

Abstract
We propose a concept for the rational design and synthesis of highly selective chemotherapeutic agents that makes direct use of genetic information about the disease state. The key idea is to use the mRNA or DNA specific to the disease state to trigger the catalytic release of a cytotoxic drug by promoting the association of a prodrug with a catalyst capable of releasing the drug. We demonstrate the feasibility of such an approach in vitro with a model system that is based on the hydrolysis of p-nitrophenyl esters by imidazole. In our model system, the catalytic component consists of an imidazole group linked to the 5′ end of a 15-mer that is complementary to the 5′ end of the triggering oligodeoxynucleotide. The corresponding prodrug component consists of a p-nitrophenol ester linked to the 3′ end of an 8-mer oligodeoxynucleotide that is complementary to the 3′ end of the triggering sequence. We show that this system efficiently releases p-nitrophenol in the presence of all three components and that the reaction is catalytic and undergoes multiple turnovers. We also show that the complex between the catalytic component and the triggering oligodeoxynucleotide behaves like an enzyme and follows Michaelis–Menten kinetics, with a KM of 22 μM and a kcat of 0.018 min−1. Most importantly, we show that catalytic release of p-nitrophenol is sensitive to the presence of a single base-pair mismatch.
Chemotherapeutic approaches to curing infectious diseases and cancer depend on drugs that are selectively toxic to the disease-causing organism or the diseased cell (1–3). Viral infections and cancer pose the greatest challenge for chemotherapy because there is little biochemically to distinguish an infected or cancerous cell from a normal cell, and as a result many currently used drugs show little selectivity (4). There have been a number of approaches to increasing the selectivity of anticancer agents through the use of immunoconjugates, antibody-, gene-, and bacterial-directed enzymatic activation of prodrugs and by capitalizing on elevated levels of certain enzymes and receptors within cancer cells (5, 6). Other approaches have sought to exploit what is known about the molecular mechanisms of cancer to identify new biochemical targets for drugs (7). Although all of these methods can in principle lead to more selective chemotherapeutic agents, they are by no means easy to implement.
Recent advances in genomic sequencing and DNA chip technology now make it possible to determine the genetic makeup of diseases such as cancer (8, 9). This, together with the ability to bind specific mRNA or DNA sequences with oligodeoxynucleotides (ODNs) or analogs such as peptide nucleic acids via simple base-pairing rules (10–12), or DNA with polyamides via its own set of rules (13), has opened the door for new approaches to chemotherapy that make direct use of genetic information. Current approaches in this category can be classified as anti-sense or antigene and are based on specifically binding to, and either interfering with or damaging, the targeted nucleic acid sequence (14). What makes these approaches so attractive is the ease by which it would seem possible to tailor chemotherapeutic agents for individual patients based on genetic information that could be obtained about their disease states from DNA chips. As promising as both approaches are, it is difficult to predict the therapeutic effect of targeting a viral or cancer-specific nucleic acid sequence, and in many such applications of antisense technology, the therapeutic effect has been found not to involve an antisense mechanism (15).
Herein, we propose a general concept for the rational design of highly selective chemotherapeutic agents that also makes use of the ease by which molecules can be synthesized to recognize specific nucleic acid sequences. In this approach, which we refer to as nucleic acid-triggered catalytic drug release, a disease-specific nucleic acid sequence is used not as a chemotherapeutic target but rather as a trigger to cause the catalytic release of a cytotoxic drug. The key idea is to use the mRNA or DNA sequence specific to a disease as a template to promote the association of a prodrug and a catalytic component capable of converting the prodrug to a cytotoxic drug, which then kills the diseased cell. In this approach, the disease-specific sequence could either be a unique sequence or an overexpressed sequence. Unlike antisense and antigene approaches, the therapeutic effectiveness of this approach depends only on the presence of a disease-specific nucleic acid sequence and not on its biological activity.
In one formulation of this approach (Fig. 1), the prodrug consists of a cytotoxic drug that is attached to a molecule that binds reversibly to an mRNA or DNA sequence that is specific to the disease (the triggering sequence). The corresponding catalytic component consists of a catalyst attached to another molecule that binds tightly to the site on the triggering sequence that is adjacent to the prodrug binding site. In the diseased cell, the catalytic component will bind to the disease-specific mRNA or DNA sequence to form an enzyme-like complex that contains a prodrug binding site in addition to the catalytic site. Once formed, this catalytic complex will carry out multiple turnovers of prodrug to drug, which will result in the death of the diseased cell. In a nondiseased cell lacking the triggering sequence, association of the catalyst with the prodrug will not occur efficiently, and the cell will survive. This approach should also be applicable to diseases in which the triggering sequence differs from the normal one by a single nucleotide, as would be the case for many cancers, because binding of molecules such as ODNs are known to be sensitive to centrally located single base-pair mismatches (16). In this paper, we will demonstrate the feasibility of this approach with a simple model system that is based on the ability of imidazole to catalyze the hydrolysis of p-nitrophenyl esters.
Figure 1.

General concept of nucleic acid-triggered catalytic drug release. The triggering nucleic acid sequence could in principle be either an mRNA or duplex DNA sequence specific to the disease state. The catalyst and drug could be attached to any sequence-specific single- or double-strand binding agent, such as an ODN or analog such as peptide nucleic acid, or a minor groove-binding polyamide. Ideally, the drug-releasing catalytic component binds tightly to the triggering sequence to form an enzyme-like catalyst, whereas the prodrug binds reversibly, so that it can be exchanged for another prodrug after release of the drug.
Materials and Methods
General.
Dichloromethane and triethylamine were dried by refluxing with CaH2 overnight followed by distillation. Histamine, maleic anhydride, and thionyl chloride were purchased from Aldrich. Tris(2-carboxyethyl)phosphine hydrochloride was purchased from Pierce. Reagents for automatic ODN synthesis were purchased from Glen Research (Sterling, VA). ODNs were synthesized on an Applied Biosystems 380 DNA synthesizer using phosphoramidite chemistry and recommended protocols (dimethoxytrityl off synthesis). 1H NMR, 13C NMR, and 31P NMR spectra were obtained on a Varian UnityPlus-300 (300 MHz) or a Varian Mercury-300 (300 MHz) spectrometer. The chemical shifts are expressed in ppm from tetramethylsilane using residual chloroform (δ = 7.24) and acetone (δ = 2.04) as an internal standard. 31P NMR spectra were referenced against 85% H3PO4 in a coaxial insert. Flash chromatography was performed on Selecto Scientific silica gel. TLC was run on precoated 254-nm fluorescent silica gel sheets manufactured by Alltech Associates. UV spectral data were acquired on a Bausch and Lomb Spectronic 1001 spectrophotometer or Varian Cary 1E UV-Vis spectrophotometer. Matrix-assisted laser desorption ionization (MALDI) mass spectra of ODNs were measured on PerSeptive Voyager RP MALDI–time of flight (TOF) mass spectrometer.
Synthesis of Im-15-mer 2.
Imidazole was linked to the 5′ end of d(ATTACGCTGGACTCT) by a previously described procedure (17). Phosphoramidite 11 was used in the last coupling step (0.1 M phosphoramidite 11 in acetonitrile, coupling time 30 min). After oxidation with 0.1 M iodine, the protected ODN was treated with 0.2 M histamine 12 in water for 6 h. Complete deprotection was carried out in concentrated aqueous ammonia at 55°C for 8 h. The ammonia solution was evaporated to dryness on a Savant Speedvac, first under water aspirator pressure and then under high vacuum to yield the crude oligomer 2. It was then dissolved in doubly distilled water and purified by reversed-phase HPLC on a Rainin Dynamax column (C-18, 5 μm, 4.6 × 250 mm) using buffer A [50/50 (vol/vol) 100 mM triethyl ammonium acetate buffer, pH 7.0/water] and buffer B [50/50 (vol/vol) 100 mM triethyl ammonium acetate buffer, pH 7.0/acetonitrile]. A linear gradient was run from 0 to 30% buffer B in 30 min at a flow rate of 1.0 ml/min and the effluent monitored at 260 nm. The desired fraction was collected, concentrated, and desalted by loading onto the same column in pure water, washing with excess doubly distilled water, and eluting with 50:50 acetonitrile/water. The desalted fractions were combined and concentrated to dryness in vacuo. The purified product oligomer 2 was analyzed by MALDI–TOF, [M-H+] 4771.8, found 4773.4.
Synthesis of Maleamic Acid of D-Valine 6.
D-Valine (11.7 g, 100 mmol) was dissolved in 10 ml of water, and then maleic anhydride (9.8 g, 100 mmol) was added all at once and stirred for 4 h at ambient temperature. The resulting white powder was filtered, washed with water (3 × 10 ml), followed by anhydrous ethanol (3 × 10 ml) and then anhydrous ether (3 × 10 ml) to give 11.6 g (69%) of the maleamic acid 6 (18). [α]D = −15.0° (c 1.1, acetone); 1H NMR (300 MHz, CD3COCD3) δ 1.01 (d, J = 4.9 Hz, 6H) 2.05 (m, 1H), 4.53 (m, 1H), 6.32 (d, J = 12.9 Hz, 1H), 6.76 (d, J = 12.9 Hz, 1H).
Synthesis of N-Maleoyl-D-Valine Ester 8.
The maleamic acid of D-valine 6 (500 mg, 2.54 mmol) was dissolved in 10 ml of thionyl chloride and heated at reflux until gas evolution had ceased. The excess thionyl chloride was evaporated under reduced pressure. Carbon tetrachloride was added to the remaining material, and the resulting solution was evaporated under reduced pressure to ensure complete removal of thionyl chloride. The resulting product was dissolved in 10 ml of CH2Cl2 and was slowly added to a stirred mixture of 4-nitrophenol 7 (353 mg, 2.54 mmol) and triethylamine (0.71 ml, 5.08 mmol) in 10 ml of CH2Cl2 at 0°C. The reaction mixture was allowed to warm to room temperature. After stirring an additional 30 min at room temperature, the reaction mixture was diluted with 200 ml of CH2Cl2, washed with brine and water, dried over Na2SO4, and concentrated in vacuo. The residue was flash chromatographed on silica gel (1:4 ethyl acetate/hexane) to afford the N-maleoyl-D-valine ester 8, 246 mg (33%). [α]D = +33.0° (c 0.9, acetone); 1H NMR (300 MHz, CD3COCD3) δ 0.93 (d, J = 6.6 Hz, 3H), 1.11 (d, J = 6.8 Hz, 3H), 2.60∼2.64 (m, 1H), 4.82 (d, J = 7.1 Hz, 1H), 7.05 (s, 2H), 7.36∼7.39 (m, 2H), 8.30∼8.32 (m, 2H); 13C NMR (300 MHz, CD3COCD3) δ 19.9, 20.8, 57.2, 123.6, 123.8, 125.8, 135.3, 146.3, 156.0, 167.4, 170.9; high-resolution mass spectrometry calculated for C15H15N2O6 [M + H+] 319.0930, found 319.0931.
Synthesis of pNP-8-mer 1.
The conjugation of a 3′-thiolated ODN with a maleimide group was carried out according to a general procedure (19). The ODN 9 bearing a 3′-terminal disulfide group (100 nmol) (20) was reduced to one with a 3′-terminal thiol group 10 with Tris(2-carboxyethyl)phosphine (150 nmol) in 500 μl of 0.1 M sodium phosphate buffer, pH 7.0, for 2 h at room temperature under argon. The N-maleoyl-D-valine ester 8 (1 μmol, in 50 μl of acetonitrile) was then added to thiol group 10 to give pNP-8-mer 1, which was purified by reversed-phase HPLC on a Rainin Dynamax column (C-18, 5 μm, 4.6 × 250 mm) using buffers A [90/10 (vol/vol) 75 mM sodium phosphate buffer, pH 7.0/10% methanol] and B (50/50, 75 mM sodium phosphate buffer, pH 7.0/methanol). A 30-min 1 ml/min linear gradient was run from 0 to 100% buffer B in buffer A, and the effluent was monitored at 260 nm. The desired fraction was collected, concentrated, and desalted by loading onto the same column, washing with excess doubly distilled water, and eluting with 50:50 acetonitrile/water. The desalted fractions were combined and concentrated to dryness in vacuo. The product was analyzed by MALDI–TOF, calculated [M-H+] 2803.2, found 2802.9.
Kinetics.
For typical assays, pNP-8-mer 1 and Im-15-mer 2 were incubated in 10 mM sodium phosphate, pH 7.0, which contained 0.1 M or 1.0 M NaCl in an ultra-micro (10 μl) UV cell (Varian). The reaction temperature was maintained at 20°C, and the production of p-nitrophenolate was monitored by UV absorbance at 400 nm (ɛ400 = 6.26 × 103). Initial velocities of the reaction, obtained for each substrate concentration, were fitted to the Michaelis–Menten equation by a nonlinear least squares method with kaleidagraph software. The inhibition constant KI was determined in the presence of 20 μM d(TCCTGTCA) in 10 mM sodium phosphate buffer, pH 7.0, containing 1.0 M NaCl by plotting 1/v vs. 1/[S] and calculating KI from the slope of the line according following equation:
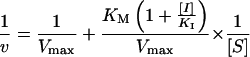 |
1 |
where v is the velocity of the reaction, Vmax is the maximum velocity, KM is the substrate concentration at half-maximal reaction rate, [I] is the inhibitor concentration, and [S] is the substrate concentration. For comparison purposes, the reaction rate constant kIm for the imidazole-catalyzed hydrolysis of N-maleoyl-D-valine p-nitrophenyl ester 8 was determined in imidazole buffer at pH 7.0, 1.0 M NaCl at 20°C by a previously described procedure (21).
Results and Discussion
Design.
To demonstrate the feasibility of nucleic acid-triggered catalytic drug release, we designed a model system (Fig. 2) based on the well known ability of imidazole to catalyze the release of p-nitrophenol from p-nitrophenyl esters (22–24). Release of a substituted o- or p-nitrophenol is also the key step in the activation of a recently reported class of prodrugs of the chemotherapeutic drugs daunorubicin, phenol nitrogen mustard, and 5-fluorouracil (25–27). The model system consists of three components: a prodrug, a catalyst, and a triggering ODN. The catalytic component, Im-15-mer 2, consists of an imidazole group linked to the 5′ end of a 15-mer that is complementary to the 5′ end of the triggering 23-mer ODN 3. The 15-mer sequence was chosen because it was calculated to have a TM of 60°C under the assay conditions and therefore was expected to form a stable complex with the triggering ODN. The corresponding prodrug component, pNP-8-mer 1, consists of a p-nitrophenol ester linked to the 3′ end of an 8-mer ODN that is complementary to 3′ end of a 23-mer ODN 3, representing the triggering sequence. The 8-mer sequence was chosen because it was calculated to have a TM of 45°C under the assay conditions and was expected to form a less stable complex that would bind reversibly to the triggering sequence at 20°C.
Figure 2.

Three-component model system used to validate the approach, which consists of a prodrug 1, a catalyst 2, and a triggering sequence 3.
Synthesis.
The two components were synthesized as shown in Fig. 3. Attachment of the imidazole group was carried out according to a previously described method (17) by coupling a benzyl thioester phosphoramidite 11 to the 5′ end of a controlled pore glass-supported d(ATTACGCTGGACTCT), followed by reaction with histamine 12 to give Im-15-mer 2. In contrast, attachment of a p-nitrophenyl ester to an ODN required a significant amount of experimentation due to the lability of the p-nitrophenyl ester group at pH values higher than 7 that are typically required for many conjugation reactions. Eventually, we found that a p-nitrophenyl ester could be linked to an ODN at pH 7 by conjugation of the N-maleoyl derivative 8 to 3′-thiolated d(TCCTGTCA) 10 to give pNP-8-mer 1 (19). In anticipation of future in vivo experiments, we made use of the N-maleoyl derivative of D-valine p-nitrophenyl ester because the D-forms of amino acid esters with branched side chains are generally poor substrates for esterases, lipases, and peptidases (28–31). The required N-maleoyl-D-valine ester 8 was prepared by a two-step reaction from maleic anhydride 5 and D-valine 4 that proceeds via the maleamic acid intermediate 6, which is cyclized and then esterified with p-nitrophenol 7 in one pot after treatment with thionyl chloride (32).
Figure 3.

Synthetic scheme for the preparation of the prodrug and catalyst components.
Experimental Evaluation of the Model System.
The model system was then examined for the three requisite features of nucleic acid-triggered catalytic drug release: (i) that efficient and catalytic release of the drug from the prodrug only takes place in the presence of both the catalytic component and triggering sequence, (ii) that the complex formed between the catalytic component and the triggering sequence behaves like an enzyme, and (iii) that the rate of drug release is selective for the fully complementary triggering sequence. These features were verified by spectroscopically monitoring the release of p-nitrophenolate at 400 nm under a variety of conditions as described in the following paragraphs.
Requirement for All Three Components.
The requirement that both the catalytic component and triggering sequence be present for efficient drug release from the prodrug component was examined by monitoring the release of p-nitrophenol from pNP-8-mer with various combinations of Im-15-mer, a 15-mer lacking the imidazole group, and the triggering 23-mer in 1 M NaCl, pH 7, at 20°C. Only when Im-15-mer and the triggering 23-mer were present did the release of p-nitrophenol occur at a significant rate above background (Fig. 4). Whereas the initial rate of p-nitrophenol release was 0.057 μM/min in the presence of all three components, it was 0.002 μM/min in the absence of the 23-mer or Im-15-mer, and 0.0008 μM/min in the presence of a 15-mer lacking an appended imidazole group. Most significantly, p-nitrophenol was released catalytically as evidenced by the formation of about 15 μM p-nitrophenol during the course of the reaction in the presence of only 5 μM of the catalytic ODN and 5 μM of the triggering ODN.
Figure 4.

Efficient and catalytic release of the drug requires both the catalytic component and the triggering sequence. Im-15-mer or the 15-mer lacking the imidazole group (5 μM) was incubated with pNP-8-mer (20 μM) at 20°C in the presence or absence of triggering 23-mer (5 μM) in a 10-μl, 1-cm path-length cell at pH 7 in a 1 M NaCl, 10 mM sodium phosphate buffer. The release of p-nitrophenol as a function of time was monitored by UV/Vis spectroscopy at 400 nm on a Cary 1E UV/Vis spectrophotometer and the absorbance readings converted to μM p-nitrophenol based on its molar extinction coefficient in the buffer system used. The rate of release in the absence of Im-15-mer (not shown) was similar to that observed in the absence of the 23-mer.
Enzyme-Like Behavior.
The expectation that the complex formed between the catalytic component and the triggering sequence should behave like an enzyme was investigated by determining the initial rate of drug release as a function of substrate concentration and a competitor ODN (Fig. 5). As expected for an enzyme-like system, release of p-nitrophenol from pNP-8-mer was found to follow simple Michaelis–Menten kinetics (Fig. 4), with a Vmax of 0.09 μM min−1 (kcat of 0.018 min−1) and a KM of 22 μM. This corresponds to a 976-fold rate acceleration over the hydrolysis of the p-nitrophenyl ester of valine maleimide 8 catalyzed by free imidazole (kIm = 0.014 M−1 min−1) as calculated by (kcat/KM)/kIm. In a study of a related system in which the valine ester was replaced by a β-aminopropionate ester, kcat was 10-fold higher (data not shown), indicating that the reaction is quite sensitive to the presence of α-substituents, as has been found for other p-nitrophenyl esters (33). To further investigate the analogy with an enzymatic system, we examined the effect of adding an 8-mer lacking the p-nitrophenyl ester group on the rate of p-nitrophenol release and found that it behaves like a competitive inhibitor with a KI of 1.7 μM.
Figure 5.

The complex formed between the catalytic component and the triggering sequence behaves like an enzyme. The kinetics of p-nitrophenol release from the three-component system was analyzed according to a Michaelis–Menten mechanism. Shown are Eadie–Hofstee plots of the initial rate data in the presence and absence of a competitor ODN. The initial rates were obtained by analysis of the time dependence of p-nitrophenol release as a function of pNP-8-mer concentration in the presence of 5 μM Im-15-mer and 5 μM 8-mer lacking the p-nitrophenyl ester attachment that were carried out under the conditions described in the Fig. 4 legend. The extrapolated lines were derived from KM and Vmax data obtained from nonlinear least-squares fit of the kinetic data.
Sensitivity to Mismatches.
The requirement that drug release is selective for the fully complementary triggering sequence was tested by monitoring the release of p-nitrophenol from pNP-8-mer in the presence of the fully complementary 23-mer or one in which the T at position 20 was replaced with a C (Fig. 6). Release of p-nitrophenol was 7.5-fold more efficient in the presence of the fully matched template than the mismatched template at 20°C and a physiologically relevant salt concentration of 0.1 M NaCl. Under these conditions, the initial rate of release of p-nitrophenol was 0.028 μM/min in the presence of the fully matched sequence, but only 0.0037 μM/min in the presence of the mismatched sequence. These results are consistent with the prediction that the TM for the matched duplex d(TCCTGTCA)⋅d(TGACAGGA) under these conditions is 34°C, whereas that of the corresponding duplex containing the T⋅C mismatch (hyther Version 1.0, provided by N. Peyret & J. SantaLucia, Wayne State University, Detroit, MI) is only 6°C (16, 34). At a higher salt concentration of 1 M NaCl, there was only a 1.4-fold difference in rate, which was consistent with the predicted TM values of 44°C and 18°C for the matched and mismatched duplexes, respectively. These results suggest that it would be possible to optimize the selectivity of drug release for a given triggering sequence by changing the length or other properties of the prodrug so as to optimize the differences in stability between the matched and mismatched duplexes.
Figure 6.

Catalytic drug release is most efficient for the fully complementary triggering sequence. Im-15-mer (5 μM) was incubated with pNP-8-mer (20 μM) at 20°C in the presence of the fully matched and singly mismatched 23-mer (5 μM) at pH 7 in a 0.1 M NaCl, 10 mM sodium phosphate buffer.
Conclusion
We have demonstrated that genetic information can be used to trigger the catalytic release of a drug in a highly sequence-specific manner by an approach that could potentially be used to selectively kill disease-causing organisms or diseased cells. Most significantly, catalytic drug release was shown to be sensitive to even a single base-pair mismatch, suggesting that this approach could even be used against diseases like cancer, in which there may only be a single base-pair difference between the cancer cell and a normal cell. The ability to discriminate between nucleic acids differing in a single base is not a requirement for treating cancer, however, as one could also make use of a overexpressed mRNA unique to the cancer as a trigger. This same approach could also be used for the cell-specific release of therapeutically useful drugs for other types of diseases or for the creation of in vitro or in vivo diagnostic agents, in which genetic information could be used to trigger the release of reporter molecules.
Acknowledgments
We thank Dewey Holten for a critical review of this manuscript. This work was supported in part by National Institutes of Health Grant CA40463. The assistance of the Washington University High Resolution NMR Facility, funded in part through National Institutes of Health Biomedical Research Support Shared Instrument Grants RR-02004, RR-05018, and RR-07155, is gratefully acknowledged, as is the Washington University Mass Spectrometry Resource, a National Institutes of Health Research Resource (Grant P41RR0954).
Abbreviations
- MALDI–TOF
matrix-assisted laser desorption ionization–time of flight
- ODN
oligodeoxynucleotide
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Albert A. Selective Toxicity: The Physico-chemical Basis of Therapy. London: Chapman and Hall; 1979. [Google Scholar]
- 2.Drews J. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 3.Sykes R. Int J Antimicrob Agents. 2000;14:1–12. doi: 10.1016/s0924-8579(99)00141-7. [DOI] [PubMed] [Google Scholar]
- 4.Hardman J G, Limbird L E, Molinoff R W, Ruddon R W, Gilman A G, editors. Goodman & Gilman's The Pharmacological Basis of Therapeutics. New York: McGraw-Hill; 1996. [Google Scholar]
- 5.Duncan R, Connors T A, Meada H. J Drug Target. 1996;3:317–319. doi: 10.3109/10611869608996823. [DOI] [PubMed] [Google Scholar]
- 6.Dubowchik G M, Walker M A. Pharmacol Ther. 1999;83:67–123. doi: 10.1016/s0163-7258(99)00018-2. [DOI] [PubMed] [Google Scholar]
- 7.Gibbs J B. Science. 2000;287:1969–1973. doi: 10.1126/science.287.5460.1969. [DOI] [PubMed] [Google Scholar]
- 8.Golub T R, Slonim D K, Tamayo P, Huard C, Gaasenbeek M, Mesirov J P, Coller H, Loh M L, Downing J R, Caligiuri M A, et al. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 9.Gerhold D, Rushmore T, Caskey C T. Trends Biochem Sci. 1999;24:168–173. doi: 10.1016/s0968-0004(99)01382-1. [DOI] [PubMed] [Google Scholar]
- 10.Uhlmann E. Biol Chem. 1998;379:1045–1052. [PubMed] [Google Scholar]
- 11.Nielsen P E. Curr Opin Struct Biol. 1999;9:353–357. doi: 10.1016/S0959-440X(99)80047-5. [DOI] [PubMed] [Google Scholar]
- 12.Fox K R. Curr Med Chem. 2000;7:17–37. doi: 10.2174/0929867003375506. [DOI] [PubMed] [Google Scholar]
- 13.Dervan P B, Burli R W. Curr Opin Chem Biol. 1999;3:688–693. doi: 10.1016/s1367-5931(99)00027-7. [DOI] [PubMed] [Google Scholar]
- 14.Thurston D E. Br J Cancer. 1999;80:65–85. [PubMed] [Google Scholar]
- 15.Wagner R W, Flanagan W M. Mol Med Today. 1997;3:31–38. doi: 10.1016/S1357-4310(96)10053-8. [DOI] [PubMed] [Google Scholar]
- 16.Peyret N, Seneviratne P A, Allawi H T, SantaLucia J., Jr Biochemistry. 1999;38:3468–3477. doi: 10.1021/bi9825091. [DOI] [PubMed] [Google Scholar]
- 17.Hovinen J, Guzaev A, Azhayeva E, Azhayev A, Lonnberg H. J Org Chem. 1995;60:2205–2209. [Google Scholar]
- 18.Fox S W, Minard F N. J Am Chem Soc. 1952;74:2085–2087. [Google Scholar]
- 19.Arar K, Monsigny M, Mayer R. Tetrahedron Lett. 1993;34:8087–8090. [Google Scholar]
- 20.Asseline U, Bonfils E, Kurfurst R, Chassignol M, Roig V, Nguyen T T. Tetrahedron. 1992;48:1233–1254. [Google Scholar]
- 21.Lombardo A. J Chem Educ. 1982;59:887–888. [Google Scholar]
- 22.Bender M L. J Am Chem Soc. 1957;79:1652–1655. [Google Scholar]
- 23.Bruice T C, Schmir G L. J Am Chem Soc. 1957;79:1663–1667. [Google Scholar]
- 24.Bruice T C, Sturtevant J M. J Am Chem Soc. 1959;81:2860. [Google Scholar]
- 25.Gesson J P, Jacquesy J C, Mondon M, Petit P, Renoux B, Andrianomenjanahary S, Dufat-Trinh Van H, Koch M, Michel S, Tillequin F, et al. Anticancer Drug Des. 1994;9:409–423. [PubMed] [Google Scholar]
- 26.Lougerstay-Madec R, Florent J-C, Monneret C, Nemati F, Poupon M F. Anti-Cancer Drug Des. 1998;13:995–1007. [PubMed] [Google Scholar]
- 27.Madec-Lougerstay R, Florent J-C, Monneret C. J. Chem. Soc., Perkin Trans. 1. 1999. , 1369–1376. [Google Scholar]
- 28.Stoops J K, Horgan D J, Runnegar M T C, De Jersey J, Webb E C, Zerner B. Biochemistry. 1969;8:2026–2033. doi: 10.1021/bi00833a037. [DOI] [PubMed] [Google Scholar]
- 29.Chakravarty P K, Carl P L, Weber M J, Katzenellenbogen J A. J Med Chem. 1983;26:633–638. doi: 10.1021/jm00359a003. [DOI] [PubMed] [Google Scholar]
- 30.Campbell D A, Gong B, Kochersperger L M, Yonkovich S, Gallop M A, Schultz P G. J Am Chem Soc. 1994;116:2165–2166. [Google Scholar]
- 31.Miyazawa T. Amino Acids. 1999;16:191–213. doi: 10.1007/BF01388169. [DOI] [PubMed] [Google Scholar]
- 32.Frazier, K. A. (1990) U.S. Patent 4,980,482.
- 33.Milstien J B, Fife T H. J Am Chem Soc. 1968;90:2164–2168. doi: 10.1021/ja01010a039. [DOI] [PubMed] [Google Scholar]
- 34.SantaLucia J., Jr Proc Natl Acad Sci USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]