

Abstract
Reactive oxygen species contribute to neuronal death following cerebral ischemia. Prior studies using transgenic animals have demonstrated the neuroprotective effect of the antioxidant, copper/zinc superoxide dismutase (SOD1). In this study we investigated whether SOD1 overexpression using gene therapy techniques in non-transgenic animals would increase neuronal survival. A neurotropic, herpes simplex virus-1 (HSV-1) vector containing the SOD1 gene was injected into the striatum either before or after transient focal cerebral ischemia. Striatal neuron survival at two days was improved by 52% when vector was delivered 12–15 hours prior to ischemia and by 53% when vector delivery was delayed 2 hours following ischemia. These data add to the growing literature which suggests that an antioxidant approach, perhaps by employing gene therapy techniques, may be beneficial in the treatment of stroke. (According to the guidline, it is mandatory to include classification terms here. But I did not find them –HZ)
Keywords: copper, zinc superoxide dismutase; gene therapy; stroke, focal ischemia, cerebral ischemia
Copper/zinc superoxide dismutase (SOD1) forms part of the antioxidant machinery which detoxifies reactive oxygen species (ROS) under normal physiologic conditions. Superoxide anion is converted by SOD to hydrogen peroxide, which is subsequently detoxified to water by glutathione peroxidase (GPX) or catalase [1]. Reperfusion following cerebral ischemia leads to an overproduction of ROS and consumption of endogenous antioxidants. Excessive levels of ROS have been implicated in cell death through the direct destruction of cellular macromolecules, leading to necrotic cell death. ROS have also been postulated to participate in signaling mechanisms which result in apoptotic cell death [1]. Neurons, in particular, are vulnerable to ROS damage due to their relatively low levels of endogenous antioxidants [2]. Therefore, much research has focused on antioxidant approaches to improve cerebral ischemic injury.
Prior studies have been contradictory regarding the possible neuroprotective function of SOD1. Studies using transgenic animal models have clearly established a beneficial role of SOD1 in adult ischemia/reperfusion injury [3,4], but not in a model of permanent focal ischemia [5]. SOD1 overexpression worsens injury in a neonatal model of ischemia, secondary to the accumulation of hydrogen peroxide resulting from decreased GPX activity [6,7]. This highlights the limitations of transgenic studies, where lifelong overexpression of the gene of interest may have confounding effects on related enzymatic pathways. Furthermore, in vitro studies of SOD1 overexpression using HSV-1 gene therapy demonstrated increased neuronal damage in a hippocampal culture model of excitotoxic injury and showed no protection under oxygen-glucose deprivation (OGD) conditions [8].
In the current study, we investigated the possible neuroprotective role of SOD1 in vivo using gene therapy techniques. We previously demonstrated the protective effect of GPX and catalase using this model [9,10]. We hypothesized that overexpression of SOD1 using a herpes simplex viral vector protects striatal neurons subjected to transient focal cerebral ischemia.
Amplicon plasmids were constructed containing pα22sβgalα4s (control vector containing Eschericia coli lacZ reporter gene) or pα22sβgalα4SOD1 (vector with bipromoter region expressing lacZ and SOD1) as described previously [8,11]. The plasmid was inserted into the HSV-1 amplicon by standard methods [12]. Viral titers for control vector ranged from 1.2 x 107 to 2.1 x 107/mL with a vector: helper ratio 1:1.3. For SOD1-expressing vector, viral titers ranged from 9.5 x 105 to 1.2 x 107/mL with a vector: helper ratio 1:1.5.
All animal protocols were approved by the Stanford University Administrative Panel of Laboratory Animal Care. Forty-two male Sprague-Dawley rats (Charles River, Wilmington, MA) weighing 290 to 320 grams were included in this study. Under isoflurane anesthesia, rats underwent transient focal cerebral ischemia using an intraluminal suture as previously described by our laboratory [9,10,13]. Briefly, the common carotid artery was ligated, an arteriotomy was made, and a 3.0 monofilament suture with a rounded tip was advanced through the internal carotid artery to the origin of the middle cerebral artery (MCA), thereby occluding the MCA. The suture was left in place for one hour and then removed to allow reperfusion. Temperature, heart rate, respirations, and depth of anesthesia were monitored during the procedure and kept in physiological ranges. Vector was delivered into the striatum 12–15 hours prior to ischemia or delayed 2 hours after the onset of ischemia. Under isoflurane anesthesia, the rat was placed in a stereotaxic frame and the skull was exposed. Two burr holes were drilled bilaterally with the coordinates anteroposterior 0 mm, lateral 3.5 mm relative to the bregma. The dura was kept intact. A Hamilton syringe (Hamilton Company, Reno, NV) with a 28-gauge needle was lowered to depths of 5.5mm and 4.5mm relative to the dura. Vector (2.5μ L per injection, total 5μl per striatum) was delivered at each depth with the use of a microsyringe pump controller (World Precision Instruments, Inc., Sarasota, FL) to ensure uniform delivery. Following vector administration, the wound was closed and the animals were transported to the animal facility for post-operative monitoring.
The bipromoter vector system employed in this study co-expresses both transgene and reportor gene in a similar pattern, as described previously [13–15]. The reporter gene driven by the beta-gal transcriptional unit demonstrates more than 98% covariance in expression with the transgene [16]. In addition, we have shown that HSV vector is neurotrophic[13]. Furthermore, overexpression of SOD 1 using the same vector results in increases in the corresponding protein in primary hippocampal neuronal culture[8]. Therefore, the protective effect of SOD 1 gene therapy is detected by counting the X-gal-positive neurons and compared with the number of transfected neurons in the non-ischemic cortex. Rats were sacrificed 48 hours following ischemia by isoflurane overdose and transcardially perfused with 200mL PBS followed by 200mL 4% paraformaldehyde. Brains were postfixed in 4% paraformaldehyde/20% sucrose solution for 1 to 2 days prior to frozen sectioning using a cryostat (Microm International). Sections 30μm in thickness were collected in the coronal plane at 90μm increments 1mm anterior and 1mm posterior to the injection site. Sections were stained with X-gal (5′-bromo-4-chloro-3-indoly-β-D-galactopyranoside, Molecular Probes) to visualize transfected cells, and counterstained with cresyl violet to visualize the infarct. X-gal-stained cells demonstrating neuronal morphology were counted at 40X objective magnification in each striatum in a blinded fashion as previously described [13]. The median 10 sections were included in the statistical analysis. The protective effect of SOD1 was assessed by comparing the mean percentage of surviving striatal neurons relative to the non-ischemic side in each group. Because neuron survival could be influenced by the severity of the ischemia, infarct size was assessed using a semi-quantitative scale, where 0= no infarct, 1= stroke confined to striatum, 2= striatum and some cortex, and 3= complete MCA distribution. Brains without stroke were excluded from the analysis. This scale has been previously shown to reasonably reflect the quantitative measurement of infarct size [13].
Standard statistical analysis was performed using Prism Software 3.02 (GraphPad, Inc.) Differences in neuronal survival at each time point were determined by the Student t test. Differences in infarct size were determined using the Mann-Whitney test. Results are expressed as mean ± SEM. Statistical significance was established for p < 0.05.
A representative coronal section demonstrating bilateral vector expression and striatal infarct caused by our model of transient focal cerebral ischemia is shown in Figure 1. Evidence for HSV vector gene expression using X-gal staining is shown in Figure 2. Animals treated with SOD1 vector demonstrate improved neuronal survival when delivered either before or after ischemia.
Figure 1.
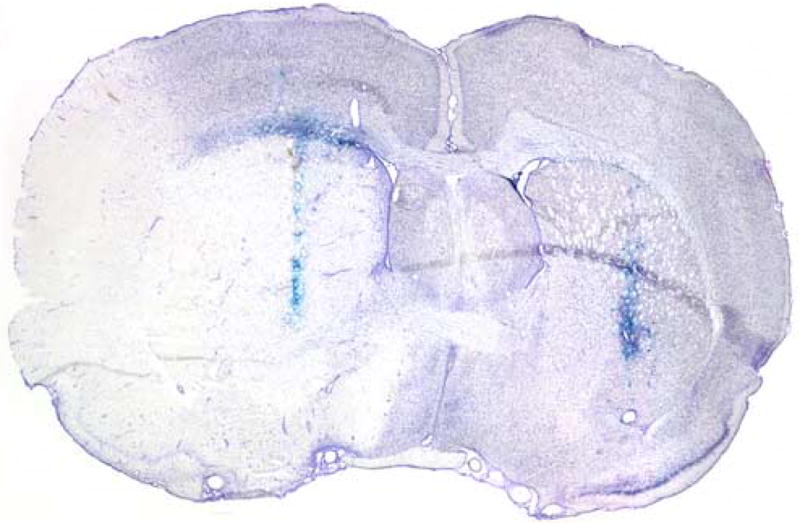
Representative coronal section at 1X magnification demonstrating bilateral HSV vector expression (X-gal staining in blue) and striatal infarct.
Figure 2.
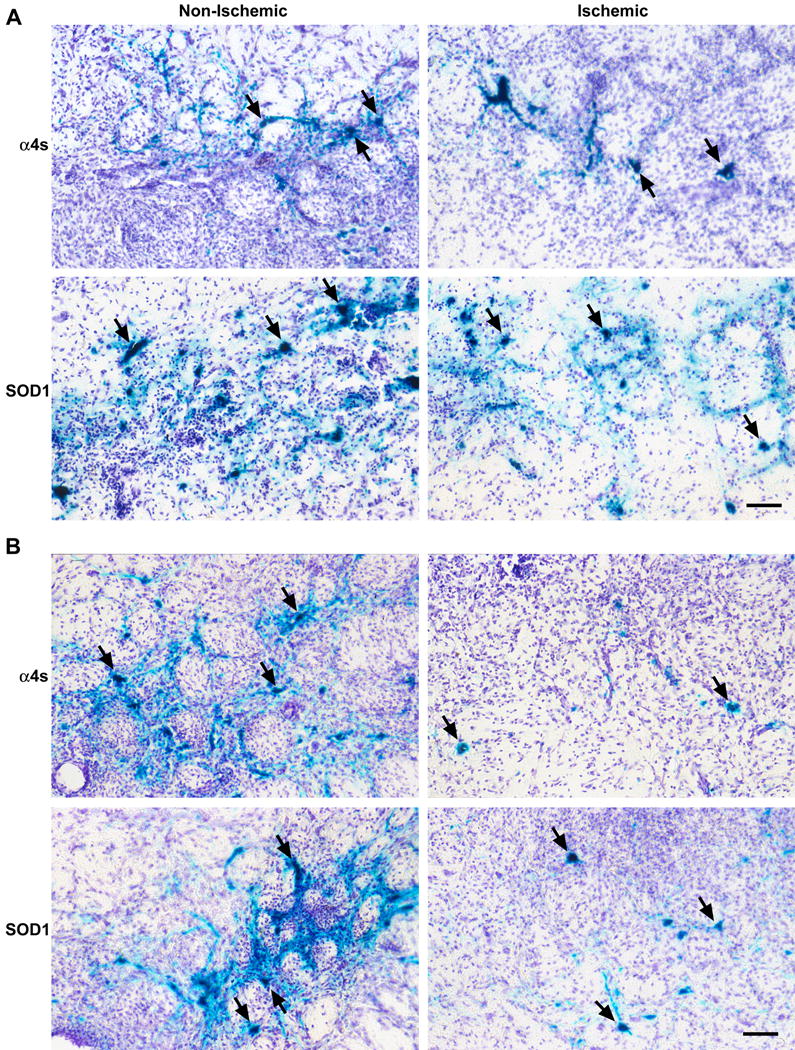
Photomicrographs at 10X objective magnification of surviving striatal neurons 48 hours following ischemia. Vector was delivered 12–15 hours prior to (A) or 2 hours following the onset of ischemia (B). Transfected neurons (see arrows) are stained with X-gal and demonstrate neuronal morphology. Increased survival is demonstrated in those neurons transfected with vector containing SOD1 compared to control (α4s) in both pre- and post-ischemia time points. Scale bar = 50 μm.
We have previously reported that gene expression mediated by HSV system starts as early as 4–6 h, peaks at 12h, and decreases thereafter [13]. The initial time point involved injection of vector 12–15 hours prior to MCA occlusion, such that peak expression of vector was occurring at the time of ischemia [13]. Transfected neuronal survival was examined at 48h but not beyond of that after stroke, since significant decreases in gene expression beyond of 2 d might undermine reliability of survival neuron estimation. Quantification of mean percentage of surviving striatal neurons at this time point demonstrated a two-fold improvement in survival among those receiving SOD1 vector (Figure 3, p < 0.05). We then investigated whether a more clinically-relevant paradigm of post-ischemic vector delivery would confer neuronal protection. Injection of vector two hours following the onset of ischemia continued to demonstrate a two-fold improvement in survival (Figure 3, p < 0.05). Since the mean number of surviving neurons between ischemic and non-ischemic striatum was compared, each animal served as its own internal control to reduce the variability of this model.
Figure 3.
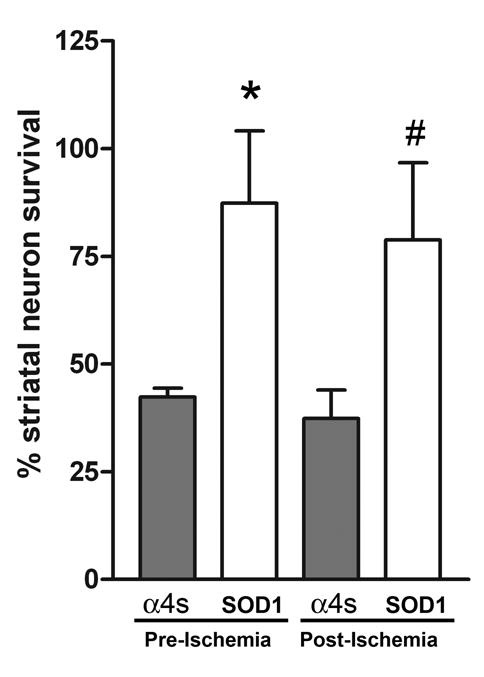
Percentage of surviving striatal neurons represented with respect to time. Survival was calculated as a ratio of surviving X-gal positive neurons in the ischemic striata to the non-ischemic striata. At the pre-ischemia time point, neurons transfected with SOD1 demonstrate a two-fold improvement in survival (87.3% ± 16.8 versus 42.3% ± 2.1, n=9/group; * p=0.02). Vector delivered following ischemia continued to demonstrate a two-fold increase in survival (78.8% ± 17.9 versus 37.3% ± 6.7, n=12/group; # p=0.04). However, a greater number of animals were required to achieve this endpoint and there was a trend toward diminishing statistical significance.
Given the transfection efficiency of the HSV vector of approximately 10%, we did not anticipate a significant impact on infarct size as a result of viral delivery [17]. The survival of transfected neurons, however, could be influenced by extent of the infarct. Analysis of infarct size as described above revealed no difference between control- and SOD1-transfected groups, suggesting minimal impact on our data due to differences in infarct size. Prior studies using this model yielded similar results [9,10]. Accordingly, evaluation of neurobehavioral outcomes was not assessed.
An antioxidant strategy for the treatment of stroke has been investigated extensively [18]. While transgenic studies have provided valuable insight into the possible role each antioxidant plays, they are limited in that they reflect lifelong in vivo expression of the gene of interest. Exogenous delivery of individual proteins such as SOD1 and catalase has been investigated previously [19]. The systemic delivery of polyethylene glycol-conjugated SOD1 (PEG-SOD) demonstrated a protective benefit in rats; however the therapeutic dose range was limited [20]. PEG-SOD has been shown to have limited penetration into brain parenchyma, suggesting that its effect is mediated at the vascular, not cellular, level [21]. The use of liposomal SOD1 demonstrated a protective effect, but this study only examined pre-ischemia treatment [22].
The mechanism by which SOD1 gene therapy confers neuronal protection likely involves both anti-necrotic and anti-apoptotic mechanisms. Direct scavenging of superoxide anion by SOD1 reduces oxidative damage to cellular macromolecules, thus decreasing necrosis. ROS generated in the mitochondria also lead to the release of cytochrome c, initiating the caspase-dependent apoptotic pathway [1]. Overexpression of SOD1 in transgenic animals resulted in decreased cytochrome c release [23] and attenuated activation of caspase-9 and caspase-3 [24]. SOD1 has also been implicated in caspase-independent pathways, as evidenced by reduced translocation of apoptosis-inducing factor following gene therapy with SOD1 in vitro (unpublished data, RMS).
This study demonstrates that increasing endogenous expression of SOD1 through gene therapy improves striatal neuron survival following transient focal cerebral ischemia. Because SOD1 overexpression is induced through viral transfection and not transgenic manipulation, this study permitted investigation of the protective effect of SOD1 following ischemia over time. The half-life of SOD1 is short (~ 6 minutes), and endogenously-generated enzyme is quickly consumed during an oxidative burst such as ischemia-reperfusion [25]. Increasing the supply of endogenously-generated SOD1 through gene therapy is one approach to overcome this challenge. Vector expression begins as early as four hours following injection, peaks at approximately 12 hours, and is significantly decreased by 48 hours [13]. The majority of reactive oxygen species generated following ischemia-reperfusion occurs in the first 2 hours following reperfusion [26]. Our data indicate that SOD1 continues to confer neuronal protection when the vector is administered at two hours, corresponding to expression of SOD1 as early as 6 hours following the onset of ischemia. Given the kinetics of the oxidative burst following ischemia/reperfusion and the decreasing statistical significance at two hours, an extended therapeutic window was not pursued. A similar result of decreasing protection post-ischemia was observed when gene therapy using either catalase or HSP72 was administered up to five hours after ischemia [10,17].
The data reported in this study conflict with recent in vitro reports which showed no protection from SOD1 gene therapy following oxygen-glucose deprivation [8]. The rationale for this lack of protection was that SOD1 overexpression could not take place in the face of decreased protein synthesis during a necrotic insult. SOD1 gene therapy in vivo presumably generates endogenous levels of a sufficient magnitude to effectively scavenge ROS during subsequent ischemia, accounting for the increased survival of SOD1-transfected neurons. Furthermore, the post-ischemia neuroprotection observed in this study may reflect a milder insult produced by a one-hour MCA occlusion compared to the in vitro model of OGD, allowing for protein synthesis to occur. Thus, it is possible that SOD1 gene therapy would not be protective with a more severe insult. The use of a gene therapy model in vivo may also support the benefits of studying an intact system, where GPX and catalase have been reported to be upregulated during an ischemic insult [27]. Thus, there may be improved scavenging of hydroxyl radicals by these downstream enzymes which facilitated the observed neuroprotection. We were also surprised to see that the magnitude of neuroprotection was similar in pre- and post-ischemia time points. However, there is a trend of decreasing statistical significance at the post-ischemia time point, and the similar degree of protection may be due to the small sample size.
There is evidence that non-neuronal cells may also participate in ROS scavenging during ischemia/reperfusion [28]. The role of supporting cells such as astrocytes and microglia in contributing to neuronal protection was not evaluated and is beyond the scope of the current study; however, it should be noted that while HSV-1 is a neurotropic virus, it is able to infect a certain number of non-neuronal cells [13]. Finally, although a neuroprotective effect of antioxidant therapy using SOD1 in vivo is demonstrated in our study, the lack of effect on overall infarct size limits the translation of SOD1 gene therapy into a clinical setting.
In conclusion, increasing endogenous expression of SOD1 through gene therapy provides neuroprotection following experimental stroke, including post-ischemic protection. However, the invasive method of vector delivery and the kinetics of vector expression may limit application of this technique in the clinical setting.
Acknowledgments
The authors would like to thank Beth Hoyte and Dr. Raymond Sobel for assistance in figure preparation, and Dr. Bruce Schaar for critical reading of the manuscript. Funding for this project was provided by NIH NINDS P01 37520 (GKS, RMS), and Bernard and Ronni Lacroute (GKS).
References
- 1.Sugawara T, Chan P. Reactive Oxygen Radicals and Pathogenesis of Neuronal Death After Cerebral Ischemia. Antioxidants & Redox Signaling. 2003;5:597–607. doi: 10.1089/152308603770310266. [DOI] [PubMed] [Google Scholar]
- 2.Halliwell B, Gutteridge J. Lipid peroxidation, oxygen radicals, cell damage, and antioxidant therapy. Lancet. 1984;1:1396–1397. doi: 10.1016/s0140-6736(84)91886-5. [DOI] [PubMed] [Google Scholar]
- 3.Kinouchi H, Epstein C, Mizui T, Carlson E, Chen S, Chan P. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proceedings of the National Academy of Science. 1991;88:11158–11162. doi: 10.1073/pnas.88.24.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan P, Kawase M, Murakami K, et al. Overexpression of SOD1 in Transgenic Rats Protects Vulnerable Neurons Against Ischemic Damage After Global Cerebral Ischemia and Reperfusion. The Journal of Neuroscience. 1998;18(20):8292–8299. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan P, Kamii H, Yang G, et al. Brain infarction is not reduced in SOD-1 transgenic mice after a permanent focal cerebral ischemia. NeuroReport. 1993;5:293–296. doi: 10.1097/00001756-199312000-00028. [DOI] [PubMed] [Google Scholar]
- 6.Ditelberg J, Sheldon R, Epstein C, Ferriero D. Brain injury after perinatal hypoxia-ischemia is exacerbated in copper/zinc superoxide dismutase transgenic mice. Pediatric Research. 1996;39:204–208. doi: 10.1203/00006450-199602000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Fullerton H, Ditelberg J, Chen S, et al. Copper/zinc superoxide dismutase transgenic brain accumulates hydrogen peroxide after perinatal hypoxia ischemia. Annals of Neurology. 1998;44:357–364. doi: 10.1002/ana.410440311. [DOI] [PubMed] [Google Scholar]
- 8.Zemlyak I, Nimon V, Brooke S, Moore T, McLaughlin J, Sapolsky R. Gene therapy in the nervous system with superoxide dismutase. Brain Research. 2006;1088:12–18. doi: 10.1016/j.brainres.2006.02.109. [DOI] [PubMed] [Google Scholar]
- 9.Hoehn B, Yenari M, Sapolsky R, Steinberg G. Glutathione Peroxidase Overexpression Inhibits Cytochrome c Release and Proapoptotic Mediators to Protect Neurons from Experimental Stroke. Stroke. 2003;34:2489–2494. doi: 10.1161/01.STR.0000091268.25816.19. [DOI] [PubMed] [Google Scholar]
- 10.Gu W, Zhao H, Yenari M, Sapolsky R, Steinberg G. Catalase over-expression protects striatal neurons from transient focal cerebral ischemia. NeuroReport. 2003;15:413–416. doi: 10.1097/00001756-200403010-00006. [DOI] [PubMed] [Google Scholar]
- 11.Wang H, Cheng E, Brooke S, Chang P, Sapolsky R. Over-expression of antioxidant enzymes protects cultured hippocampal and cortical neurons from necrotic insults. Journal of Neurochemistry. 2003;87(6):1527–1534. doi: 10.1046/j.1471-4159.2003.02123.x. [DOI] [PubMed] [Google Scholar]
- 12.Ho D. Amplicon-based herpes simplex virus vectors. Methods in Cell Biology. 1994;43 (pt A):191–210. doi: 10.1016/s0091-679x(08)60604-4. [DOI] [PubMed] [Google Scholar]
- 13.Yenari M, Minami M, Sun G, et al. Calbindin D28K Overexpression Protects Striatal Neurons From Transient Focal Cerebral Ischemia. Stroke. 2001;32:1028–1035. doi: 10.1161/01.str.32.4.1028. [DOI] [PubMed] [Google Scholar]
- 14.Lawrence MS, Ho DY, Sun GH, Steinberg GK, Sapolsky RM. Overexpression of Bcl-2 with herpes simplex virus vectors protects CNS neurons against neurological insults in vitro and in vivo. J Neurosci. 1996;16(2):486–496. doi: 10.1523/JNEUROSCI.16-02-00486.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLaughlin J, Roozendaal B, Dumas T, et al. Sparing of neuronal function postseizure with gene therapy. Proc Natl Acad Sci U S A. 2000;97(23):12804–12809. doi: 10.1073/pnas.210350097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fink SL, Chang LK, Ho DY, Sapolsky RM. Defective herpes simplex virus vectors expressing the rat brain stress-inducible heat shock protein 72 protect cultured neurons from severe heat shock. J Neurochem. 1997;68(3):961–969. doi: 10.1046/j.1471-4159.1997.68030961.x. [DOI] [PubMed] [Google Scholar]
- 17.Hoehn B, Ringer T, Xu L, et al. Overexpression of HSP72 After Induction of Experimental Stroke Protects Neurons From Ischemic Damage. Journal of Cerebral Blood Flow and Metabolism. 2001;21:1303–1309. doi: 10.1097/00004647-200111000-00006. [DOI] [PubMed] [Google Scholar]
- 18.Margaill I, Plotkine M, Lerouet D. Antioxidant strategies in the treatment of stroke. Free Radical Biology & Medicine. 2005;39:429–443. doi: 10.1016/j.freeradbiomed.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 19.Liu T, Beckman J, Freeman B, Hogan E, Hsu C. Polyethylene glycol-conjugated superoxide dismutase and catalase reduce ischemic brain injury. American Journal of Physiology. 1989;256:H589–H593. doi: 10.1152/ajpheart.1989.256.2.H589. [DOI] [PubMed] [Google Scholar]
- 20.He Y, Hsu C, Ezrin A, Miller M. Polyethylene glycol-conjugated superoxide dismutase in focal cerebral ischemia-reperfusion. American Journal of Physiology. 1993;265:H252–H256. doi: 10.1152/ajpheart.1993.265.1.H252. [DOI] [PubMed] [Google Scholar]
- 21.Kirsch J, Helfaer M, Lange D, Traystman R. Evidence for Free Radical Mechanisms of Brain Injury Resulting from Ischemia/Reperfusion-Induced Events. Journal of Neurotrauma. 1992;9:S157–S163. [PubMed] [Google Scholar]
- 22.Imaizumi S, Woolworth V, Fishman R, Chan P. Liposome-entrapped superoxide dismutase reduces cerebral infarction in cerebral ischemia in rats. Stroke. 1990;21:1312–1317. doi: 10.1161/01.str.21.9.1312. [DOI] [PubMed] [Google Scholar]
- 23.Fujimura M, Morita-Fujimura Y, Noshita N, Sugawara T, Kawase M, Chan P. The Cytosolic Antioxidant Copper/Zinc-Superoxide Dismutase Prevents the Early Release of Mitochondrial Cytochrome c in Ischemic Brain after Transient Focal Cerebral Ischemia in Mice. The Journal of Neuroscience. 2000;20(8):2817–2824. doi: 10.1523/JNEUROSCI.20-08-02817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugawara T, Noshita N, Lewen A, et al. Overexpression of Copper/Zinc Superoxide Dismutase in Transgenic Rats Protects Vulnerable Neurons against Ischemic Damage by Blocking the Mitochondrial Pathway of Caspase Activation. The Journal of Neuroscience. 2002;22(1):209–217. doi: 10.1523/JNEUROSCI.22-01-00209.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turrens J, Crapo J, Freeman B. Protection against Oxygen Toxicity by Intravenous Injection of Liposome-entrapped Catalase and Superoxide Dismutase. Journal of Clinical Investigation. 1984;73:87–95. doi: 10.1172/JCI111210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peters O, Back T, Lindauer U, et al. Increased Formation of Reactive Oxygen Species After Permanent and Cerebral Artery Occlusion in the Rat. Journal of Cerebral Blood Flow and Metabolism. 1998;18(2):196–205. doi: 10.1097/00004647-199802000-00011. [DOI] [PubMed] [Google Scholar]
- 27.Azbill R, Mu X, Bruce-Keller A, Mattson M, JE S. Impaired mitochondrial function, oxidative stress and altered antioxidant enzyme activities following traumatic spinal cord injury. Brain Research. 1997;765:283–290. doi: 10.1016/s0006-8993(97)00573-8. [DOI] [PubMed] [Google Scholar]
- 28.Wilson J. Antioxidant defense of the brain: a role for astrocytes. Canadian Journal of Physiology and Pharmacology. 1997;75:1149–1163. [PubMed] [Google Scholar]