

Abstract
Apoptosis is a fundamental biologic process by which metazoan cells orchestrate their own self-demise. Genetic analyses of the nematode C elegans identified three core components of the suicide apparatus which include CED-3, CED-4, and CED-9. An analogous set of core constituents exists in mammalian cells and includes caspase-9, Apaf-1, and bcl-2/xl, respectively. CED-3 and CED-4, along with their mammalian counterparts, function to kill cells, whereas CED-9 and its mammalian equivalents protect cells from death. These central components biochemically intermingle in a ternary complex recently dubbed the “apoptosome.” The C elegans protein EGL-1 and its mammalian counterparts, pro-apoptotic members of the bcl-2 family, induce cell death by disrupting apoptosome interactions. Thus, EGL-1 may represent a primordial signal integrator for the apoptosome. Various biochemical processes including oligomerization, adenosine triphosphate ATP/dATP binding, and cytochrome c interaction play a role in regulating the ternary death complex. Recent studies suggest that cell death receptors, such as CD95, may amplify their suicide signal by activating the apoptosome. These mutual associations by core components of the suicide apparatus provide a molecular framework in which diverse death signals likely interface. Understanding the apoptosome and its cellular connections will facilitate the design of novel therapeutic strategies for cancer and other disease states in which apoptosis plays a pivotal role.
Keywords: apoptosis, apoptosome, cell death, death receptor
Introduction
Neoplasia literally means new growth and can emanate from either increased cell proliferation or decreased cell death. Malignant neoplasms are the consequence of accumulated mutations in genes that regulate the birth or death of cells [1]. Whereas mitosis is the organized process by which individual cells replicate, its antithesis, apoptosis, is the equally methodical process by which cells disintegrate.
Components of the apoptotic pathway are conserved throughout evolution in organisms as diverse as worms, flies, mammals, and possibly even plants [2–4]. Apoptosis plays a fundamental role in the development of multicellular organisms, eliminating excess cells and allowing for tissue remodeling, a vivid example of which is the assimilation of a tadpole's tail during metamorphosis into a frog. In nematodes, which are the subject of extensive developmental research, each cell can be accounted for and monitored, thus revealing a predictable pattern of programmed cell death. Similarly, in the developing vertebrate nervous system, numerous identical ganglia containing vast numbers of cells are generated, but only the ganglia serving the many muscles and sensory receptors of the arms and legs survive; in the remaining ganglia, supernumerary cells are faithfully eliminated. In essence, apoptosis is a form of premeditated cell murder that is genetically encoded and, generally, benefits the metazoan organism.
Derangements of apoptosis, however, do occur and can have deleterious consequences as exemplified by several human diseases including cancer, neurodegenerative disorders, and acquired immunodeficiency syndrome [5]. In the case of cancer, a neoplasm may form by an aberrant overproliferation of cells. Conversely, a defect in the cell death machinery may promote a net increase in cell survival and thus disrupt overall homeostasis, leading to cancer. In fact, the first component of the cell death pathway identified was bcl-2, which was discovered due to its role in B-cell malignancies, in which a chromosomal abnormality causes overexpression of the protein [6]. The identification of bcl-2 defined a new class of proto-oncogene, which, instead of enhancing cell proliferation, functioned to block cell death. Therefore, considering the role of apoptosis in human disease, and preeminently cancer, modulation of the suicide threshold may have immense therapeutic potential.
Recently, there has been a tremendous growth in the number of molecular players in the cell death arena. These include, for example, over 14 caspases [7], at least 23 bcl-2 family members [6], and 6 death receptor/ligand pairs (with a few decoys tossed in to make it interesting) [8,9]. What are we to make of this apparent cacophony of suicide regulators? In The Selfish Gene, Dawkins described multicellular organisms as “survival machines,” built and programmed by the self-seeking, self-replicating, and self-sufficient hereditary particles we commonly refer to as genes. Inspired by this concept, I use the metaphor of a machine to try to understand, describe, and categorize the inner workings of the apoptosis pathway [10,11]. In this regard, the cell death machine comprises an engine (or effectors), an ignition (or activators), and a set of brakes (or inhibitors).
In brief, it is becoming apparent that caspases, a family of proapoptotic cysteine proteases, make up the effector arm or engine of the cell death machine. They are expressed as zymogens, and upon protolytic processing, generate active dimeric species. The ignition or activators of the pathway are quite diverse and include cell-surface death receptors (such as CD95[Fas/APO-1]) or the apoptotic protease activating factor-1 (Apaf-1) (or its C elegans counterpart CED-4). And finally, the brakes of the cell death machine are primarily divided into two groups: 1) prosurvival members of the bcl-2 family, and 2) more direct caspase regulators such as the inhibitor of apoptosis (IAP) family. Although a survey of the various components of the death pathway is beyond the scope of this article, please refer to other recent review articles [6–8,11–13].
Although numerous suicide regulators continue to be discovered, all yearning for the spotlight, the “heart and soul” of the cell death machine has essentially been established. Pioneering genetic studies using the C elegans model system have opened up the field of apoptosis, once harboring phenomenology, to hard-core molecular biology [14–16]. These powerful genetic screens identified three genes, ced-3, ced-4, and ced-9, that are important in the regulation of nematode cell death. Mutations in ced-3 or ced-4 abolish all somatic cell deaths that normally occur during development, suggesting that these genes encode activators or effectors of the cell death apparatus. By contrast, ced-9 functions as a negative regulator of ced-3 and ced-4, preventing activation of the suicide response.
This cadre of cell death regulators adhere surprisingly well to the metaphor of a suicide machine. The pro-apoptotic CED-3 protein is the C elegans caspase, homolgous to the larger family of mammalian caspases, and functions as an effector of cell death in the nematode [16]. The CED-4 protein, although also a cold-blooded killer, functions as an activator of CED-3 and thus, in effect, activates the cell death machine [4,17,18]. Importantly, CED-4 possesses homology to the mammalian caspase activator, Apaf-1 [19]. And finally, the CED-9 protein, which is a homologue of bcl-2, functions as an inhibitor of cell death in the nematode [14,20]. Emphasizing the evolutionary conservation of the cell death pathway, bcl-2 can substitute functionally for CED-9 in the nematode, thus partially blocking developmental cell death [20].
Dying worms also provided some insight into a molecular order for the cell death pathway. Epistasis analyses in the nematode suggested that ced-9 functions genetically upstream of ced-3 and ced-4 [21]. Consistent with this notion, bcl-2 has been shown to function biochemically upstream of certain mammalian caspases [22,23]. Ectopic expression of CED-4 in C elegans neurons induces cell death, which requires CED-3 activity, suggesting that CED-4 may function upstream of CED-3 [24]. Furthermore, the CED-4 product is required for CED-9 to block apoptosis, suggesting that CED-9 may directly or indirectly regulate CED-4 activity [24].
Whereas it was known that CED-3, CED-4, and CED-9 were critical components of the cell death machinery, it was unclear how they were connected. Studies by several groups showed that these core constituents interact and function in a ternary death complex aptly named the apoptosome [17,18,22,25–27] (Figure 1, left). Similarly, in mammals, the respective counterparts of the C elegans proteins, caspase-9, Apaf-1, and bcl-2/xl, mutually associate [28,29] (Figure 1, right). This cadre of core components in both C elegans and mammals establishes a fundamental paradigm that is likely recapitulated in the suicide pathway. In the context of this review, the word apoptosome serves as a theoretical construct referring to the sometimes transient associations that constitute the ternary death apparatus, rather than the assembly of smaller protein components into a larger functional complex (i.e., ribosome, spliceosome, proteosome, etc.). Understanding the anatomy and regulation of the apoptosome is a primary objective of this article. In addition, how diverse death signals, specifically death receptors, engage the apoptosome are also discussed.
Figure 1.
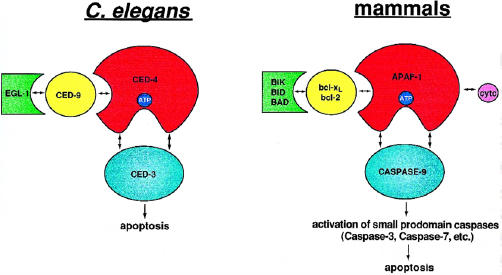
The apoptosome: A molecular framework for cell death C elegans (left) and mammals (right). In C elegans, CED-4 plays a crucial role as a molecular death switch, interacting with and assisting in the activation of the latent death protease CED-3. CED-9 presumably interacts with and negatively regulates CED-4 function. EGL-1 can disrupt the apoptosome by sequestering CED-9. With some added complexity, this primordial paradigm is likely recapitulated in mammals. Unlike C elegans, the mammalian death machinery is complicated by redundancy. Bcl-xl, bcl-2, and other death suppressors of the bcl-2 family inhibit apoptosis by negatively regulating Apaf-1. By contrast, death-promoting members of the bcl-2 family such as bid, bik, and bad antagonize the interactions between bcl-2/xl and Apaf-1. In the absence of negative regulation, Apaf-1 is free to engage and activate the death protease, caspase-9. Cytochrome c and ATP/dATP likely function as cofactors for the apoptosome. Once triggered, apical caspases such as caspase-9 activate downstream small prodomain caspases.
Anatomy of the Apoptosome
Studies by several groups using various systems showed a physical association between core components of the C elegans death machine. In transfected mammalian cells, insect cells, and yeast, CED-4 was shown to biochemically interact with CED-9 and its mammalian counterparts bcl-2 and bcl-xl [25–27,30,31] (Figure 1). There are four distinct bcl-2 homology (BH) regions that define the bcl-2 family [6] (Figure 2). CED-9 has distinct BH1, BH2, and BH4 domains, but not a BH3 domain [6] (Figure 2). Using two-hybrid analysis, Ottilie et al. showed that all 3 BH domains found in CED-9 are necessary for the CED-9:CED-4 binding [32]. Similarly, alteration of the BH2 domain or the BH4 domain in bcl-xl and bcl-2, respectively, blocks their ability to bind CED-4 [30,31]. Taken together, this would suggest that prosurvival bcl-2 family members require intact BH1, BH2, and BH4 domains to engage CED-4-like molecules.
Figure 2.
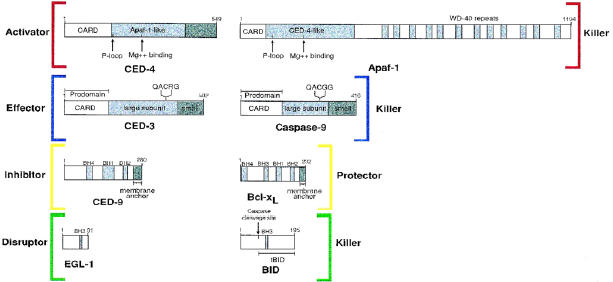
Schematic representation of apoptosome constituents.
Correlating with the biochemical interaction data, CED-9 and bcl-2/xl negatively regulate CED-4 function as assessed by CED-4-induced apoptosis or caspase activation [17,18,30]. Inactivating mutations in CED-9 or bcl-2/xl attentuated their ability to interact with CED-4. This linked an inducer of cell death, CED-4, with classic inhibitors of the suicide pathway. Importantly, this provided the first evidence that the bcl-2 family may mechanistically function by negatively regulating CED-4-like molecules. CED-9, like bcl-2 and bcl-xl, has a C-terminal hydrophobic tail and is thought to reside on intracellular membranes (including mitochondria). Thus it was shown that CED-9 regulates CED-4 subcellular localization, causing both molecules to colocalize on membranes [25,33].
Having seen that CED-4 interacted with CED-9, the next logical step was to determine whether CED-4 could also engage CED-3, the C elegans caspase. As with the CED-4:CED-9 interaction, a number of investigators reported the biochemical association between CED-4 and CED-3 [18,30,34] (Figure 1). Both molecules have a shared region of homology in their N terminus called a CARD domain (or caspase recruitment domain), which mediates homophilic associations between death adaptors and caspases (Figure 2). However, the interaction between CED-4 and CED-3 is somewhat complex with several sites of contact and cannot merely be explained by a CARD-CARD interaction [33]. As it was previously shown that CED-4 functioned upstream of CED-3 [24], it made good sense to assume that CED-4 may function as a direct activator of CED-3. Like the many mammalian caspases, CED-3 is expressed as a single polypeptide zymogen, and, upon proteolytic processing, gets converted into an active dimeric protease. Consistent with this notion, CED-4 was shown to both enhance CED-3-induced apoptosis and stimulate CED-3 processing [4,17,18]. This ascribed CED-4 activity could be negated by coexpression of CED-9.
Interestingly, CED-4 can interact independently and simultaneously with CED-3 and CED-9, forming a ternary death complex now known as the apoptosome [30] (Figure 1). This triad of death conspirators was also found to colocalize in cells [33], with CED-9 (and its hydrophobic C terminus) dictating a membrane localization. Mutual interactions between these core constituents provided a fundamental molecular framework for the cell death pathway.
Insight into the mammalian apoptosome comes not only from analogy to the C elegans model, but also from an in vitro apoptosis system used by Wang and coworkers [35]. Addition of dATP to cell-free extracts derived from naive Hela cells generated an apoptotic extract as assessed by its ability to induce DNA fragmentation, elicit morphological changes in nuclei, and activate the proapoptotic protease caspase-3. Biochemical fractionation of these extracts revealed 3 crucial apoptotic factors designated Apaf-1, Apaf-2, and Apaf-3. The first identity revealed was Apaf-2, which proved to be cytochrome c, now known as an important regulator of mammalian apoptosome function [35] (discussed later). Apaf-1 and Apaf-3 were revealed to be a mammalian CED-4 and CED-3 homologue, respectively [19,36]. Although the designation for Apaf-1 has not changed, Apaf-3, being a member of the growing caspase family, is more commonly referred to as caspase-9. As predicted by the molecular framework established in worms, an equivalent ternary complex existed in mammals, composed of caspase-9, Apaf-1, and bcl-xl [28] (Figure 1). One caveat to this, however, is that full-length Apaf-1 interacts with caspase-9 only in the presence of the apoptosome cofactors, cytochrome c and dATP [36] (discussed later). The observed coimmunoprecipitation of Apaf-1 and caspase-9 in vivo [28] suggests that these cofactors were present in this system.
What then happens when selected components of the mammalian apoptosome are disrupted? Inactivation of caspase-9 results in embryonic lethality and derangements in brain development caused by decreased apoptosis [37,38]. Caspase-9 -/- embryonic stem cells are resistant to various forms of apoptosis and fail to activate the downstream proapoptotic protease caspase-3. Furthermore, a number of groups have shown that a dominant negative version of capase-9, in which the catalytic cysteine is altered, blocks diverse death signals including ultraviolet radiation, death receptors, and proapoptotic bcl-2 family members [28,39].
Apaf-1-deficient mice had an even more profound apoptosis-defective phenotype. In addition to being embryonic lethal, inactivation of Apaf-1 caused a dramatic halt in developmental cell death, leading to neuronal hyperplasia, persistence of interdigital webs, dysregulation of eye development, and severe craniofacial abnormalities [40,41]. As in caspase-9-deficient mice, cells from Apaf-1-deficient mice were resistant to a variety of apoptotic stimuli. It is important to note, however, that the phenotype of the caspase-9 -/- mice is not identical to the Apaf-1 -/- mice. In fact, it appears that the defects in brain development seen in the caspase-9 -/- mice may represent a subset of the phenotype seen in the Apaf-1 -/- mice, in which developmental apoptosis is more significantly perturbed. An attractive hypothesis to explain this discrepancy is that Apaf-1 may function to activate more than one caspase, and thus its absence would be equivalent to missing several caspases. Of the caspases tested thus far, however, only caspase-9 has been shown to be autoproteolytically activated by Apaf-1 [39].
Although disrupting the killer genes (caspase-9 and Apaf-1) causes decreased apoptosis in mice, inactivating the prosurvival genes bcl-2 and bcl-xl causes inappropriate cellular suicide. Bcl-2-deficient mice develop normally but later in life exhibit increased lymphoid apoptosis among various other tissue abnormalities [6,42]. By contrast, inactivation of bcl-xl leads to embryonic lethality consequent to massive neuronal and erythroid cell apoptosis [43]. Taken together, inactivation of apoptosome components in mammals generally correlates with the phenotype predicted by nematode genetics. One caveat to this statement is the added complexity in mammals due to a redundancy inherent in their cell death pathway.
EGL-1 As a Signal Integrator for the Apoptosome
If prosurvival members of the bcl-2 family function by interacting with and inhibiting CED-4-like molecules, then how do proapoptotic bcl-2 members, such as bax, bik, and bid, function? These death-inducing bcl-2 family members all share a region of homology called the BH3 domain, which is important both for their interaction with bcl-2-like molecules and for their ability to trigger cell death. One hypothesis is that pro-apoptotic bcl-2 family members may function by interacting with bcl-2/xl and preventing it from binding CED-4-like molecules. By disengaging a CED-4-like killer, the death pathway would then be activated. In support of this hypothesis, expression of the death-promoting bcl-2 family members, bax, bak, or bik, was shown to abrogate the bcl-xl association with either CED-4 [30] or Apaf-1 [28]. A dominant negative version of caspase-9 inhibited bax-, bak-, and bik-induced cell death, suggesting that pro-apoptotic bcl-2 family members transmit a death signal via the apoptosome [28]. In sum total, this would suggest that BH3-containing killer molecules induce cell death by commandeering bcl-2/xl and thus releasing Apaf-1 to activate procaspase-9.
Proof of concept came with studies in the nematode in which EGL-1 was identified as a C elegans protein that contains a BH3 domain and is functionally similar to other proapoptotic bcl-2 family members [44] (Figure 2). Mutation of the egl-1 gene in worms blocks most if not all somatic cell death during nematode development. Gain of function mutations in egl-1 results in apoptosis of a specific neuronal population in nematodes [44]. Ectopic expression of EGL-1 in C elegans mechanosensory cells induces cell death, which requires CED-3 and CED-4 activity, suggesting that EGL-1 functions upstream of both these molecules. In addition, the CED-9 product is required for EGL-1 to induce apoptosis, intimating that EGL-1 may function by directly or indirectly inactivating CED-9. Taken together, this would suggest that EGL-1 functions upstream of the apoptosome in general, making it one of the earliest acting components of the nematode death apparatus thus far identified.
Biochemical interaction data on EGL-1 confirmed the predictions made genetically. EGL-1 was shown to physically associate with CED-9 [44,45]. By doing so, EGL-1 disrupts the apoptosome, thus interfering with the CED-9:CED-4 association, an activity that required the BH3 domain of EGL-1 [45]. This in effect activates the apoptosome by disengaging CED-4, allowing it to promote CED-3 processing and thus induce cell death (Figure 3). EGL-1 did not have independent associations with either CED-3 or CED-4 and functioned as an inducer of apoptosis only through its interactions with CED-9. In the presence of CED-9, CED-4 localizes to intracellular membranes. However, when EGL-1 is added to the mix, CED-4 (and presumably associated CED-3) gets shifted to the cytosol [45]. In a latent, inactive state, the ternary suicide complex is assembled on intracellular membranes (including on mitochondria). Expression of EGL-1 would then negate CED-9 function and thus liberate CED-4 and CED-3, allowing them to induce cell death.
Figure 3.
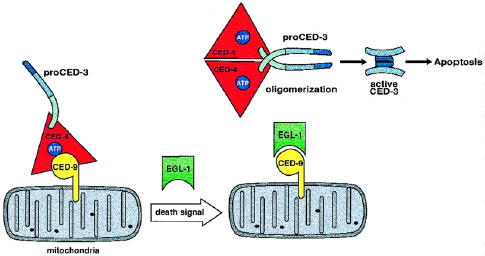
The C elegans apoptosome and its mechanism of activation. In the inactive state the ternary complex (CED-3, CED-4, and CED-9) is assembled on mitochondria. An unspecified death signal mobilizes EGL-1. In turn, EGL-1 binds CED-9, disengaging the apoptosome. CED-4 proceeds to oligomerize and induce the proximity and consequent activation of proCED-3 molecules.
The identification and characterization of EGL-1 in C elegans define an emerging class of cell death activators that function by disrupting the mutual associations present in the apoptosome. Importantly, EGL-1 appears to be a required element of the nematode suicide appartus. Worms with defective EGL-1 are unable to undego normal developmental cell death, suggesting that EGL-1 activity is crucial in unleashing the death potential of the apoptosome. The requirement of an intact BH3 domain makes EGL-1 functionally analogous to other proapoptotic bcl-2 family members such as bax, bid, or bik. Interestingly, egl-1 was found to function downstream of two genes involved in specifying nematode cell death fate, ces-1 and ces-2. [44]. Thus, this would suggest that EGL-1 may represent a primordial integrator of the suicide signal. It will be important to determine how these and other death-inducing signals funnel into the pathway of cell death regulated by EGL-1. If a similar analogy is applied to mammals, one could speculate that the growing family of proapoptotic bcl-2 family members may each transduce differing death signals. Bid, for example, has been shown to in transmit the suicide signal from cell death receptors to the apoptosome [46–48] (discussed later). The existence of numerous proapoptotic bcl-2 family members (or EGL-1-like molecules) emphasizes the redundancy present in mammalian apoptosis pathways.
An Induced Proximity Model for Apoptosome Function
Although it appears that EGL-1 functions to liberate the killer proteins (CED-3 and CED-4) from sequestration by the protective protein (CED-9), the mechanism by which the killers are then activated remains to be determined. Studies of the proximal signaling machinery of the cell surface death receptor CD95 (Fas/Apo-1) provided the answer [8,9]. In this pathway, CD95 gets aggregated extracellularly by its natural ligand, CD95L, or agonist antibody. This then recruits the cytoplasmic death adpator molecule FADD (Fas-associated death domain protein) to the cell surface signaling complex. Through homophilic DED (death effector domain) associations, FADD then oligomerizes and activates the proapoptoic protease caspase-8 (FLICE/MACH) (Figure 4, top). Aggregation of FADD increases the local concentration of caspase-8 in a microenvironment, harnessing the low proteolytic activity inherent in the caspase-8 zymogen to promote intermolecular processing [49]. In other words, by inducing a proximity among caspase-8 molecules, FADD likely enhances proteolytic self-activation of procaspase-8 [49,50].
Figure 4.
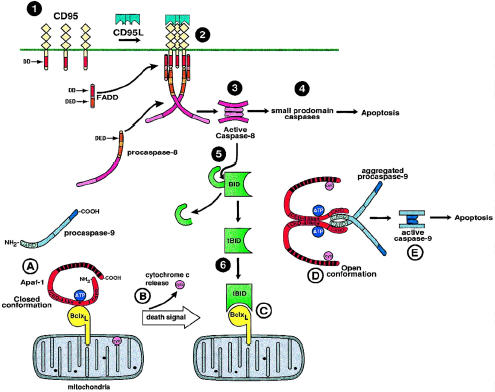
The mammalian apoptosome and its interface with the CD95 pathway. 1: Unactivated CD95(Fas/APO-1) expressed on the cell surface. DD, death domain. DED, death effector domain. 2: Formation of the CD95 death-signaling complex after trimerization by CD95L. Initially, the death adaptor FADD is recruited the cytoplasmic domain of CD95. FADD in turn induces proximity among procaspase-8 molecules, leading to 3: proteolytic activation. 4: Active caspase-8 then cleaves small-prodomain caspases, leading to cell death. 5: Alternatively, caspase-i can cleave the bcl-2 homologue Bid. 6: Once Bid is proteolytically cleaved to tBid (truncated) it can bind bcl-xl. In the inactive state (A), Apaf-1 is in a closed conformation bound to bcl-xl. The asterisk represents the P-loop motif. CARD, caspase recruitment domain. Upon death stimulus, cytochrome c is released from mitochondria (B). Likewise, death signals mobilize proapoptotic members of the bcl-2 family, such as Bid, to disrupt the latent apoptosome (C). Cytochrome c and/or dATP/ATP hydrolysis induces a conformational change in Apaf-1, negating the inhibitory effect of the WD-40 repeats (black boxes in the Apaf-1 molecule). In the open conformation and liberated of bcl-xl inhibition, Apaf-1 is free to self-associate via its CED-4-like domain (D). CARD-CARD associations link Apaf-1 to procaspase-9. Oligomerization of Apaf-1 aggregates procaspase-9, leading to its proteolytic activation (E) and consequent induction of cell death.
This paradigm for caspase activation is used by and likely originates from the apoptosome. Studies that used the C elegans components showed that CED-4 can self-associate through amino acids 171–435 [51]. CED-9 functioned to prevent CED-4 oligomerization. As the caspase CED-3 is tethered to CED-4, oligomerization presumably induces proximity of CED-3 zymogens favoring proteolytic self-activation (see Figure 3). Mutations that abolished CED-4 oligomerization also prevented CED-4-mediated activation of CED-3.
A somewhat more complex scenario exists in mammals. Full-length Apaf-1 requires dATP and cytochrome c to promote caspase-9 activation in vitro [36]. Unlike CED-4, the C-terminal half of Apaf-1 contains a stretch of 12 WD-40 repeats, which is a motif often involved in protein-protein interactions (see Figure 2). A truncated Apaf-1 molecule (N-Apaf-1, a.a.1-559), in which the WD-40 repeats are absent, constitutively activates caspase-9 in vitro (without cytochrome c and dATP dependence) [39,52]. This truncated Apaf-1 was shown to oligomerize via its CED-4 homology domain and, analogous to CED-4, was thought to activate procaspase-9 by inducing proximity (Figure 4). Disruption of the self-association domain in Apaf-1 abrogated its ability to activate procaspase-9. The association between Apaf-1 and caspase-9 is mediated by CARD-CARD interactions [36] (see Figure 2). Expressed alone, the Apaf-1 CARD domain, which does not oligomerize, failed to induce caspase-9 processing [39,52]. As predicted, however, if this CARD domain is aggregated (by using various oligomerization systems), procaspase-9 can then be efficiently activated [52].
Whereas N-Apaf-1 appears to function similarly to other oligomerizing molecules such as FADD or CED-4, the role of the C-terminal half of Apaf-1, which encompasses the WD-40 repeats, remains to be deciphered (see Figure 2). Unlike the N-Apaf-1, full-length Apaf-1 fails to interact with and activate caspase-9 alone but requires the presence of dATP and cytochrome c [36]. This suggested that the WD-40 repeats may function as a negative regulator of Apaf-1. Consistent with this, Hu et al. show that the WD-40 repeats bind to and negatively regulate N-Apaf-1, negating its ability to oligomerize or activate procaspase-9 [52] (Figure 4). Although it is evident that the WD-40 repeats bind the N terminus of Apaf-1, it remains to be determined whether they function competitively, directly binding to the Apaf-1 oligomeri0zation domain. Alternatively, by binding to an adjacent site, they may mask the oligomerization domain via steric hinderance. As suggested by Hu et al., these findings support an attractive model in which Apaf-1 exists in 2 distinct conformations [52] (Figure 4). In the closed conformation, the WD-40 repeats bind to the N terminus of Apaf-1, keeping it in an inactive, latent state. By contrast, in the open conformation, the N terminus of Apaf-1 is liberated and can subsequently self-oligomerize and aggregate pro-caspase-9 (Figure 4). Interestingly, because dATP and cytochrome c are required for full-length Apaf-1 activity, they may serve as important cofactors that facilitate the shift from the closed to open conformation.
Cofactors of Apoptosome Function: ATP/dATP
The three known CED-4 sequences (C elegans, C briggsae, and C vulgaris) and Apaf-1 contain a nucleotide-binding phosphate-loop, (P-loop) motif characteristic of adenosine triphosphatases (ATPases) and guanosine triphosphatases (GTPases) [53]. In addition, downstream of each P-loop exists a potential Mg2+ binding motif typical of a large class of ATPases [53] and consisting of two aspartic acid residues preceded by 4 bulky hydrophobic residues (see Figure 2). Interestingly, database searches with CED-4 and Apaf-1 identified a class of plant resistance (R) gene products [2,4]. These R gene products are important components of the hypersensitive response, which is a mechanism induced by certain plant pathogens and involving cell death to restrict pathogen spread [54]. In addition to being homologous in the P-loop and Mg2+ binding domain, these plant resistance response genes (including PRF, RPM1, and L6) also have several other short conserved regions homologous to CED-4/Apaf-1 [2]. Of note, a number of these R gene products have C terminal leucine-rich repeats, which, analogous to the WD-40 repeats of Apaf-1, are involved in protein-protein interactions [2]. The presence of a conserved ATPase domain in CED-4/Apaf-1 and the R response proteins suggests there may be mechanistic similarities between the suicide apparatus in animals and plants. In support of this somewhat stunning notion, a caspase-like activity has been identified in hypersensitive response-related plant cell death [3].
Whereas CED-4 has been shown to bind ATP analogues [4], no one has been able to formally show ATPase activity for the molecule. Mutation of the P-loop in CED-4 abolishes its ability to induce cell death and stimulate CED-3 processing, verifying the functional relevance of this domain [4,17,26,30]. The CED-4 P-loop mutants fail to both bind CED-9 and interact with the prodomain (or CARD) of CED-3 [33]. Interestingly, altering the P-loop in CED-4, does not affect CED-4 self-association [51]. The P-loop, however, does appear to be an important point of regulation in the CED-4 molecule.
By contrast to CED-4, alteration of the P-loop in N-Apaf-1 abolishes its ability to oligomerize [52]. The reason for this difference is unclear but may have to do with varied levels of expression between the two proteins in mammalian trasfection systems. Interestingly, mutation of the P-loop abrogates the activity of the mammalian expressed protein [52] but only slightly diminishes the activity of the bacterially expressed protein [39]. This discrepancy could be due to the higher expression levels achieved in bacteria, forcing aggregation independent of self-association. Thus at higher expression levels, N-Apaf-1 may bypass the requirement of nucleotide binding and still oligomerize. This also suggests that the nucleotide-binding pocket of Apaf-1, which spans the CED-4-like homology region, may contribute to the structure of the oligomerization surface (see Figure 2).
What is the role of dATP or ATP hydrolysis in apoptosome function? In the original in vitro model of apoptosis [35], dATP was found to be a required component. Furthermore, studies suggest that dATP or ATP hydrolysis is necessary for caspase-9 activation by full-length Apaf-1 [36], but not by the truncated derivative N-Apaf-1 [39,52]. Thus, ATP/dATP hydrolysis could provide the energy required to overcome negative regulation of Apaf-1 by the WD-40 repeats, promoting an open conformation over a closed one [52]. Alternatively, ATP/dATP hydrolysis may be needed to disengage the proteolytically activated caspase-9 from the apoptosome. Consistent with this hypothesis, Srinivasula et al. demonstrated that N-Apaf-1-activated caspase-9 remains bound to N-Apaf-1 and is unable to cleave its down-stream target caspase-3 [39].
Cofactors of Apoptosome Function: Cytochrome c
Discovery of cytochrome c as a component of the cell death pathway sent shock waves through the field of apoptosis research [35]. Traditionally thought of as an integral part of electron transport chain, cytochrome c resides in the space between the inner and outer mitochondrial membranes. During apoptosis, cytochrome c is translocated, almost entirely, to the cell cytosol by a thus-far unknown mechanism. Once in the cytosol, cytochrome c serves as important instigator of the cell death pathway by binding to and enabling the central apoptosome component Apaf-1 [19]. Like dATP, cytochrome c is not a requirement for the functionality of the constitutively active N-Apaf-1 [39]. This would suggest that, alone or in combination with dATP, cytochrome c facilitates the conversion of Apaf-1 from the inactive, closed conformation to the active, open conformation, freeing the N terminus of Apaf-1 from negative regulation by the WD-40 repeats (Figure 4). Where cytochrome c precisely interacts with Apaf-1 is unclear, but a purely speculative model would suggest it interacts with and negates the function of the WD-40 repeats (Figure 4). Cytochrome c, which is positively charged, physically interacts with its electron transport partners cytochrome reductase and cytochrome oxidase, which are negatively charged [55]. It would be interesting to determine whether analogous electrostatic interactions exist between cytochrome c and Apaf-1.
The finding that cytochrome c is an important apoptosis-mediator opens up a can of worms in the study of cell death (no pun intended). This electron carrier is present in all organisms that have mitochondrial respiratory chains: plants, animals, and eucaryotic organisms. It is strictly conserved among various species, with 26 of 104 residues being invariant for over 1.5 billion years (before the divergence of plants and animals) [55]. In terms of function, cytochrome c from any eukaryotic species can interact with any cytochrome oxidase [55]. Thus it is intriguing that cytochrome c from several vertebrate species, but not from yeast, was able to initiate an in vitro system of apoptosis [56]. It is unclear, whether C elegans cytochrome c is released during apoptosis and whether it binds to CED-4. Furthermore the electron transport activity of cytochrome c is not required for its proapoptoic activity [56].
Cytochrome c, while still being translocated to the cytosol, is unable to trigger the apoptotic pathway in the absence of Apaf-1 or caspase-9 [38,41], suggesting that it functions upstream of these molecules. Furthermore, in the presence of the broad spectrum caspase inhibitor z-VAD-fmk, cytochrome c is still released from mitochondria [57]. Hence, although key components of the apoptosome are missing or inactivated, cytochrome c can still be liberated, intimating that death signals in general must have some cytochrome c-releasing mechanism, independent of the apoptosome. Importantly, overexpression of bcl-2 has been shown to block cytochrome c release in response to a variety of death signals [58,59]. How bcl-2 manages to block cytochrome c release and keep the apoptosome inactive is unclear. Some clues may come from studies examining the proapoptotic bcl-2 family member bid (discussed in more detail in the following section).
Death Receptors Interface With the Apoptosome
As described earlier, CD95 is a prototypic member of the death receptor family and signals apoptosis via engagement of the death adaptor FADD which in turn recruits the death protease caspase-8. Once caspase-8 is activated it can then proteolytically activate a cascade of caspases leading to the phenotype of apoptosis (Figure 4). Recently, several groups identified Bid, a previously characterized proapoptotic molecule of bcl-2 family, as a novel substrate for caspase-8. Bid, a bcl-2 interacting protein, is a mammalian counterpart to C elegans EGL-1 and one of the “BH3-only” death-promoting bcl-2 family members [46,47,60]. Interestingly, when cytosolic Bid is cleaved by caspase-8 it is translocated to mitochondria and mediates the release of cytochrome c (Figure 4). Bid-induced apoptosis is inhibited by bcl-2, bcl-xl, and a dominant negative derivative of caspase-9.
When recombinant truncated Bid (tBid) is added to purified mitochondria in vitro, cytochrome c release is induced [46]. The mechanism by which Bid achieves this is unclear. Some have suggested that gross mitochondrial swelling and permeability transition, which occur in the late phases of the apoptotic phenotype, may be the cause of cytochrome c release [61–63]. In the case of Bid, however, this does not occur, because cytochrome c can be liberated without the loss of mitochondrial membrane potential [46,47]. It will be interesting to determine whether this phenomena of cytochrome c release in vitro, can be recapitulated by other death-promoting bcl-2 family members. In addition, Bid can presumably also function like EGL-1 (and killer bcl-2 family members) to disrupt the apoptosome. By binding to bcl-2/xl, Bid likely disengages the apoptosome, allowing Apaf-1 to oligomerize and activate caspase-9 (Figure 4).
Bid needs to be cleaved in order to gain full apoptotic potential [46]. The C terminal fragment Bid (Figure 2), which contains the BH3 domain, then gets recruited to mitochondria. Previous studies have shown that Bax similarly gets translocated to mitochondria after a death stimulus [64]. This suggests that death-promoting members of the bcl-2 family (EGL-1 included) have an inactive state which has to undergo a molecular alteration, either removal of an inhibitory domain or conformational change, in order to be activated.
Bid links the cell surface death receptors to mitochondria and the apoptosome. Death receptors such as CD95 likely signal apoptosis by 2 distinct pathways. One is by activation of caspase-8, which in turn activates downstream caspases by proteolytic cleavage ultimately resulting in cell suicide. Alternatively, caspase-8 can cleave Bid, which functions to release cytochrome c and disrupt the apoptosome resulting in caspase-9 activation and cell death (Figure 4). Thus, in the former scenario, death receptors use Bid to amplify the suicide signal. This was initially suggested by in vitro studies that showed that caspase-8-induced apoptosis can be amplified through mitochondrial release of cytochrome c [65]. Depending on the cell type, one death receptor pathway may be preferred over the other [66]. Bcl-2 and bcl-xl inhibit death receptor-induced apoptosis in some cells but not in others [66]. One would predict that bcl-2 and bcl-xl may exert their protective effect in cells where the Bid signaling pathway predominates. For example, in MCF7 breast carcinoma cells, bcl-xl inhibits CD95- and tumor necrosis factor receptor 1-induced apoptosis downstream of caspase-8 activation [67], presumably by inhibiting the ability of Bid to disengage the apoptosome and release cytochrome c.
Conclusions
Apoptosis as a biological process was first defined years ago by Wyllie and colleagues. Until recently, little was known about the molecular mechanism behind this elegant phenomena. Over the past few years many important advances have been made in understanding the inner workings of the suicide apparatus.
The nematode model system laid the foundation for much of what is known about the cell death pathway today. By using this powerful genetic screen, three core components of the suicide apparatus were identified CED-3, CED-4 and CED-9. The caspase CED-3 is the effector of cell death in the nematode and acts as a molecular executioner. A coconspirator to murder, CED-4, functions to activate CED-3. By contrast, the law enforcement authority, CED-9, inhibits the death pathway by keeping CED-4 in check. These three components mutually associate in the molecular dance of death called the apoptosome. EGL-1, a new suspect in this murder mystery, functions by binding CED-9 and disrupting the interactions of the ternary death complex. Thus, in the C elegans paradigm of death, EGL-1 can be mobilized by various unspecified suicide signals. By binding CED-9, EGL-1 disrupts the latent, inactive apoptosome. Once CED-4 is disengaged from CED-9-inhibition, it oligomerizes and induces proximity among CED-3 molecules causing auto-proteolytic activation and consequent cell death.
If the cell death pathway is conserved throughout evolution, then it is reasonable to postulate that the primordial suicide paradigm established in nematodes would be recapitulated in higher organisms. The mammalian apoptosome is made of a similar cast of characters including an effector, caspase-9; an activator, Apaf-1; and an inhibitor, bcl-2/xl. Death-promoting members of the bcl-2 family such as bax, bid, and bik likely function as disruptors of the apoptosome, analogous to C elegans EGL-1. In summary, bcl-2 maintains an inactive, latent apoptosome. Upon death stimulus, a bcl-2 antagonist presumably liberates Apaf-1 and releases cytochrome c from mitochondria. Once disengaged, Apaf-1 is free to oliogmerize and thus aggregate and activate parocaspase-9, ultimately resulting in cell suicide.
Whereas the basic molecular framework has been installed, numerous questions still lurk in the background. For example, what is the mechanism of cytochrome c release from mitochondria? How do bcl-2 antagonists, such as Bid, mediate release of cytochrome c, in addition to disrupting the apoptosome? Do the other pro-apoptotic members of the bcl-2 family mimic the activity of Bid? What exactly is the role of cytochrome c and ATP/dATP in apoptosome function? These and other questions will continue to fuel the morbid curiosity of cell death researchers.
In the big picture, however, it will be important to determine how diverse death signals impinge upon the “heart and soul” of the cell death machine. In C elegans it is unclear how the regulators of cell death fate engage EGL-1 and thus activate the apoptosome. Although a number of apoptotic triggers are well characterized, including death receptors, growth factor withdrawal, p53 and chemotherapeutics, it is unclear how they are connected to the core machinery. A model for how death receptors interface with the apoptosome has already been proposed: activation of the bcl-2 antagonist, Bid. It is tempting to speculate that each of the pro-apopotic bcl-2 family members may couple to a different upstream death signaling pathway, thus acting as distinct signal integrators. Characterizing the precise molecular circuitry of the cell death pathway will facilitate the design of rational therapies for cancer and other human disease states in which apoptosis plays an important role.
Acknowledgements
Space limitations precluded comprehensive referencing for all work discussed. I thank Robin Kunkel for assistance in the preparation of the figures for this manuscript; Peter Ward for encouragement and support of this project, and Al Rehemtulla, Dan Hamstra, Neelam Taneja, Dot Oyedijo, Murthy Shaniah, and Uttara Prasad for helpful suggestions.
References
- 1.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1988;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 2.van der Biezen EA, Jones JD. The NB-ARC domain: A novel signalling motif shared by plant resistance gene products and regulators of cell death in animals. Curr Biol. 1998;8:R226–R227. doi: 10.1016/s0960-9822(98)70145-9. [letter] [DOI] [PubMed] [Google Scholar]
- 3.del Pozo O, Lam E. Caspases and programmed cell death in the hypersensitive response of plants to pathogens. Curr Biol. 1998;8:R896. doi: 10.1016/s0960-9822(07)00555-6. [In Process Citation] [DOI] [PubMed] [Google Scholar]
- 4.Chinnaiyan AM, Chaudhary D, O'Rourke K, Koonin EV, Dixit VM. Role of CED-4 in the activation of CED-3. Nature. 1997;388:728–729. doi: 10.1038/41913. [letter] [see comments] [DOI] [PubMed] [Google Scholar]
- 5.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 6.Adams JM, Cory S. The Bcl-2 protein family: Arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 7.Thornberry NA, Lazebnik Y. Caspases: Enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 8.Ashkenazi A, Dixit VM. Death receptors: Signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 9.Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME. Apoptosis signaling by death receptors. Eur J Biochem. 1998;254:439–459. doi: 10.1046/j.1432-1327.1998.2540439.x. [DOI] [PubMed] [Google Scholar]
- 10.Chinnaiyan AM, Dixit VM. The cell-death machine. Curr Biol. 1996;6:555–562. doi: 10.1016/s0960-9822(02)00541-9. [DOI] [PubMed] [Google Scholar]
- 11.Raff M. Cell suicide for beginners. Nature. 1998;396:119–122. doi: 10.1038/24055. [news] [DOI] [PubMed] [Google Scholar]
- 12.Evan G, Littlewood T. A matter of life and cell death. Science. 1998;281:1317–1322. doi: 10.1126/science.281.5381.1317. [DOI] [PubMed] [Google Scholar]
- 13.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 14.Hengartner MO, Ellis RE, Horovitz HR. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature. 1992;356:494–499. doi: 10.1038/356494a0. [DOI] [PubMed] [Google Scholar]
- 15.Yuan J, Horvitz HR. The Caenorhabditis elegans cell death gene ced-4 encodes a novel protein and is expressed during the period of extensive programmed cell death. Development. 1992;116:309–320. doi: 10.1242/dev.116.2.309. [DOI] [PubMed] [Google Scholar]
- 16.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1b-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 17.Seshagiri S, Miller L. Caenorhabditis elegans CED-4 stimulates CED-3 processing and CED-3-induced apoptosis. Curr Biol. 1997;7:455–460. doi: 10.1016/s0960-9822(06)00216-8. [DOI] [PubMed] [Google Scholar]
- 18.Wu D, Wallen HD, Inohara N, Nunez G. Interaction and regulation of the Caenorhabditis elegans death protease CED-3 by CED-4 and CED-9. J Biol Chem. 1997;272:21449–21454. doi: 10.1074/jbc.272.34.21449. [DOI] [PubMed] [Google Scholar]
- 19.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 20.Hengartner MO, Horvitz HR. C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell. 1994;76:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- 21.Ellis RE, Yuan J, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991;7:663–698. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- 22.Chinnaiyan AM, Orth K, O'Rourke K, Duan H, Poirier GG, Dixit VM. Molecular ordering of the cell death pathway: bcl-2 and bcl-xl function upstream of the CED-3-like apoptotic proteases. J Biol Chem. 1996;271:4573–4576. doi: 10.1074/jbc.271.9.4573. [DOI] [PubMed] [Google Scholar]
- 23.Armstrong RC, Aja T, Xiang J, Gaur S, Krebs JF, Hoang K, Bai X, Korsmeyer SJ, Karanewsky DS, Fritz LC, Tomaselli KJ. Fas-induced activation of the cell death-related protease CPP32 Is Inhibited by Bcl-2 and ICE family protease inhibitors. JBC. 1996;271:16850–16855. doi: 10.1074/jbc.271.28.16850. [DOI] [PubMed] [Google Scholar]
- 24.Shaham S, Horvitz HR. Developing Caenorhabditis elegans neurons may contain both cell-death protective and killer activities. Genes Dev. 1996;10:578–591. doi: 10.1101/gad.10.5.578. [DOI] [PubMed] [Google Scholar]
- 25.Wu D, Wallen HD, Nunez G. Interaction and regulation of subcellular localization of CED-4 by CED-9. Science. 1997;275:1126–1129. doi: 10.1126/science.275.5303.1126. [see comments] [DOI] [PubMed] [Google Scholar]
- 26.James C, Gschmeissner S, Fraser A, Evan GI. CED-4 induces chromatin condensation in Schizosaccharaomyces pombe and is inhibited by direct physical association with CED-9. Curr Biol. 1997;7:246–252. doi: 10.1016/s0960-9822(06)00120-5. [DOI] [PubMed] [Google Scholar]
- 27.Spector MS, Desnoyers S, Hoeppner DJ, Hengartner MO. Interaction between the C. elegans cell-death regulators CED-9 and CED-4. Nature. 1997;385:653–656. doi: 10.1038/385653a0. [DOI] [PubMed] [Google Scholar]
- 28.Pan G, O'Rourke K, Dixit VM. Caspase-9, Bcl-Xl, and Apaf-1 form a ternary complex. J Biol Chem. 1998;273:5841–5845. doi: 10.1074/jbc.273.10.5841. [DOI] [PubMed] [Google Scholar]
- 29.Hu Y, Benedict MA, Wu D, Inohara N, Nunez G. Bcl-Xl interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc Natl Acad Sci USA. 1998;95:4386–4391. doi: 10.1073/pnas.95.8.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chinnaiyan AM, O'Rourke K, Lane BR, Dixit VM. Interaction of CED-4 with CED-3 and CED-9: a molecular framework for cell death. Science. 1997;275:1122–1126. doi: 10.1126/science.275.5303.1122. [DOI] [PubMed] [Google Scholar]
- 31.Huang DC, Adams JM, Cory S. The conserved N-terminal BH4 domain of Bcl-2 homologues is essential for inhibition of apoptosis and interaction with CED-4. EMBO J. 1998;17:1029–1039. doi: 10.1093/emboj/17.4.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ottilie S, Wang Y, Banks S, Chang J, Vigna NJ, Weeks S, Armstrong RC, Fritz LC, Oltersdorf T. Mutational analysis of of the interacting cell death regulators CED-9 and CED-4. Cell Death and Differentiation. 1997;4:526–533. doi: 10.1038/sj.cdd.4400288. [DOI] [PubMed] [Google Scholar]
- 33.Chaudhary D, O'Rourke K, Chinnaiyan AM, Dixit VM. The death inhibitory molecules CED-9 and CED-4L use a common mechanism to inhibit the CED-3 death protease. J Biol Chem. 1998;273:17708–17712. doi: 10.1074/jbc.273.28.17708. [DOI] [PubMed] [Google Scholar]
- 34.Irmler M, Hofmann K, Vaux D, Tschopp J. Direct physical interaction between the Caenorhabditis elegans ‘death proteins’ CED-3 and CED-4. FEBS Lett. 1997;406:189–190. doi: 10.1016/s0014-5793(97)00271-8. [DOI] [PubMed] [Google Scholar]
- 35.Liu X, Kim CN, Yang J, Temmerson R, Wang X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 36.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 37.Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 38.Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- 39.Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- 40.Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- 41.Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- 42.Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycyctic kidneys and hypopigmented hair. Cell. 1993;75:229–240. doi: 10.1016/0092-8674(93)80065-m. [DOI] [PubMed] [Google Scholar]
- 43.Motoyama N, Wang F, Roth KA, Sawa H, Negishi I, Senju S, Zhang Q, Fujii S, Loh D. Massive cell death of immature hematopoeitic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- 44.Conradt B, Horvitz HR. The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell. 1998;93:519–529. doi: 10.1016/s0092-8674(00)81182-4. [DOI] [PubMed] [Google Scholar]
- 45.del Peso L, Gonzalez VM, Nunez G. Caenorhabditis elegans EGL-1 disrupts the interaction of CED-9 with CED-4 and promotes CED-3 activation. J Biol Chem. 1998;273:33495–33500. doi: 10.1074/jbc.273.50.33495. [In Process Citation] [DOI] [PubMed] [Google Scholar]
- 46.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 47.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 48.Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-Xl prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999;274:1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- 49.Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM. An induced proximity model for caspase-8 activation. J Biol Chem. 1998;273:2926–2930. doi: 10.1074/jbc.273.5.2926. [DOI] [PubMed] [Google Scholar]
- 50.Martin DA, Siegel RM, Zheng L, Lenardo MJ. Membrane oligomerization and cleavage activates the caspase-8 (FLICE/MACHalpha1) death signal. J Biol Chem. 1998;273:4345–4349. doi: 10.1074/jbc.273.8.4345. [DOI] [PubMed] [Google Scholar]
- 51.Yang X, Chang HY, Baltimore D. Essential role of CED-4 oligomerization in CED-3 activation and apoptosis. Science. 1998;281:1355–1357. doi: 10.1126/science.281.5381.1355. [see comments] [DOI] [PubMed] [Google Scholar]
- 52.Hu Y, Ding L, Spence DM, Nunez G. WD-40 repeat region regulates apaf-1 self-association and procaspase-9 activation. J Biol Chem. 1998;273:33489–33494. doi: 10.1074/jbc.273.50.33489. [In Process Citation] [DOI] [PubMed] [Google Scholar]
- 53.Gorbalenya AE, Koonin EV. Viral proteins containing the purine NTP-binding sequence pattern. Nucleic Acids Res. 1989;17:8413–8440. doi: 10.1093/nar/17.21.8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Greenberg JT. Programmed cell death: A way of life for plants. Proc Natl Acad Sci USA. 1996;93:12094–12097. doi: 10.1073/pnas.93.22.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stryer L. Biochemistry. New York: WH Freeman & Co; 1988. [Google Scholar]
- 56.Kluck R, Martin S, Hoffman B, Zhou J, Green D, Newmeyer D. Cytochrome c activation of CPP32-like proteolysis plays a critical tole in Xenopus cell-free apoptosis system. EMBO J. 1997;16:4639–4649. doi: 10.1093/emboj/16.15.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bossy-Wetzel E, Newmeyer D, Green D. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochon-drial transmembrane depolarization. EMBO J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 59.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng T-I, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 60.Gross A, Yin X-M, Wang K, Wei M, Jockel J, Milliman C, Erdjument-Bromage H, Tempst P, Korsmeyer S. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, Bcl-Xl prevents this release but not tumor necrosis factor R1/Fas Death. J Biol Chem. 1999;274:1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- 61.Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [see comments] [DOI] [PubMed] [Google Scholar]
- 62.Susin SA, Zamzami N, Kroemer G. Mitochondria as regulators of apoptosis: doubt no more. Biochim Biophys Acta. 1998;1366:151–165. doi: 10.1016/s0005-2728(98)00110-8. [DOI] [PubMed] [Google Scholar]
- 63.Susin SA, Zamzami N, Castedo M, Daugas E, Wang HG, Geley S, Fassy F, Reed JC, Kroemer G. The central executioner of apoptosis: Multiple connections between protease activation and mitochondria in Fas/APO-1/CD95- and ceramide-induced apoptosis. J Exp Med. 1997;186:25–37. doi: 10.1084/jem.186.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gross A, Jockel J, Wei MC, Korsmeyer SJ. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 1998;17:3878–3885. doi: 10.1093/emboj/17.14.3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kuwana T, Smith JJ, Muzio M, Dixit V, Newmeyer DD, Kornbluth S. Apoptosis induction by caspase-8 is amplified through the mitochondrial release of cytochrome c. J Biol Chem. 1998;273:16589–16594. doi: 10.1074/jbc.273.26.16589. [DOI] [PubMed] [Google Scholar]
- 66.Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Srinivasan A, Li F, Wong A, Kodandapani L, Smidt R, Jr, Krebs JF, Fritz LC, Wu JC, Tomaselli KJ. Bcl-xl functions down-stream of caspase-8 to inhibit Fas- and tumor necrosis factor receptor 1-induced apoptosis of MCF7 breast carcinoma cells. J Biol Chem. 1998;273:4523–4529. doi: 10.1074/jbc.273.8.4523. [DOI] [PubMed] [Google Scholar]