons 2 and 3′, and the stop codons were located in exons 14 and 15. The structure determined in this study was consistent with the one determined by Yang et al. [7]. The putative transactivation domain, DNA binding domain, and oligomerization domain were encoded in exons 2 to 3, exons 4 to 8, and exons 9 to 10, respectively. Two major splicing forms of the p51 gene, p51A and p51B (also designated as TAp63γ and TAp63α), were derived from exons 2 to 10 and 15, and exons 2 to 14, respectively. The sequences of whole exons in the BAC clone were identical to those of the p51 cDNAs, and all exon-intron junctions followed the GT-AG rule [21]. The boundary sequences of each exon-intron are shown in Figure 1. Comparison of the genomic organization between p51 and the other two p53 family genes revealed that the exon boundary positions and exon sizes of the p51 gene were highly homologous to those of the p53 and p73 genes. In contrast, the homologies in the size and sequence of introns were not so high among these three genes [22,23].
Figure 1.
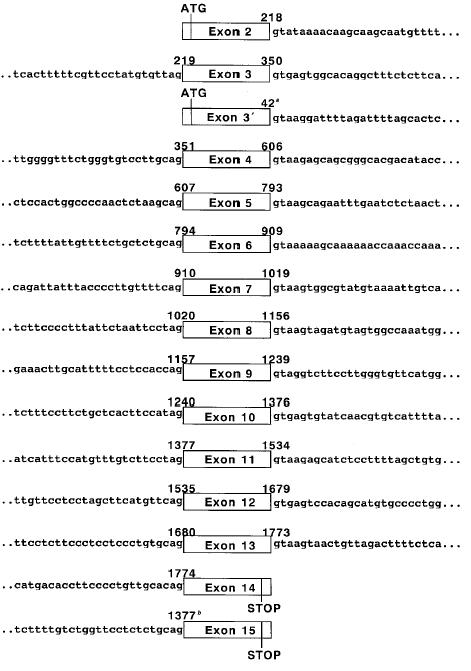
Intronic sequences surrounding the p51 coding exon. Nucleotide numbers corresponding to the sequences of p51B, aΔNp63γ, or b51A cDNA (Genbank Accession Nos. AB016073, AF075429, and AB016072) are shown above the boxes. More detailed information of these sequences have been submitted to Genbank/EMBL/DDBJ and have been assigned the Accession Nos. AF116756 to AF116770.
Genetic Alterations of the p51 Gene in Lung Cancers
PCR amplification of genomic DNA from cell lines and primary tumors was performed by using the intron-based primers for each of the 15 coding exons (Table 1). The size of each PCR fragment was less than 500 bp and was suitable for SSCP analysis in the low pH buffer system [16]. Forty-four lung cancer cell lines and 45 primary lung cancers were analyzed for p51 mutations and polymorphisms. A mutation was detected in a squamous cell carcinoma cell line, EBC1 (Figure 2). Sequence analysis of DNA from EBC1 revealed one base insertion of adenine at codons 193–194 (AAAAAA to AAAAAAA). This insertion produces a frameshift mutation in the DNA binding domain, resulting in predicted synthesis of truncated p51 of 195 amino acids. The wild type allele was retained in this cell line. The p53 gene was also mutated in EBC1 cells (Table 3).
Figure 2.
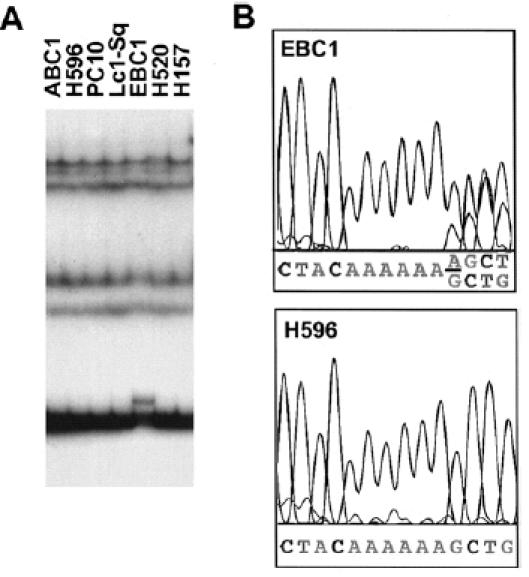
Mutation of the p51 gene in a lung cancer cell line. (A) Mutation of the p51 gene detected by PCR-SSCP. In exon 5, an SSCP variant was detected in a squamous cell carcinoma cell line, EBC1. (B) Mutation of the p51 gene detected by direct sequencing. In EBC1, one base of adenine was inserted in codons 193–194 of exon 5, resulting in a frameshift of one allele (underlined). Normal sequence in H596 is shown as a control.
Table 3.
Analysis of p51 Transcripts in Human Lung Cancer Cell Lines.
| Sample | Histology | Level of Expressiona | Size of mRNA (kb) | Pattern of Expressionb | p53 Statusc | |||||
| TA | ΔN | α | β | γ | δ | |||||
| A427 | AdC | - | - | - | - | - | - | - | wt | |
| A549 | AdC | - | ± | - | - | - | - | - | mut | |
| PC3 | AdC | + | 3.8 | + | + | + | + | + | + | mut |
| PC7 | AdC | + | 4.4 | + | - | ± | - | ± | + | mut |
| PC9 | AdC | - | ± | - | - | - | - | - | mut | |
| PC14 | AdC | - | - | - | - | - | - | - | mut | |
| LCMS | AdC | - | - | - | - | - | - | - | mut | |
| H23 | AdC | - | ± | - | - | - | ± | - | mut | |
| H441 | AdC | + | 4.4 | + | ± | + | + | + | + | mut |
| H322 | AdC | ++ | 4.4 | ± | + | + | + | + | + | mut |
| Ma1 | AdC | - | ± | - | - | - | ± | - | mut | |
| Ma3 | AdC | ++ | 3.8 | + | + | + | + | + | + | mut |
| Ma10 | AdC | ++ | 3.8 | ± | + | + | - | + | + | mut |
| Ma12 | AdC | + | 4.4 | ± | - | - | - | ± | - | wt |
| Ma17 | AdC | - | - | - | - | - | - | - | wt | |
| Ma24 | AdC | ++ | 4.4 | + | + | + | + | + | + | mut |
| Ma26 | AdC | + | 4.4 | + | - | ± | - | ± | + | mut |
| Ma29 | AdC | + | 4.4 | + | - | ± | + | + | + | mut |
| RERF-LCOK | AdC | + | 4.4 | ± | - | ± | - | ± | + | mut |
| VMRC-LCD | AdC | - | ± | - | - | - | - | - | mut | |
| ABC1 | AdC | + | 3.8 | + | + | + | ± | + | + | mut |
| H596 | ASC | ++ | 4.4 | + | + | + | + | + | + | mut |
| PC10 | SqC | ++ | 4.4 | + | + | + | + | + | + | mut |
| LC1-Sq | SqC | ++ | 4.4 | + | + | + | + | + | + | mut |
| EBC1 | SqC | + | 4.4 | ± | - | - | - | - | - | mut |
| H520 | SqC | - | ± | - | - | - | - | - | mut | |
| H157 | SqC | - | ± | - | - | - | - | - | mut | |
| H1155 | LCC | - | - | - | - | - | - | - | mut | |
| Ma2 | LCC | - | - | - | - | - | - | - | mut | |
| PC13 | LCC | ND | - | - | - | - | - | - | mut | |
| Lu65 | LCC | + | 4.4 | + | - | ± | - | - | ± | mut |
| Lu99A | LCC | - | - | - | - | - | - | - | wt | |
| Ma25 | LCC | - | ± | - | - | - | - | - | mut | |
| Lu24 | SCC | - | ± | - | ± | - | - | ± | mut | |
| Lu130 | SCC | + | 4.4 | ± | - | - | ± | ± | - | mut |
| Lu134 | SCC | + | 4.4 | ± | - | ± | - | - | ± | mut |
| Lu135 | SCC | - | ± | - | - | - | - | - | mut | |
| Lu139 | SCC | + | 4.4 | - | - | - | - | - | - | mut |
| H69 | SCC | + | 4.4 | ± | - | - | - | - | - | mut |
| H82 | SCC | - | ± | - | - | - | - | - | mut | |
| N417 | SCC | - | - | - | - | - | - | - | mut | |
| SBC5 | SCC | + | 4.4 | + | ± | ± | - | - | ± | mut |
| H526 | SCC | + | 4.4 | - | - | - | - | - | - | mut |
| H209 | SCC | - | - | - | - | - | - | - | wt | |
| NL1 | Normal | ND | + | + | - | - | ± | ± | ||
| NL2 | Normal | ND | ± | ± | - | - | ± | - | ||
| NL3 | Normal | ND | ± | ± | - | - | + | + | ||
Level of expression was analyzed by Northern blot analysis. ND, not done.
Pattern of expression was analyzed by RT-PCR.
These data were obtained from IARC p53 database [24]. Our unpublished data are also included.
AdC, adenocarcinoma; ASC, adenosquamous cell carcinoma; SqC, squamous cell carcinoma; LCC, large cell carcinoma; SCC, small cell carcinoma; Normal, mRNAs of normal lung tissues were obtained from Clontech (NL1) and patients with lung cancer (NL2 and NL3). wt, wild type; mut, mutated.
Four different types of genetic polymorphisms were detected. A silent polymorphism was found at codon 248 in exon 6, and three others were located in introns 5 and 8 (Table 2). Thus, there was no genetic polymorphism by which p51 protein with different amino acid sequence could be expressed.
Table 2.
Polymorphisms of the p51 Gene.
| Location | Sequence | Codon | Amino Acid | Allele Frequencya |
| Intron 5 (+34 bp from exon 5) | T/G | 0.89/0.11 | ||
| Intron 5 (+42 bp from exon 5) | G/A | 0.92/0.08 | ||
| Exon 6 | C/T | 248 | Val/Val | 0.98/0.02 |
| Intron 8 (-22 bp from exon 9) | A/G | 0.88/0.12 |
Allele frequency was determined in 44 lung cancer cell lines.
We further searched for genetic alterations in the p51 locus by Southern blot analysis. There were no amplification, homozygous deletion, and rearrangement of the p51 gene in 42 lung cancer cell lines (data not shown).
Expression of the p51 Transcripts in Lung Cancers
We first examined the expression of the p51 gene in lung cancer cell lines by Northern blot analysis. p51 transcripts were detected in 23 of 43 cell lines. In each cell line, major transcripts of either 4.4 kb or 3.8 kb in size were detected. The levels of p51 expression were variable among cell lines and relatively high in H322, Ma3, Ma10, Ma24, H596, PC10, and LC1-Sq (Figure 3A, Table 3). Cell lines with high levels of p51 expression were derived either from adenocarcinoma or squamous cell carcinoma and were not from large cell carcinoma or small cell carcinoma. However, apparently no correlation was observed between the level of expression and the histology of cell lines. The p53 gene is not mutated in 7 (A427, A549, Ma12, Ma17, Lu99A, Lu24, and H209) of the 43 cell lines examined (Table 3). Among them, a low level of p51 expression was detected in Ma12 only, and p51 transcripts were not detected in the remaining 6 cell lines. Thus, it is possible that the p51 gene is not expressed in most of lung cancer cells with wild type p53 expression.
Figure 3.
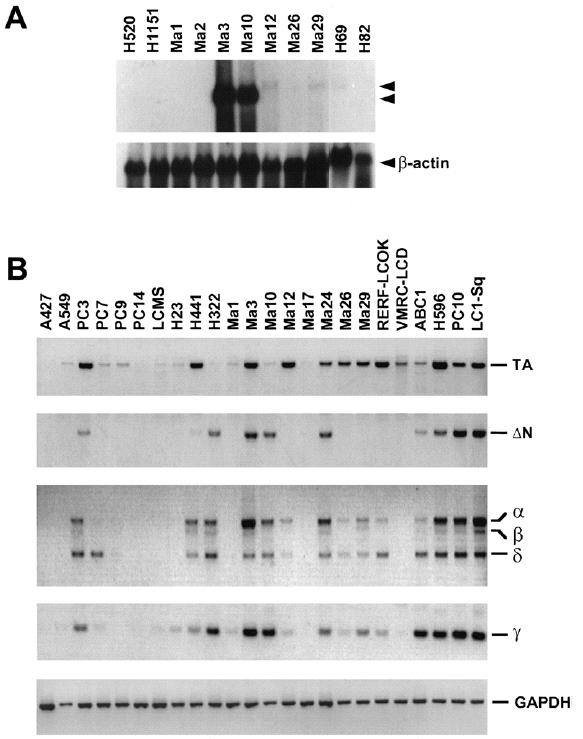
Expression of the p51 gene in lung cancers. (A) Northern blot analysis of the p51 gene in lung cancer cell lines. Two major transcripts (4.4 kb and 3.8 kb) are indicated by arrowheads. The membrane was rehybridized with a human β-actin probe (Clontech). (B) RT-PCR analysis using primers that specifically amplified the TA, ΔN, α and β, and γ isotypes of p51 transcripts. By using these primers, the TA, ΔN, α, β, and γ types were detected as PCR products of 629 bp, 440 bp, 702 bp, 608 bp, and 191 bp, respectively. A novel isotype, δ, was found as a PCR product of 453 bp. Expression of a control housekeeping gene (GAPDH) is shown in the bottom.
Up to the present, six of alternatively spliced p51 transcripts have been identified, and they encode proteins with two different N termini (TA and ΔN) and three different C termini (α, β, and γ) [7]. To obtain more detail information on p51 expression in lung cancers, we performed RT-PCR analysis with 4 sets of primers that specifically amplified the TA, ΔN, α and β, and γ isotypes (Figure 3B). Expression patterns of these isotypes in each cell line are summarized in Table 3. The result of RT-PCR analysis was almost consistent with and similar to that of Northern blot analysis. Either one or more p51 isotypes were detected in 34 of 44 cell lines. p51 transcripts were also detected by RT-PCR analysis in all three of normal lung tissues. There was no correlation between the isotypes expressed and histology of cell lines. Interestingly, all of 7 cell lines expressing high levels of p51 expressed the ΔN isotype that inactivates the function of the TA isotype transdominantly [7]. The α and β isotypes were not detected in normal lung tissues, although those two isotypes were detected in most lung cancer cell lines.
We found a novel p51 variant that was amplified by using a set of primers specific for the α and β isotypes (Figure 3B, Table 3). This variant was derived from an alternative splicing from exon 11 to exon 14, thus, lacked exons 12 and 13, and was designated the δ isotype (Figure 4A). The δ isotype was expressed in 18 of 44 lung cancer cell lines and 2 of 3 normal lung tissues. To examine the tissue distribution of the δ isotype, we performed PCR analysis with cDNAs from various normal human tissues. Expression of the δ isotype was detected in uterus, thymus, placenta, mammary gland, and fetal lung, and not detected in kidney, stomach, small intestine, spleen, bone marrow, brain, spinal cord, testis, skeletal muscle, adrenal grand, salivary grand, pituitary gland, fetal liver, and fetal kidney (Figure 4b).
Figure 4.

(A) Schematic representation of the isotypes of p51 transcripts. The positions of primers for RT-PCR analysis are indicated by arrows. (B) Tissue distribution of the p51δ isotype. Expression of the δ isotype in normal human tissues analyzed by RT-PCR is also shown.
Among the 7 cell lines without p53 mutations, RT-PCR products were detected in 3 cell lines, A549, Ma12, and Lu24. In all the 3 cell lines, the TA isotype but not the ΔN isotype was detected, suggesting that the function of wild-type p53 protein is not suppressed by expression of dominant-negative p51 protein in these cell lines. Absence of transcription would be a more common phenomenon in lung cancer cells with the wild-type p53 gene, as suggested by Northern blot analysis. In contrast, expression of the ΔN isotype was detected in 11 cell lines, and all of them had mutated p53 gene. Thus, in cell lines with p53 mutations, the function of the p51 gene might be also inactivated in part by expression of dominant-negative ΔN isotype p51 protein.
Although major transcripts of either 4.4 kb or 3.8 kb were detected by Northern blot analysis, the association between the sizes and the isotypes of mRNA transcripts is presently unknown.
Discussion
Here we determined the genomic structure of the p51 gene and, based on this data, we designed 15 sets of primers for mutation analysis of all the coding sequences of the p51 gene. Previous mutation analysis of the p51 gene showed that the p51 gene was mutated in a lung squamous cell carcinoma, a head and neck cancer cell line (Ho-1-u-1), and a cervical cancer cell line (SKG-III) [3]. We therefore performed mutation analysis of the p51 gene in human lung cancer. A mutation was detected in only one of 89 (1.1%) lung cancers. Thus, mutation of the p51 gene rarely occurs in lung cancer cells. Mutations detected in the previous study were located in exon 4 (a lung squamous cell carcinoma and Ho-1-u-1) or exon 5 (SKG-III). A mutation we found in this study was also located in exon 5, which encodes the putative DNA binding domain. These results suggest that mutations of the p51 gene cluster in the DNA binding domain, as in the case of mutations in the p53 gene [24]. The result may also suggest that p51 mutations occur in a small subset of squamous cell carcinoma and not in other histological types of lung cancer. Although the present result indicated that the p51 gene is infrequently mutated in lung cancers, intron-based primers designed in this study will be useful for the molecular analysis of the p51 gene in a variety of human cancers.
Amplification of genes in the 3q28 region has been suggested to occur in lung cancer cells by the CGH analysis [11–16], however, amplification of the p51 gene at 3q28 was not detected by Southern blot analysis. Thus, the p51 gene would not be a target of gene amplification at 3q28 in lung cancer cells.
Northern blot analysis as well as RT-PCR analysis revealed that the p51 gene is expressed in a large subset of lung cancer cells and the levels of p51 expression vary considerably among lung cancer cells. Thus, it would be of great interest to clarify the association of p51 expression with biological properties of lung cancer cells. It is possible that expression of the p51 gene is silenced in a subset of lung cancer cells by several mechanisms, including DNA methylation in the promoter region [25,26]. If so, transcriptional silencing would be one of the mechanisms for p51 inactivation. For this reason, molecular analysis of the promoter region is now in progress in our laboratories.
It is noted that the ΔN isotype of mRNA transcripts was expressed in cell lines with high levels of p51 expression. Because the ΔN isotype encodes a protein that transdominantly suppresses the transactivation function of p51A (TAp63γ) as well as p53, it was suggested that the p51 gene, and possibly the p53 gene, is functionally inactivated in lung cancer cell lines in part by expression of the dominant-negative ΔNp51 protein. However, because all the cell lines that express the ΔN isotype of mRNA transcripts carry mutated p53 genes, the interaction between the ΔN isotype of p51 protein and mutated p53 protein remains unclear. Further studies are required to resolve this question.
The α and β isotypes were not detected in normal lung tissues, whereas they were detected in most lung cancer cell lines. However, the biological significance of this finding is unclear at present because the functional differences among α, β, γ, and δ isotypes are unknown.
Presence of a novel isotype of p51 mRNA transcripts also indicated that the complexed functions of the p51 gene can be regulated partly by alternative splicing of the p51 transcripts. Recently, two novel p73 splicing variants, p73γ and p73δ, were identified in addition to p73α and p73β. It has been suggested that p73 provides a complex regulatory mechanism to the control of cell growth, because of different expression patterns for the four splicing variants in normal and neoplastic cells, and of homodimeric and hetrodimeric interactions of various p73 isotypes and p53 [27]. Understanding the interaction between various isotypes of p51 protein, as well as p53 and p73, would be of great importance in clarifying the mechanisms for regulation of cell growth and apoptosis in cancer cells.
Acknowledgements
We thank the following scientists for providing cell lines: Dr. Y. Hayata of Tokyo Medical College, Drs T. Terasaki and S. Hirohashi of National Cancer Center Research Institute, Dr. M. Takada of Izumisano Municipal Hospital, Drs. A. F. Gazdar and J. D. Minna of the University of Texas, Southwestern Medical Center. Cell lines were also obtained from ATCC. We also thank Dr. H. Sasaki of National Cancer Center Research Institute for providing cDNAs from various human normal tissues.
Footnotes
This work was supported in part by Grants-in-Aid from the Ministry of Health and Welfare, from the Ministry of Education, Science, Sports and Culture, from the Foundation for Promotion of Cancer Research, and from the Naito Foundation of Japan. K. Shimizu is a recipient of the Research Resident Fellowship from the Foundation for Promotion of Cancer Research.
These authors contributed equally to this work.
References
- 1.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 2.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 3.Osada M, Ohba M, Kawahara C, Ishioka C, Kanamaru R, Katoh I, Nimura Y, Nakagawara A, Obinata M, Ikawa S. Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat Med. 1998;4:839–843. doi: 10.1038/nm0798-839. [DOI] [PubMed] [Google Scholar]
- 4.Kaghad M, Bonnet H, Yang Y, Creancier L, Biscan J-C, Valent A, Minty A, Chalon P, Lelias J-M, Dumont X, Ferrara P, McKeon F, Caput D. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other cancers. Cell. 1997;90:809–819. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 5.Jost CA, Marin MC, Kaelin WG., Jr p73 is a human p53-related protein that can induce apoptosis. Nature. 1997;389:191–194. doi: 10.1038/38298. [DOI] [PubMed] [Google Scholar]
- 6.Trink B, Okami K, Wu L, Spiuranpong V, Jen J, Sidransky D. A new human p53 homolog. Nat Med. 1998;4:747–748. doi: 10.1038/nm0798-747. [DOI] [PubMed] [Google Scholar]
- 7.Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dötsch V, Andrews NC, Caput D, McKeon F. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2:305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 8.Senoo M, Seki N, Ohira M, Sugano S, Watanabe M, Tachibana M, Tanaka T, Shinkai Y, Kato H. A second p53-related protein, p73L, with high homology to p73. Biochem Biophys Res Commun. 1998;248:603–607. doi: 10.1006/bbrc.1998.9013. [DOI] [PubMed] [Google Scholar]
- 9.Nomoto S, Haruki N, Kondo M, Konishi H, Takahashi T, Takahashi T, Takahashi T. Search for mutations and examination of allelic expression imbalance of the p73 gene at 1p36.33 in human lung cancers. Cancer Res. 1998;58:1390–1383. [PubMed] [Google Scholar]
- 10.Takahashi H, Ichimiya S, Nimura Y, Watanabe M, Furusato M, Wakui S, Yatani R, Aizawa S, Nakagawara A. Mutation, allelotyping, and transcription analyses of the p73 gene in prostatic carcinoma. Cancer Res. 1998;58:2076–2077. [PubMed] [Google Scholar]
- 11.Reid T, Petersen I, Holtgreve-Grez H, Speicher MR, Schröck E, du Manoir S, Cremer T. Mapping of multiple DNA gain and losses in primary small cell lung carcinomas by comparative ge-nomic hybridization. Cancer Res. 1994;54:1801–1806. [PubMed] [Google Scholar]
- 12.Levin NA, Brzoska P, Gupta N, Minna JD, Gray JW, Christman MF. Identification of frequent novel genetic alterations in small cell lung tumors. Cancer Res. 1995;54:5086–5091. [PubMed] [Google Scholar]
- 13.Levin NA, Brzoska PM, Warnock ML, Gray JW, Christman MF. Identification of novel regions of altered DNA copy number in small cell lung tumors. Genes, Chromosomes Cancer. 1995;13:175–185. doi: 10.1002/gcc.2870130307. [DOI] [PubMed] [Google Scholar]
- 14.Schwendel A, Langreck H, Reichel M, Schröck E, Reid T, Dietel M, Petersen I. Primary small-cell lung carcinomas and their metastases are characterized by a recurrent pattern of genetic alterations. Int J Cancer. 1997;74:86–93. doi: 10.1002/(sici)1097-0215(19970220)74:1<86::aid-ijc15>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 15.Balsara BR, Sonoda G, du Manoir S, Siegfried JM, Gabrielson E, Testa JR. Comparative genomic hybridization analysis detects frequent, often high-level, overrepresentation of DNA sequences at 3q, 5p, 7p, and 8q in human non-small cell lung carcinomas. Cancer Res. 1997;57:2116–2120. [PubMed] [Google Scholar]
- 16.Bjorkqvist AM, Husgafvel-Pursiainen K, Anttila S, Karjalainen A, Tammilehto L, Mattson K, Vainio H, Knuutila S. DNA gains in 3q occur frequently in squamous cell carcinoma of the lung, but not in adenocarcinoma. Genes, Chromosomes Cancer. 1998;22:79–82. [PubMed] [Google Scholar]
- 17.Kohno T, Otsuka T, Takano H, Yamamoto Y, Hamaguchi M, Terada M, Yokota J. Identification of a novel phospholipase C family gene at chromosome 2q33 that is homozygously deleted in human small cell lung carcinoma. Hum Mol Genet. 1995;4:667–674. doi: 10.1093/hmg/4.4.667. [DOI] [PubMed] [Google Scholar]
- 18.Sakamoto H, Mori M, Taira M, Yoshida T, Matsukawa S, Shimizu K, Sekiguchi M, Terada M, Sugimura T. Transforming gene from human stomach cancers and a noncancerous portion of stomach mucosa. Proc Natl Acad Sci USA. 1986;83:3997–4001. doi: 10.1073/pnas.83.11.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kukita Y, Tahira T, Sommer SS, Hayashi K. SSCP analysis of long DNA fragments in low pH gel. Hum Mutat. 1997;10:400–407. doi: 10.1002/(SICI)1098-1004(1997)10:5<400::AID-HUMU11>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 20.Tani M, Shindo-Okada N, Hashimoto Y, Shiroishi T, Takenoshita S, Nagamachi Y, Yokota J. Isolation of a novel Sry-related gene that is expressed in high-metastatic K-1735 murine melanoma cells. Genomics. 1997;39:30–37. doi: 10.1006/geno.1996.4483. [DOI] [PubMed] [Google Scholar]
- 21.Breathnach R, Benoist C, O'Hare K, Gannon F, Chambon P. Ovaibumin gene: Evidence for a leader sequence in mRNA and DNA sequences at the exon-intron boundaries. Proc Natl Acad Sci USA. 1978;75:4853–4857. doi: 10.1073/pnas.75.10.4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mai M, Huang H, Reed C, Qian C, Smith JS, Alderete B, Jenkins R, Smith DI, Liu W. Genomic organization and mutation analysis of p73 in oligodendrogliomas with chromosome 1 p-arm deletions. Genomics. 1998;51:359–363. doi: 10.1006/geno.1998.5387. [DOI] [PubMed] [Google Scholar]
- 23.Lamb, Crawford L. Characterization of the human p53 gene. Mol Cell Biol. 1986;6:1379–1385. doi: 10.1128/mcb.6.5.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hainaut P, Hernandez T, Robinson A, Rodriguez-Tome P, Flores T, Hollstein M, Harris CC, Montesano R. IARC Database of p53 gene mutations in human tumors and cell lines: Updated compilation, revised formats and new visualisation tools. Nucleic Acids Res. 1998;26:205–213. doi: 10.1093/nar/26.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nature Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 26.Tornaletti S, Pfeifer GP. Complete and tissue-independent methylation of CpG sites in the p53 gene: Implications for mutations in human cancers. Oncogene. 1995;10:1493–1499. [PubMed] [Google Scholar]
- 27.Laurenzi VD, Costanzo A, Barcaroli B, Terrinoni A, Falco M, Annicchiarico-Petruzzelli M, Levrero M, Melino G. Two new p73 splice variants, γ and δ, with different transcriptional activity. J Exp Med. 1998;188:1763–1768. doi: 10.1084/jem.188.9.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]