

Abstract
VP16 is a virion phosphoprotein of herpes simplex virus and a transcriptional activator of the viral immediate-early (IE) genes. We identified four novel VP16 phosphorylation sites (Ser18, Ser353, Ser411, and Ser452) at late times in infection, but found no evidence of phosphorylation of Ser375, a residue reportedly phosphorylated when VP16 is expressed from a transfected plasmid. A virus carrying a S375A mutation of VP16 was viable in cell culture but with a slow growth rate. The association of the mutant VP16 protein with IE gene promoters and subsequent IE gene expression was markedly reduced during infection, consistent with prior transfection and in vitro results. Surprisingly, the association of Oct-1 with IE promoters was also diminished during infection by the mutant strain. We propose that Ser375 is important for the interaction of VP16 with Oct-1, and that the interaction is required to enable both proteins to bind to IE promoters.
Keywords: transcriptional activation, Vwm65, α-trans-inducing factor, Oct-1, immediate-early gene, chromatin immunoprecipitation
INTRODUCTION
The virion protein VP16 of herpes simplex virus type 1 (HSV-1) serves multiple functions during lytic infection. VP16 (also known as Vmw65 and α-TIF) is most prominently known as a transcriptional activator of the viral immediate early (IE) genes (O’Hare, 1993; Triezenberg, 1995). Two types of cis-acting elements in IE promoters mediate activation by VP16. A motif rich in guanosine and adenosine nucleotides binds the host heterodimeric protein GABP (LaMarco and McKnight, 1989; Triezenberg, LaMarco, and McKnight, 1988), but no direct interaction of GABP with VP16 has yet been demonstrated. A second element, with a consensus sequence of TAATGARAT (where R represents either purine nucleotide), is bound by a protein complex comprising VP16 and two cellular proteins, the DNA binding protein Oct-1 and an accessory protein HCF-1 (Gerster and Roeder, 1988; Katan et al., 1990; Kristie, LeBowitz, and Sharp, 1989; O’Hare and Goding, 1988; Xiao and Capone, 1990). VP16 interacts directly with both Oct-1 and HCF-1 through adjacent regions in the core domain of VP16. The interaction with Oct-1 is mediated by a region spanning amino acids 370–390 (Greaves and O’Hare, 1990; Lai and Herr, 1997) of VP16, and the interaction with HCF-1 is mediated by VP16 residues 355–370 (Lai and Herr, 1997; Shaw, Knez, and Capone, 1995; Simmen et al., 1997; Wu et al., 1994). Although crystallographic and small-angle X-ray scattering structures of the core domain (residues 49–410) of VP16 have been described (Grossmann et al., 2001; Liu et al., 1999), the region spanning these two protein interaction motifs was disordered in that structure.
The transcriptional activation domain (TAD) of VP16 resides within the carboxyl-terminal residues 411–490 (Greaves and O’Hare, 1989; Triezenberg, Kingsbury, and McKnight, 1988; Werstuck and Capone, 1989). Within that domain are two subregions, spanning 411–456 and 450–490, either of which can function to activate expression of target genes (Regier, Shen, and Triezenberg, 1993; Tal-Singer et al., 1999; Walker, Greaves, and O’Hare, 1993). Although each of these subregions is rich in acidic amino acids, mutational analyses have demonstrated that bulky hydrophobic residues are particularly critical to their transcriptional function (Cress and Triezenberg, 1991b; Regier, Shen, and Triezenberg, 1993; Sullivan et al., 1998). The TAD of VP16 can interact with a number of basal transcription factors including TBP and TAF9 from TFIID, TFIIA, TFIIB, and TFIIH (Triezenberg, 1995). Furthermore, the VP16 TAD can interact genetically and physically with coactivator protein complexes including the Mediator complex of the RNA polymerase II holoenzyme (Mittler et al., 2003; Yang et al., 2004), histone acetyltransferases (Berger et al., 1992; Memedula and Belmont, 2003; Utley et al., 1998; Wang, Grossman, and Kieff, 2000), and ATP-dependent remodeling enzymes homologous to the yeast SWI/SNF complex (Memedula and Belmont, 2003; Neely et al., 1999). Recent evidence directly implicates the VP16 AD in recruiting the mammalian histone acetyltransferases p300 and CBP and the remodeling enzymes BRG1 and hBRM to viral IE gene promoters during lytic infection (Herrera and Triezenberg, 2004).
In addition to its well-known transcriptional function, VP16 also plays other roles in the infectious cycle of HSV-1. VP16 is an abundant component of the tegument compartment of the virion. An HSV-1 strain from which the entire VP16 gene had been deleted is not viable in noncomplementing cells, an indication that VP16 is an essential protein for virion assembly (Weinheimer et al., 1992). A temperature-sensitive mutant of HSV-2 has a conditional defect in virion stability that maps to the VP16 gene (Moss, 1989). Temperature-sensitive mutants of HSV-1 have also been constructed by replacing specific cysteine residues of VP16 with glycine (Poon and Roizman, 1995), resulting in defects in virion maturation. In contrast, deletion of only the VP16 activation domain resulted in efficient production of virions that are nonetheless poorly infectious because of the reduced expression of IE genes (Smiley and Duncan, 1997; Tal-Singer et al., 1999). VP16 interacts with other virion proteins including the vhs protein, which induces degradation of mRNAs in infected cells (Strand, Vanheyningen, and Leib, 2004). Several lines of evidence suggest that this activity is likely regulated at least in part by interaction with VP16 (Elliott, Mouzakitis, and O’Hare, 1995; Knez, Bilan, and Capone, 2003; Lam et al., 1996; Smibert et al., 1994; Strand and Leib, 2004). VP16 also interacts with another virion protein, VP22, (Elliott, Mouzakitis, and O’Hare, 1995), likely through sequences within the VP16 TAD, but the relevance of this interaction in the context of lytic infection have yet to be defined.
VP16 has long been known as a phosphoprotein in infected cells and in virions (Gibson and Roizman, 1974; Lemaster and Roizman, 1980), although the regulatory role (if any) of this modification has yet to be defined. VP16 phosphorylation upon entry into the infected cell may facilitate its dissociation from the virion and its intracellular localization (Morrison et al., 1998; Morrison, Wang, and Meredith, 1998). One predominant phosphorylation site, Ser375, was identified in VP16 protein expressed from a transfected plasmid (O’Reilly, Hanscombe, and O’Hare, 1997). This site is particularly interesting because it lies within the region implicated in binding to Oct-1. Moreover, mutation of Ser375 to alanine disrupted interaction with Oct-1 and thereby ablated the ability of VP16 to activate expression of reporter genes. However, whether phosphorylation per se modifies the interaction of VP16 with Oct-1 has not been definitively established, nor was this site of phosphorylation confirmed in infected cell lysates.
To further advance our understanding of VP16 phosphorylation and the potential regulatory implications thereof, we mapped phosphorylation sites in VP16 protein isolated from cells at late times of infection. We identified four phosphorylated Ser residues, but saw no evidence of phosphorylation of Ser375. A recombinant virus bearing a Ser375Ala mutation in VP16 was viable but showed diminished growth kinetics following either high or low multiplicity infections. This defect corresponds to a significant reduction in IE gene expression during infections and to a diminished presence of both VP16 and Oct-1 proteins on IE gene promoters. These results support a model in which Ser375 is essential for Oct-1 interaction and IE gene activation, but do not directly implicate phosphorylation in regulating that activity.
RESULTS
Sites of VP16 phosphorylation at late times post-infection
To establish the phosphorylation status of VP16 in infected cells, HeLa cells were infected with wild type HSV-1 (KOS) at a multiplicity of infection (moi) of 5. [32P]-orthophosphate was added to the culture medium at 1.5 hours post-infection (hpi). Infected cells was harvested and lysed at 8 hpi. VP16 was isolated by immunoprecipitation (IP) using the monoclonal antibody LP1, which recognizes an epitope near the amino terminus of VP16. The proteins in the IP pellets were separated by SDS-PAGE and radiolabeled proteins were visualized by autoradiography. A [32P]-labeled band with an apparent molecular weight of 65 kDa was observed for samples from the IP pellet (Fig. 1 panel B). Lysates of non-infected cells, processed through a parallel radiolabeling and IP protocol, did not display a comparable band (data not shown). The radiolabeled species co-migrated with VP16 visualized by immunoblotting (Fig. 1C). These results confirm the presence of phosphorylated VP16 in infected cell lysates. Samples of immunoprecipitated material from virions harvested at 14 hpi and purified by density gradient centrifugation showed a comparable radiolabeled band (data not shown), indicating that the VP16 protein in virions is also phosphorylated.
Figure 1.
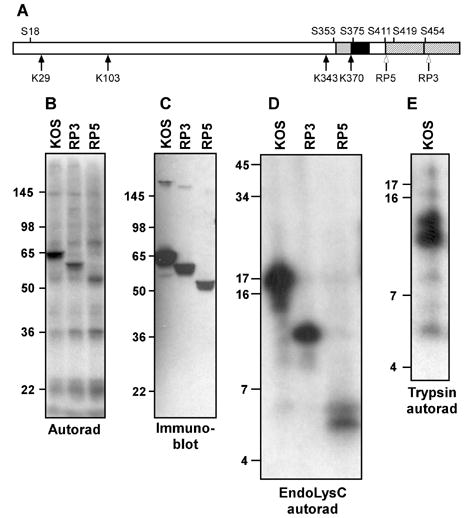
Mapping VP16 phosphorylation sites using deletion mutants and peptide mapping. A. Schematic diagram of VP16. The two subregions of the C-terminal activation domain are denoted by hatching. Regions implicated in interaction with Oct-1 and HCF-1 are indicated by black and grey boxes, respectively. The four lysine residues in VP16 (K) and some of the serine residues (S) are indicated. Open arrows mark the truncations of the VP16 open reading frame in the viral strains RP3 and RP5. B. Autoradiogram of material precipitated using a VP16-specific monoclonal antibody (LP1) from HeLa cells infected by virus strains KOS, RP3 and RP5 and radiolabeled with [32P]-orthophosphate from 1.5 to 8 hour post-infection. The relative positions of protein molecular weight standards following separation on a 10% SDS-PAGE gel are indicated (in kDa). C. Immunoblot of a gel in parallel to that shown in panel B, probed with a VP16-specific polyclonal antibody (C8). D. Autoradiogram of radiolabeled VP16 fragments following digestion by lysyl endopeptidase (LysC), separated on a 16% polyacrylamide gel. E. Autoradiogram of radiolabeled VP16 fragments following digestion by trypsin and separation on a 16% polyacrylamide gel.
Two viral strains encoding truncation mutants of VP16 (Tal-Singer et al., 1999) were used initially to map the location of the phosphorylation site(s). The RP3 strain encodes a VP16 protein truncated at amino acid 456 (Fig. 1A) and thus lacks the carboxyl terminal half of the transcriptional activation domain. The RP5 strain encodes a protein truncated at residue 412 and thus lacks the entire activation domain. Both of these strains are viable without complementation, but RP5 is less efficient at initiating infection (Tal-Singer et al., 1999). Cells infected with KOS, RP3 or RP5 were radiolabeled as described above. Cell lysates were harvested at 8 hpi and were immunoprecipitated using the VP16-specific antibody LP1. Infection by RP3 and RP5 gave rise to a phosphorylated species that corresponded to the expected apparent molecular weight of the truncated proteins (58 KDa and 52 KDa respectively; Fig. 1B). The identity of the labeled species was confirmed by subsequent immunoblotting of the labeled extract (Fig. 1C). These results indicate that one or more phosphorylation sites exist within the N-terminal 412 amino acids of VP16, but do not preclude the possibility of additional phosphorylation sites within the C-terminal 88 residues. Moreover, these results indicate that the activation domain is not necessary for phosphorylation of VP16.
To further define the site or sites of phosphorylation, [32P]-labeled VP16 was immunoprecipitated from lysates of cells infected with KOS, RP3 and RP5 and then digested with lysylendopeptidase (LysC). Given that the four lysine residues in the VP16 protein sequence are located at positions 29, 103, 343 and 370 (Fig. 1A), complete digestion with LysC is expected to produce five fragments of approximate molecular weight 3.0 kDa (aa 1–29), 8.0 kDa (aa 30–103), 28 kDa (aa 104–343), 3.1 kDa (aa 344–370) and 13 kDa (aa371–490). The VP16 proteins from strains RP3 and RP5 differ from full-length VP16 only in the latter fragment. Digestion of the sample from KOS-infected cells yielded a single predominant phosphorylated species migrating with an apparent molecular weight of 17 kDa (Fig 1D). The truncated VP16 from RP3 gave rise to a single radioactive band that migrated at 11 kDa. RP5 gave rise to a prominent band migrating at 5.4 kDa. Each lane contains a faint band with a mobility corresponding to a protein of approximately 6.0 kDa. The predominant bands in both the RP3 and the RP5 samples migrated as would be predicted for the carboxyl-terminal fragment of the LysC digestion. These observations indicate that the major phosphorylation sites of VP16 are present within the C-terminal fragment, with at least one prominent site of VP16 phosphorylation within the fragment spanning aa 371–412 (as observed for RP5). Serine residues are present within this fragment at positions 375, 400, and 411 (Fig. 1A). No radiolabeled bands were present that correspond to the 28 kDa and 8 kDa fragments expected from LysC digestions, which are common to the three forms of VP16 tested. This indicates that phosphorylation is not present in VP16 between amino acids 30 and 343. The identity of the fragment giving rise to the relatively weak signal at 6.0 kDa is ambiguous; the two smallest products expected from LysC digestion are 3.0 kDa (aa 1–29) and 3.1 kDa (aa 344–370), and the mobility of such small polypeptides can vary significantly in SDS-PAGE gels.
To further define the positions at which VP16 is phosphorylated, the radiolabeled and immunoprecipitated VP16 protein was digested with trypsin. Complete digestion of VP16 with trypsin is expected to yield some 43 fragments, all but one with molecular weights less than 4 kDa. The exception is the C-terminal fragment of 8.5 kDa, which includes the acidic transcriptional activation domain. Trypsin digestion of VP16 immunoprecipitated from KOS-infected HeLa cells gave rise to a major phosphorylated species that migrated with apparent molecular weight of 11 kDa and a second labeled fragment of approximately 12 kDa (Fig. 1E). These bands likely correspond to the C-terminal fragment (410–490) and a partial digestion product that includes an additional 11 residues. This result implies that one or more of the Ser residues C-terminal to amino acid 410 (i.e., Ser411, 419, 452, and 462) are phosphorylated at late times in infected cells. A fainter band with mobility corresponding to approximately 5 kDa is also apparent in this sample; the identity of the peptide that this band represents is uncertain.
To determine the phosphoamino acid complement of VP16, [32P]-labeled protein was immunoprecipitated from HeLa cells infected with KOS, RP3 or RP5. The proteins in the IP pellet were acid-hydrolyzed and the component amino acids were resolved by thin layer electrophoresis (Fig. 2). In all three cases, a radioactive species was found which co-migrated with the phosphoserine standard. No labeled species were found to correlate with the phosphothreonine or phosphotyrosine samples. These results indicate serine residues but not tyrosine or threonine residues are phosphorylated in VP16 derived from late stages of infection of HeLa cells.
Figure 2.
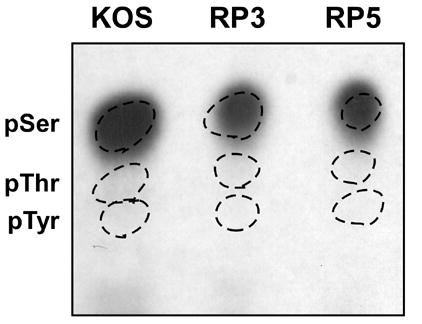
Phosphoamino acid analysis of VP16 proteins radiolabeled from 1.5 to 8 hours following infection with virus strains KOS, RP3 and RP5. The VP16 protein was isolated from infected cell lysates by immunoprecipitation with monoclonal antibody LP1 followed by SDS-PAGE. Proteins were transferred to a PVDF membrane and the band corresponding to VP16 was excised. Proteins were hydrolyzed in 6 N HCl for 1 hour at 100° C. Samples were mixed with unlabeled phosphoserine, phosphothreonine and phosphotyrosine standards and separated by thin-layer electrophoresis on a cellulose plate at pH 2.5. Phosphoamino acid standards were visualized using ninhydrin (dotted circles) and radiolabeled amino acids were detected by autoradiography.
Mass spectrometric analysis of phosphorylated peptides
The VP16 phosphorylation sites were mapped more precisely by proteolytic digestion and mass spectrometry. Non-radiolabeled VP16 was purified by immunoprecipitation from 109 HeLa cells infected with wildtype virus. The protein was eluted from the immunoprecipitation pellet at high pH and the resulting eluate was separated on a 10% SDS-PAGE gel. The gel was stained with Coomassie Blue and the band corresponding to VP16 was excised, washed in acetonitrile, dried and frozen at −80°C. Separate samples were digested with trypsin, chymotrypsin and endopeptidase AspN and subjected to microcapillary HPLC coupled with tandem mass spectrometry (μLC/MS/MS).
A summary of the results from all digestions is given in Figure 3 and Table 2. Fragments spanning approximately 79% of the VP16 protein were observed in the MS/MS spectra (Fig. 3, bold and underlined type). MS/MS spectra were consistent with the presence of both phosphorylated and the unphosphorylated peptides encompassing Ser18, Ser353 and Ser452 (Table 2). Ser18 and Ser353 reside in the two smallest LysC fragments that may have escaped detection on our SDS-PAGE gels (Fig.1). No evidence was found for phosphorylation at Ser44, Ser51, Ser90, Ser106, Ser110, Se154, Ser167, Ser309, and Ser333, consistent with the absence of radiolabeling in the LysC fragments in which these amino acids reside. Only spectra consistent with absence of phosphorylation were observed for Ser346, Ser348, Ser349, or Ser355; these residues all fall within one of the small LysC fragments. Six serine residues were not covered by fragments observed by MS. Four of these six (Ser72, Ser177, Ser186 and Ser262) are almost certainly unphosphorylated, since they reside in LysC fragments that were not radiolabeled. The remaining two, Ser 411 and Ser419 both reside within the [32P]-labeled carboxyterminal trypsin fragment (aa411–490). Their phosphorylation status could not be definitively determined by the available peptide mapping and MS data.
Figure 3.

Peptide sequences identified following LC-MS/MS analysis of VP16 proteins isolated at 8 hour post-infection with HSV-1 strain KOS. VP16 was isolated by immunoprecipitation and SDS-PAGE and then digested with trypsin or endoproteinase AspN. The entire VP16 amino acid sequence is shown. Boldface and underlined type indicates residues within peptide fragments identified following μLC-MS/MS. Asterisks indicate Ser residues found to be phosphorylated. A carat (^) indicates Ser375, for which no phosphorylation was observed. A hatch (#) indicates Ser411, the phosphorylation of which was implicated by peptide mapping experiments but not confirmed by μLC-MS/MS.
TABLE 2.
Phosphorylated and non-phosphorylated peptides spanning Ser18, Ser353, Ser375 or Ser452 as detected by MS/MS
| Residue | Coverage | Representative spectra (peptide)a |
|---|---|---|
| Serine 18 | 4–29 | LVDELFADMDADGASPPPPRPAGGPK |
| 5–29 | VDELFADMDADGASPPPPRPAGGPK | |
| 5–29 | VDELFADMDADGASPPPPRPAGGPK | |
| 6–29 | DELFADMDADGASPPPPRPAGGPK | |
| Serine 353 | 342–360 | AKLDSYSSFTTSPSEAVMR |
| 344–360 | LDSYSSFTTSPSEAVMR | |
| 344–360 | LDSYSSFTTSPSEAVMR | |
| 345–360 | DSYSSFTTSPSEAVMR | |
| 348–360 | SSFTTSPSEAVMR | |
| 351–359 | TTSPSEAVM | |
| Serine 375 | 369–398 | TKNNYGSTIEGLLDLPDDDAPEEAGLAAPR |
| 369–401 | TKNNYGSTIEGLLDLPDDDAPEEAGLAAPRLSF | |
| 371–398 | NNYGSTIEGLLDLPDDDAPEEAGLAAPR | |
| 372–398 | NYGSTIEGLLDLPDDDAPEEAGLAAPR | |
| 374–401 | GSTIEGLLDLPDDDAPEEAGLAAPRLSF | |
| Serine 452 | 445–460 | DMLGDGDSPGPGFTPH |
| 449–460 | DGDSPGPGFTPH | |
| 449–460 | DGDSPGPGFTPH |
Bold-face underlined S indicates phosphorylation of corresponding serine
No evidence of phosphorylation at Ser375 was found. All five spectra for peptides encompassing this serine correspond to the unphosphorylated form (Table 2). To explore this question more thoroughly, the analysis was repeated using targeted ion tandem mass spectrometry. In this method, peptides corresponding to the expected phosphorylated and non-phosphorylated masses of a particular digestion were selected in the first dimension MS and then subjected to fragmentation and sequencing by the second dimension MS. Although this more focused analysis should increase the likelihood of detecting low-abundance species, no phosphorylated fragments corresponding to phosphorylation at Ser375 were observed (data not shown).
Mutation of Ser375 affects IE gene expression and viral growth
The results described in the preceding paragraphs indicate that although proteolytic fragments encompassing Ser375 can be phosphorylated, no phosphorylation of that residue itself is evident in mass spectra. However, this site had previously been shown to be critical for the interaction of VP16 with Oct-1 (Greaves and O’Hare, 1990; Lai and Herr, 1997; O’Reilly, Hanscombe, and O’Hare, 1997) in vitro and for transcriptional activation in transfected cells. To probe the role of Ser375 in the function of VP16 during infection by HSV-1, a virus strain (designated SJO2) was generated in which codon 375 of VP16 was altered to specify alanine rather than serine. The presence of the mutation in the recombinant viral genome was confirmed by sequencing a PCR product spanning the site of interest amplified from the viral DNA. Southern blotting with a probe spanning the open reading frame and parts of the 3′- and 5′-UTR confirmed that the mutant VP16 gene had been inserted into the native VP16 locus (data not shown).
To determine whether the S375A mutation affected the phosphorylation of VP16, a radiolabeled immunoprecipitate from HeLa cells infected with SJO2 was compared to that from KOS-infected cells. The autoradiogram in Fig. 4A reveals in both samples a phosphorylated band at 65 KDa, corresponding to the expected size for VP16 as detected in the immunoblot (Fig 4B). The intensity of the signal is comparable in the two samples, suggesting no dramatic change in the level of phosphorylation of the S375A version of VP16. Phosphoamino acid analysis (data not shown) indicated that the only radiolabeled amino acid in the SJO2 VP16 sample was phosphoserine, indicating that the S375A mutation did not shift phosphorylation to an alternative amino acid (such as the adjacent Thr376). The digestion of both the KOS and the SJO2 samples by LysC gave rise to a band of an apparent molecular weight of 17 KDa (Fig. 4C). Again, the intensities of these peptide signals were similar between the two samples. From this experiment we conclude that Ser375 is not necessary for phosphorylation of VP16 and Ser375 is not a major site of phosphorylation at late times in infection.
Figure 4.
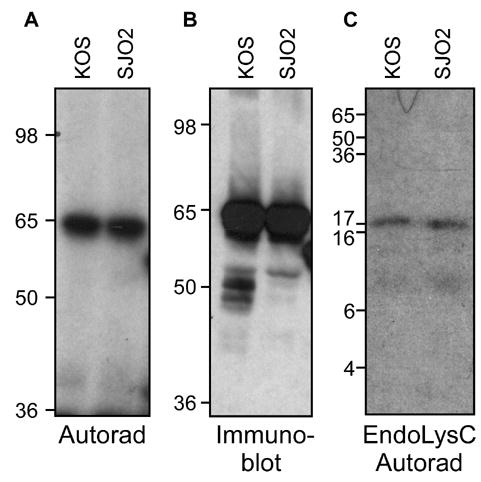
Phosphorylation of a Ser375Ala mutant of VP16. Virus strain SJO2 was constructed with an alanine codon replacing serine codon 375. HeLa cells infected with KOS or SJO2 were radiolabeled and the VP16 proteins were immunoprecipitated and separated by SDS-PAGE as described in Fig. 1. A. Autoradiogram, as described in Fig. 1. B. Immunoblot using VP16-specific antiserum C8. C. Autoradiogram of gel-isolated VP16 proteins subjected to digestion by LysC.
Since Ser375 has been implicated in processes required for IE gene expression, it was possible that the loss of the site would cause alteration in expression of these genes, regardless of phosphorylation status. To examine the hypothesis that mutation of Ser375 would diminish the VP16-mediated activation of the IE genes and would thereby result in a reduction in virus viability, the growth kinetics of SJO2 were examined. Figure 5A shows that the yield of progeny virus following high-multiplicity infection by SJO2 was reduced by approximately ten-fold relative to infection by the wildtype virus KOS. Given this modest impact on viral replication, we considered the possibility that the Ser375Ala missense mutation may be hypomorphic and thus its effect might be suppressed by the exaggerated VP16 concentration in cells infected at high multiplicity. A comparable defect in yield of infectious virus was observed following low-multiplicity infection (Fig. 5B). We conclude that the Ser375Ala mutation in VP16 affects the yield of infectious virus in a manner unaffected by multiplicity.
Figure 5.
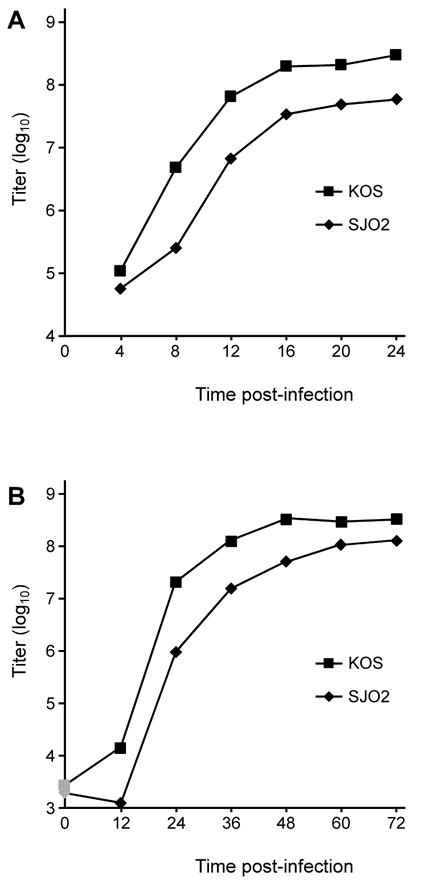
Growth curves of SJO2 and KOS. A. Vero cells were infected at high multiplicity (10 pfu/cell) with KOS or SJO2. At 4 h intervals, infected cells were lysed into the growth medium by scraping and sonication. Following centrifugation to remove cell debris, aliquots of the resulting supernatant were titered on Vero cells. B. Vero cells were infected with KOS or SJO2 at low multiplicity (0.01 pfu/cell) and harvested at 12 h intervals. Graphs represent the average of two biological replicates, which showed a range of less than 20% at every timepoint.
The mutation of Ser375 to Ala likely affects viral replication because of reduced IE gene expression arising from defective interaction of VP16 with Oct-1. This hypothesis is based on evidence of reduced promoter binding in vitro and decreased activation of IE gene promoters when VP16-Ser375SA is expressed by transfection (O’Reilly, Hanscombe, and O’Hare, 1997). Therefore, we tested both the expression of IE genes and the association of VP16 with IE gene promoters during HSV infection by SJO2. Expression of viral IE genes in HeLa cells infected by KOS, SJO2 or RP5 was measured using quantitative real-time RT-PCR assays. In Fig. 6, the expression of each IE gene transcript during infection by the mutant viruses is depicted relative to the expression detected in KOS infection. These results clearly indicate a dramatic defect in expression of all IE genes at 2 hpi during SJO2 infection, in some instances as severe as the complete deletion of the VP16 activation domain (RP5). The defect in IE gene expression in SJO2 infection is not quite as severe at 4 hpi, (depicted in Fig. 6B), suggesting that IE gene expression in SJO2 infection exceeds that in RP5 infection and perhaps accounting for the less dramatic effect on viral growth kinetics. We conclude that the mutation of Ser375 to Ala profoundly affects the ability of VP16 to activate viral IE genes during lytic infection.
Figure 6.
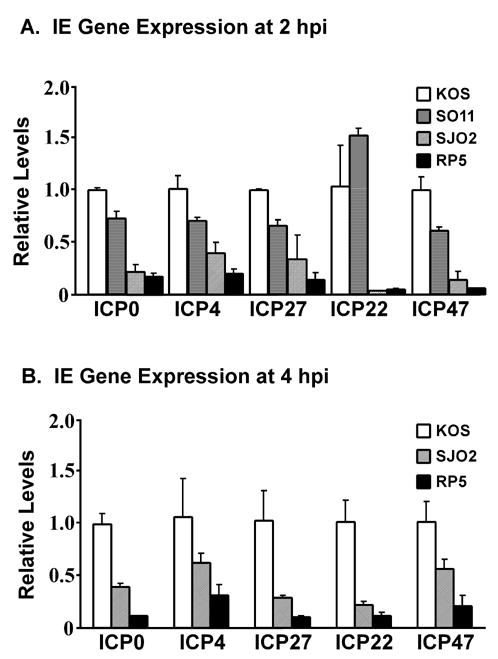
Immediate-early gene expression following infection by HSV-1 strains KOS, SJO2 and RP5. HeLa cells were infected at a multiplicity of 1 pfu/cell (KOS) or a corresponding number of virions (SJO2, RP5). RNA was harvested at 2 hpi (panel A) or 4 hpi (panel B). IE gene expression was assayed by quantitative real-time PCR following reverse transcriptase reactions. Expression levels were normalized to 18S rRNAs amplified in parallel Q-PCR reactions and are expressed relative to the levels observed in KOS infection. Error bars represent the range of two biological replicates each measured in duplicate PCRs.
Control experiments ruled out several potential alternative explanations for the reduced expression levels of IE mRNAs in SJO2-infected cells. Comparable defects in IE mRNA expression were observed when parallel experiments were performed in the presence of cycloheximide (Fig. 7A), indicating that the reduced levels are not a consequence of IE protein activity and thus are likely a direct effect of the VP16 mutation. Moreover, the effects of the SJO2 mutation were completely suppressed by biochemical complementation with wildtype VP16 protein (Fig. 7B) that was incorporated into virions of virus strain SJO2 grown in 16–8 cells, which carry a wildtype VP16 gene (Tal-Singer et al., 1999; Weinheimer et al., 1992). This result indicates that no spurious mutation in the SJO2 genome is likely to be responsible for its defects in IE gene activation.
Figure 7.
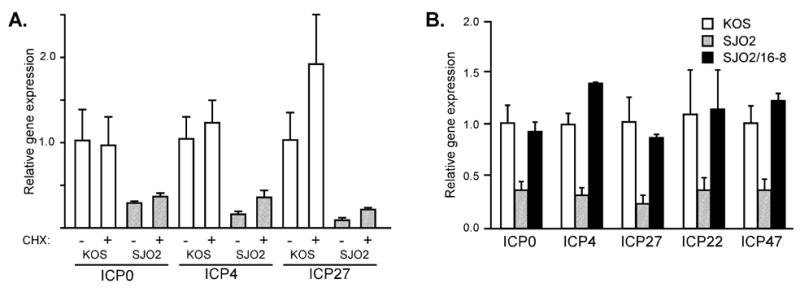
Immediate-early gene expression in cells infected with HSV-1 strains KOS and SJO2. A. HeLa cells were infected with KOS (open bars) or SJO2 (hatched bars) in the presence or absence of cycloheximide (CHX). Total cellular RNA was harvested at 2 hpi and IE mRNA levels were assayed by quantitative real-time PCR as in Fig. 6. B. HeLa cells were infected with KOS (open bars), SJO2 (hatched bars), or SJO2 virions produced in 16-8 cells (filled bars). IE mRNA levels were assayed as in Fig. 6.
Gel-shift experiments assessing the in vitro binding of VP16, Oct-1 and HCF to radiolabeled DNA fragments have indicated that the S375A mutation prevents the association of VP16 with Oct-1 and thus disrupts formation of the VP16-induced complex on the TAATGARAT elements of IE gene promoters (O’Reilly, Hanscombe, and O’Hare, 1997). To test directly whether the S375A mutation affects the association of VP16 and Oct-1 with IE promoters during lytic infection, we performed cross-linking and immunoprecipitation experiments. We have previously described the use of ChIP assays to detect VP16 and Oct-1 proteins at IE gene promoters during infection by the wildtype virus strain KOS (Herrera and Triezenberg, 2004). Two hours after HeLa cells were infected with KOS or SJO2, formaldehyde was added to the culture medium to crosslink proteins and DNA. Cell nuclei were harvested and then sonicated to shear the DNA to fragment sizes primarily within the range of 300 to 500 bp. Samples of the lysates were then immunoprecipitated with antibodies specific to VP16 or to Oct-1. After the IP pellets were resuspended, the crosslinks were reversed by incubation at 60° C. Quantitative real-time PCR assays were used to measure the amount of the ICP0 and ICP27 promoter DNA present in each immunoprecipitate. The VP16 promoter was assayed as a negative control, since neither VP16 nor Oct-1 should bind there. The cellular U3 snRNA promoter was used as a positive control for Oct-1 binding. Fig. 8A indicates that immunoprecipitation of VP16 brought down much less of the IE promoters from SJO2-infected cells than from KOS infected cells. This observation is consistent with the hypothesis that Ser375 is important for the interaction of VP16 with Oct-1 and thus with IE promoter sequences during lytic infection. Surprisingly, the association of Oct-1 with IE gene promoters was also markedly reduced in SJO2-infected cells (Fig. 8B). The presence of Oct-1 at the U3 snRNA promoter was at least as strong in the SJO2 infection as in KOS infection, confirming that the crosslinking and IP reactions occurred with comparable efficiency. We conclude that the occupancy of viral IE promoters by both VP16 and Oct-1 depends on the interaction of these two proteins, and that this interaction is disrupted by the Ser375Ala mutation. One possible explanation for this outcome is that the Ser375Ala mutation might affect some step of the infection process prior to the delivery of viral DNA to the nucleus of infected cells. To test this hypothesis, parallel ChIP assays were performed using an antibody that recognizes an epitope in the carboxyl terminus of histone H3. Comparable levels of gC promoter DNA were found in the H3 IP pellets for KOS and SJO2 infection, whereas significantly less of the ICP0 promoter DNA was found in the KOS sample (Fig. 8C). This result is consistent with our previous observation that VP16 is responsible for a diminished presence of H3 at IE promoters (Herrera and Triezenberg, 2004). Moreover, the amounts of viral DNA in the infected cell nuclei were comparable for the KOS and SJO2 infections (Fig. 8D), confirming that the mutant virus was fully capable of delivering viral DNA prior to IE gene expression.
Figure 8.
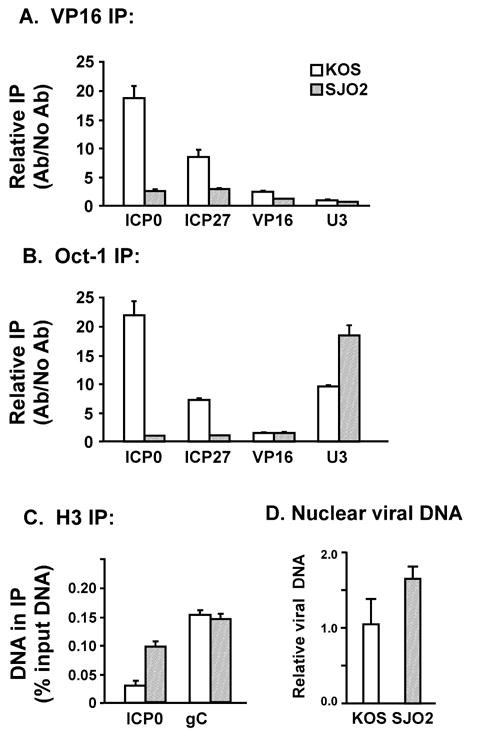
Association of VP16 and Oct-1 with IE promoters during infection by HSV-1 strains KOS and SJO2. At 2 h post-infection, proteins and nucleic acids were crosslinked by formaldehyde. Nuclei were isolated and sonicated to shear DNA to small fragments (predominantly 300–500 bp). Immunoprecipitation by antibodies specific to VP16 (panel A), Oct-1 (panel B), or H3 (panel C) was followed by quantitative real-time PCR to detect DNA fragments corresponding to IE gene promoters (ICP0 and ICP27), a late gene promoter (VP16 or gC) and a cellular promoter with an Oct-1 binding site (U3 snRNA). For panels A and B, the abundance of a given DNA in the IP pellet is indicated relative to its abundance in a control precipitation without antibody. For panel C, the abundance of a given DNA in the IP pellet is indicated relative to its abundance in input DNA (prior to IP). The amounts of viral DNA in nuclear DNA preparations from infected cells were assessed using quantitative PCR with primers specific for the VP16 gene (panel D), normalized to cellular DNA fragments from the U3 and IFN-β promoters and expressed relative to levels in wildtype-infected cells.
Mutation of Ser411 has little effect on IE gene regulation or virus growth
The phosphopeptide mapping of VP16, and particularly the radiolabeling observed for the LysC fragment of the truncated protein produced in RP5 infection, pointed to Ser411 as a site of phosphorylation, but this suggestion was neither supported nor refuted by the MS/MS results. To test whether Ser411 has a significant biological role in HSV infection, a viral strain (designated SO11) was constructed bearing an alanine substitution at this position. To facilitate the identification of the recombinant virus strains, an enhanced green fluorescent protein (EGFP) tag was added to the C-terminus of VP16 in this virus as in a counterpart wildtype virus designated DG1 (D. Greensides, M.S. thesis, 2002). The EGFP-tag had no negative effect on the growth of DG1, nor on a comparable virus constructed in the O’Hare laboratory (La Boissiere et al., 2004). To examine whether the Ser411Ala mutation in SO11 affected phosphorylation of VP16, [32P]-labeled VP16 was isolated from HeLa cells infected with KOS, SO11 and DG1. As expected, a 65 KDa [32P]-labeled band was detected in the KOS sample and a 110 KDa labeled band (representing the VP16-GFP fusion protein) was detected in the DG1 and SO11 samples (Fig 9A). An immunoblot verified the identity of the protein in all three samples (Fig. 9B). This result demonstrated that the EGFP-tag did not abolish phosphorylation of VP16. Moreover, the Ser411Ala mutation did not result in the loss of phosphorylation of VP16. Growth kinetics following infection by SO11 also did not differ significantly from those observed for KOS (data not shown). Modest changes in the expression of IE genes were observed following SO11 infection (Fig. 6A). Specifically, the steady-state RNA levels for ICP0, ICP4, ICP27 andICP47 were reduced to 60–70% of the levels observed in wildtype infections, whereas the level of ICP22 mRNA was elevated by about 50%. These results indicate that Ser 411 is not a dominant site of phosphorylation and that the role of Ser411 in regulating IE gene expression, while detectable, has no substantial effect on lytic replication.
Figure 9.
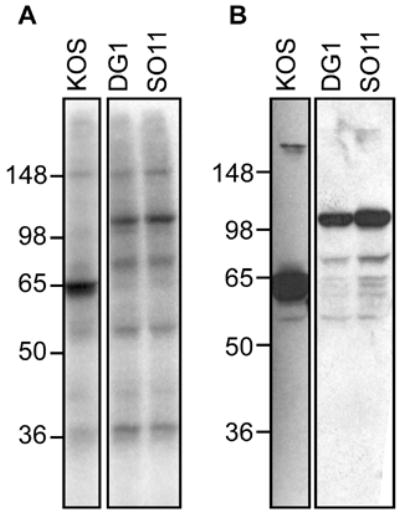
Phosphorylation of a Ser411Ala mutant of VP16. Virus strain DG1 contains a VP16 gene fused to sequences encoding green-fluorescent protein (GFP). SO11 was constructed with an alanine codon replacing serine codon 411, in the DG1 genetic background. HeLa cells infected with KOS, DG1 or SO11 were radiolabeled and the VP16 proteins were immunoprecipitated and separated by SDS-PAGE as described in Fig. 1. A. Autoradiogram, as described in Fig. 1. B. Immunoblot using VP16-specific antiserum C8.
DISCUSSION
Sites of VP16 phosphorylation at late times post-infection
The results reported here reveal several previously unknown aspects of the phosphorylation of VP16 in infected cells at late time in HSV-1 infection. Phosphoamino acid analysis indicated that Ser is the predominant phosphoamino acid in VP16, with no detectable phosphorylation at Thr or Tyr. Three phosphorylation sites (Ser18, Ser353 and Ser 452) were identified by mass spectrometry; a fourth site (Ser411) implicated by peptide mapping was neither confirmed nor refuted by mass spectrometry. None of these four sites have been previously implicated in VP16 phosphorylation. Computer programs designed to predict phosphorylation sites were more or less consistent with these observations. NetPhos 2.0 (http://www.cbs.dtu.dk/services/NetPhos/) identified many potential phosphorylation sites in VP16, but the prediction values for these four sites did not stand out markedly from others for which we observed no phosphorylation. When the DISPHOS program {http://core.ist.temple.edu/pred/pred.html; ref. (Iakoucheva et al., 2004)} was used with a human regulatory protein training set, three of these sites (Ser18, 411 and 452) were picked out and a fourth (Ser353) fell just below the threshold. However, this program also predicted phosphorylation of Thr31 and Thr458, which was not supported by our observations.
The amino acid sequences surrounding Ser18 and Ser452 are remarkably similar (ADGASPPPP and GDGDSPSPE, respectively), suggesting that they might be modified by the same protein kinase. Ser353 is also followed by Pro, but the other flanking residues show little similarity to the first sites mentioned. All three of these SP sites are consistent with the consensus sequence for phosphorylation by kinases in the JNK family. Modification by JNKs can facilitate binding of Pin1 prolylisomerases and thus conformational (and potentially functional) changes in the protein. The amino acid sequence surrounding Ser411 (TRRLSTAPP) resembles the consensus sites for modification by the RSK kinase family (Leighton et al., 1995) or by MSK kinases {reviewed in (Roux and Blenis, 2004)}. In vitro experiments to identify the kinase(s) responsible for phosphorylating VP16 have yielded conflicting results. One report indicated that casein kinase II (CKII) could modify bacterially-expressed VP16 polypeptides, and that this modification was abolished by the mutation of Ser375 to alanine (O’Reilly, Hanscombe, and O’Hare, 1997). We have also observed phosphorylation of recombinant VP16 by CKII in vitro, although in our hands mutation of Ser375 had no quantitative effect on that reaction (SO, unpublished results). In contrast, VP16 protein isolated from virions was not phosphorylated in vitro by CKII, but was modified by protein kinase A (although the site of that modification was not defined) (Morrison et al., 1998; Morrison, Wang, and Meredith, 1998). One hypothesis that might reconcile these observations is that virion-borne VP16 may be already phosphorylated at CKII sites and thus refractory to subsequent modification by CKII.
Using the presently described methods, we were unable to identify sites of phosphorylation of VP16 during the immediate-early stage of infection when VP16 activates transcription of IE genes. The lack of detection might reflect a lack or low levels of active phosphorylation of VP16 at IE times, perhaps because virion-borne VP16 is already phosphorylated and thus refractory to further modification. Alternatively, this negative observation may arise from insufficient sensitivity of the metabolic labeling methods that we employed. Others have reported that VP16 can be phosphorylated in infected cells, but those experiments employed very high infection multiplicities (50 pfu/cell) in the presence of cycloheximide and the sites of this modification were not mapped (Morrison, Wang, and Meredith, 1998). Chemical inhibitors (e.g., roscovitine) of cellular protein kinases can dramatically inhibit viral IE and E gene expression (Jordan, Schang, and Schaffer, 1999; Schang et al., 2002; Schang, Rosenberg, and Schaffer, 1999), but the relevant direct targets of those kinases have not been identified. Thus, the phosphorylation status of VP16 (and specifically Ser375) at IE times of infection remains unresolved.
Potential functions of VP16 phosphorylation sites
None of the four phosphorylation sites identified in this work have been previously implicated in any function of VP16. None of them are highly conserved within the herpes homologs and no mutations of these sites have been seen in forward or reverse genetic screens. But the sites may still offer clues to their function.
Ser18 resides within an N-terminal domain that is dispensable for both transcriptional activation and for VIC formation (Babb et al., 2001; Greaves and O’Hare, 1990; Triezenberg, Kingsbury, and McKnight, 1988; Werstuck and Capone, 1989). The amino acid sequence in this region is poorly conserved among alphaherpesvirus homologs of VP16, with a corresponding SerPro dipeptide evident only in the HSV-2 VP16 protein (Cress and Triezenberg, 1991a). When fused to a Gal4 DNA-binding domain, this region of VP16 displayed modest ability to activate transcription, although the residues most important for that activity include Leu8 and Phe9, some distance from Ser18 (Moriuchi et al., 1995). Thus, at present no clear function can be ascribed to Ser18 or its phosphorylation.
Ser353 resides near a region of VP16 implicated in interaction with HCF-1 (Lai and Herr, 1997; Shaw, Knez, and Capone, 1995; Simmen et al., 1997; Wu et al., 1994). This region was not well-ordered in the crystallographic structure of the VP16 core polypeptide {aa. 49–410; ref. (Liu et al., 1999)}. The heart of the HCF-binding motif encompasses amino acids 361–365 (EHAY) (Lai and Herr, 1997; Simmen et al., 1997; Wu et al., 1994). The associations of two human proteins with HCF-1, Luman and Zhangfei, depend on very similar motifs (Freiman and Herr, 1997; Lu and Misra, 2000; Lu et al., 1998). Using the HCF-1 binding motif as a landmark, the sequence including Ser353 of VP16 (FTTS) aligns well with both Zhangfei (LTTS) and Luman (LSSS), such that the expanded HCF-1 interaction region may have a consensus sequence Φ-S/T-S/T-S-X7–8-E/D-H-X-Y, where Φ denotes a bulky hydrophobic amino acid. Mutations within the E/D-H-x-Y motif of these three proteins disrupt interaction with HCF, but Ser353 and its neighboring residues have not been similarly tested. Nonetheless, this proximity suggests that phosphorylation of Ser353 may influence the binding ofVP16 to HCF-1.
Ser411 is not well-conserved among VP16 homologs, with corresponding Ser residues evident only in the proteins from HSV-2 and perhaps pseudorabies virus. This residue lies in the very C-terminus of the polypeptide used for crystallization of the core domain and was not well-ordered in the resulting structure (Liu et al., 1999). Ser411 also marks the N-terminal boundary of what is considered the canonical activation domain of VP16. The segment of VP16 encompassing Ser411 may be an inert linker between the DNA binding core domain and the activation domain of VP16. This residue has not been implicated in important functions of VP16 by any prior mutational analysis. Our results using a Ser411Ala mutant virus (SO11) revealed no significant changes in phosphorylation or viral growth kinetics, and modest effects on IE gene expression. Thus, phosphorylation of Ser411 apparently does not play a major regulatory role during lytic infection.
Ser452 lies between the two subdomains of the VP16 activation region, typically considered to comprise amino acids 412–456 and 450–490 (Regier, Shen, and Triezenberg, 1993; Sullivan et al., 1998; Walker, Greaves, and O’Hare, 1993). Although Ser452 may simply be part of a linker between these two activation subdomains, it is also conceivable that the relative orientations (and thus the activities) of the activation subdomains are influenced by phosphorylation in this linker. To date, no mutational tests of Ser452 have been reported.
Role of Ser375 in IE gene activation
Ser375 was identified as a site of phosphorylation of VP16 expressed in mammalian cells from transfected plasmids (O’Reilly, Hanscombe, and O’Hare, 1997). Mutations of this residue diminish the ability of VP16 to interact with Oct-1, to form the VIC complex in gel shift assays, and to activate reporter genes driven by IE promoters (Greaves and O’Hare, 1990; Lai and Herr, 1997; O’Reilly, Hanscombe, and O’Hare, 1997). Our peptide mapping and mass spectrometry data revealed no indication of Ser375 phosphorylation at late times during HSV infection. Moreover, a mutant VP16 protein with a substitution of alanine for Ser375 showed no differences in phosphorylation levels or patterns. We conclude that Ser375 is not a significant site for VP16 phosphorylation at late times in infection, when VP16 is robustly expressed.
Although our results neither support nor refute the hypothesis that Ser375 may be phosphorylated at IE stages of infection, they do confirm that Ser375 has an important role in IE gene activation and Oct-1 interaction. During infection by the SJO2 virus bearing a S375A mutation, IE gene expression was significantly diminished but not abolished as during infection by a virus lacking the VP16 activation domain (RP5). This observation is consistent with transcriptional defects observed in transfection analyses of Ser375 mutations (Greaves and O’Hare, 1990; Lai and Herr, 1997; O’Reilly, Hanscombe, and O’Hare, 1997). Thus, the S375A mutation may be considered hypomorphic with respect to transcriptional activation. If the S375A mutation disrupts the interaction of VP16 with Oct-1 (as discussed below) and thus abolishes activation through the TAATGARAT cis elements in IE gene promoters, then the slightly higher levels of IE gene expression in SJO2 infection compared to RP5 infection might arise by the residual action of VP16 through other cis elements such as the GA-rich sequences (LaMarco and McKnight, 1989; Triezenberg, LaMarco, and McKnight, 1988). This model is consistent with an observation that IE gene expression is not abolished upon high-multiplicity infection of Oct-1-deficient mouse cells (Nogueira et al., 2004).
In vitro experiments indicated a reduced ability of the S375A mutant form of VP16 to bind to Oct-1 and to participate in the VP16-induced complex in gel-shift assays (Greaves and O’Hare, 1990; Lai and Herr, 1997; O’Reilly, Hanscombe, and O’Hare, 1997). Thus, we were not surprised to find that this mutation disrupted the ability of VP16 to associate with IE gene promoters during lytic infection. The levels of IE promoter DNA fragments that co-immunoprecipitated with VP16 were markedly reduced in SJO2 infection compared with KOS infection. However, we were surprised to find that the IE promoter fragments were also less abundant in the Oct-1 immunoprecipitation pellets, indicating a reduced occupancy of these promoters by Oct-1 as well as by VP16. This observation indicates that the binding of Oct-1 to DNA at viral IE promoters depends on the interaction of Oct-1 with VP16. Although no such requirement had been revealed from in vitro gel shift experiments, there are indications that VP16 qualitatively alters the interaction of Oct-1 with DNA. For example, VP16 affects the manner in which Oct-1 binds to versions of the TAATGARAT element that do or do not have overlapping consensus octamer motifs (Herr and Cleary, 1995). The “GARAT” motif also helps dictate specificity for VP16-containing complexes (Walker, Hayes, and O’Hare, 1994). Our new observations, however, reveal a quantitative effect indicating that, in vivo, Oct-1 and VP16 together form a novel DNA-binding entity in which VP16 contributes materially to the binding of Oct-1 to viral IE promoters.
Presumably as a consequence of the reduced IE expression levels, the SJO2 virus strain showed comparable 10-fold reductions in viral growth kinetics following both low- and high-multiplicity infection (Fig. 5). The diminished growth curves for SJO2 likely arise from defects in infectivity of the progeny virions, since the yield of viral proteins and DNA (detected by Western blots and quantitative PCR, respectively; data not shown) did not differ significantly from those observed for wildtype viruses. In contrast, other VP16 mutant viruses, including the RP5 strain lacking the VP16AD altogether, show a much greater defect following low-multiplicity infection (Ace et al., 1989; Smiley and Duncan, 1997; Tal-Singer et al., 1999). Perhaps the difference in growth kinetics between SJO2 and RP5 at low multiplicity arises not solely from the modest difference in the abilities of these viruses to activate IE gene expression but from some nontranscriptional function of the VP16AD (e.g., interaction with VP22) that affects the infectivity of the virus.
Our present results do not fully address the question of whether Ser375 phosphorylation per se affects the interaction of VP16 with Oct-1 and the subsequent impact of that effect on IE gene regulation during infection. It may be that the transcriptionally active form of VP16 is phosphorylated at Ser375, and that the lack of phosphorylation at Ser375 late in infection might represent a down-regulation of that function of VP16. More sensitive assays, perhaps using antibodies specifically recognizing a phosphoserine 375 epitope, will be required to resolve this matter.
MATERIALS AND METHODS
Cells and viruses
HeLa and Vero cells obtained from the American Type Culture Collection were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 3.7 g/L sodium bicarbonate and 10% fetal calf serum (FCS; Select fetal calf serum, Atlanta Biologicals). Vero-16-8 cells (Weinheimer et al., 1992) and BHK-MMTV-VP16 cells (Hippenmeyer and Highkin, 1993) express integrated copies of the wild type VP16 gene and were used for complementation of defective virus strains. Vero-16-8 cells and BHK-MMTV-VP16 cells were grown in DMEM with FCS in the presence of Geneticin at 0.9 or 0.5 μg/mL respectively to maintain selection for the transgene.
Viral stocks were generated in Vero cells or Vero-16-8 cells. Titers of all stocks were determined by plaque assays on Vero cells. The HSV-1 strain KOS was the wild type strain used in this study. The 8MA, RP3, RP5, and DG1 strains are derivatives of KOS. In 8MA, the VP16 ORF was replaced with the LacZ gene (Weinheimer et al., 1992). The RP3 and RP5 strains carry a truncated VP16 gene comprising codons 1–454 and 1–413, respectively (Tal-Singer et al., 1999). Strain DG1 expresses full length wild-type VP16 fused at its carboxyl terminus to the enhanced green fluorescent protein (EGFP) (D. Greensides and SJT, unpublished). A virus strain expressing a similar VP16-GFP fusion has been recently described by others (La Boissiere et al., 2004).
Several additional recombinant viruses were derived from KOS for the purposes of this study, essentially as described previously (Tal-Singer et al., 1999). PCR-mediated site-directed mutagenesis was used to generate specific codon substitutions in plasmid pKOS-VP16.2. Mutagenic primer sequences are available upon request. XhoI-PstI restriction fragments bearing the VP16 coding sequences from mutant plasmids were cotransfected with genomic DNA from the VP16-null virus strain 8MA into Vero-16-8 or BHK-MMTV-VP16 cells. Recombinant virus strains were isolated through three rounds of plaque purification. For each strain, the mutation identity was confirmed by PCR amplification of a VP16 gene fragment analyzed for loss of an AccI site at codon 375 or 411, respectively, and then by direct DNA sequencing. Strain SJO2 expresses VP16 altered by a Ser375Ala mutation. Strain SO11 expresses a Ser411Ala mutant of VP16 with a carboxyl-terminal EGFP fusion.
To determine growth curves of various virus strains, Vero cells were plated at a density of 2x105 cells per 60 mm tissue culture plate. After 24 hours, cells were infected with virus at an MOI of 10 (for single-step growth curves) or 0.01 (for multiple-step growth curves). Duplicate samples were harvested at 4 hour intervals (for single-step growth curves) or 12 hour intervals (for multiple-step growth curves). The samples were harvested by dislodging cells into the overlying medium with a disposable scraper. The cells were disrupted by sonication and insoluble cell debris was removed by centrifugation. The titer of each sample was determined in triplicate by plaque assay.
Antibodies
LP1, a monoclonal antibody that recognizes an epitope near the amino terminus of VP16 (McLean et al., 1982), was generously provided by A. Minson and was used for immunoprecipitation of VP16 from infected cell extracts. C8 and LA2–3 are rabbit polyclonal antisera. C8 was raised against VP16 purified from virions (Triezenberg, Kingsbury, and McKnight, 1988). LA2–3 was raised against the bacterially expressed fusion protein Gal4VP16 and recognizes epitopes both within the Gal4 DNA-binding domain and the VP16 transcriptional activation domain (amino acids 410–490) (Sullivan et al., 1998). A rabbit polyclonal antiserum recognizing the human Oct-1 protein was a gift from Dr. Winship Herr, Cold Spring Harbor Laboratory.
Immunoblots
Wildtype or mutant VP16 proteins were detected in immunoblots using the rabbit polyclonal antisera LA2–3 or C8. Protein samples were separated by SDS-PAGE using either Laemmli buffer (10% polyacrylamide gels) or a Tris-Tricine buffer (16.5% polyacrylamide gels) and then electrophoretically transferred to nitrocellulose. The membrane was blocked for one hour at ambient temperature under constant agitation with 10% non-fat dry milk in T-TBS (125 mM Tris pH 7.5, 1.5 M sodium chloride, 0.1% Tween 20). The blot was then incubated for one hour with the primary antibody diluted in T-TBS supplemented with 10% calf serum (Gibco). All three antibodies were diluted 1:10,000 to 1:50,000. The blot was visualized, using HRP-conjugated goat-anti-rabbit IgG (BioRad, 172–1013), diluted and incubated as described for the primary antibody. The bands were visualized using a chemiluminescence detection system (LumiLight).
Immunoprecipitation
[32P]-labeled VP16 was isolated from infected cells and from solubilized virions by immunoprecipitation. HeLa cells (107 in a 150 cm2 culture flask) were infected with KOS, RP3, SJO2 or SO11 at a moi of 5–10. Infections with RP5 were done at a moi of 0.1 in order to compensate for the high particle-pfu ratio of this strain. After 1 hour, the DMEM was replaced with phosphate-free DMEM supplemented with 2% dialyzed FCS. After another 30 minutes, the medium was replaced with fresh phosphate-free DMEM supplemented with 2% dialyzed FCS and 1 mCi of [32P]-orthophosphate (NEN: specific activity 285 Ci/mg) was added.
VP16 was isolated from infected cell lysates at 8 hpi, or from virions isolated at 14 hpi. For lysates, infected cells were scraped from the culture dish and collected by centrifugation. Cell pellets were resuspended in 500 μL of IP lysis buffer {50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% SDS, 1.0% sodium deoxycholate, 1.0% Triton X-100, 2 mM phenylmethylsulfonylfluoride, 1x COMPLETE protease inhibitor mix (Roche)}, incubated on ice for 30 minutes, and then disrupted using a cup horn sonicator. Cellular debris was removed by centrifugation. Virions were isolated at 14 hpi by collecting infected cells along with the growth medium. Cells were disrupted by sonication and debris removed by centrifugation at 3000 x g for 5 min. The supernatant containing the virions was centrifuged through a 20% glycerol cushion for 90 min at 130,000 x g. The pelleted virions were resuspended in 500 μL of IP lysis buffer.
Infected cell lysates and virion extracts were precleared by adding 100 μL of a 10% suspension of heat-killed, formalin-fixed Staphylococcus aureus (SAC). The samples were incubated for 3 hours at 4°C with constant agitation and then centrifuged for 10 minutes at 16000 × g at 4°C. Five μL of the monoclonal antibody LP1 was added to the supernatant and the samples were incubated for 1 hour at 4°C with constant agitation. Fifty μL of SAC was then added and the incubation continued for another 2 hours. The precipitates were collected by centrifugation at 200×g for 2 minutes. The pellets were washed three times in lysis buffer and once briefly in 1 mM phosphate buffer, pH 8. The bound VP16 was then eluted using 50 μL of 10 mM phosphate buffer at pH 12.5 which was subsequently neutralized with 20 μL of 100 mM phosphate buffer, pH 6.8.
Peptide mapping
Immunoprecipitated [32P]-labeled VP16 isolated from infected HeLa cells at 8 hpi was separated on a 10% SDS-PAGE gel. The migration of the labeled VP16 species was determined by autoradiography of the wet gel. The corresponding band was excised from the gel and then washed twice in 0.1 M ammonium carbonate, 50% acetonitrile for 50 minutes at 30°C. The gel slice was dried under a nitrogen stream and then was rehydrated with 4 μL of 0.1 M ammonium carbonate, pH 7.5, 0.02% Tween 20. Immediately thereafter, 1 μL of digestion buffer with either 2 μg of trypsin or endolysylpeptidase C was added. The sample was digested overnight at 30°C. The protein was eluted from the gel slice by gentle sonication and was then separated on a 16.5% Tris/Tricine SDS-PAGE gel. The gel was then either transferred to nitrocellulose or soaked in several washes of 10% methanol/7% acetic acid and dried onto Whatman 3MM paper. The labeled protein on either the membrane or the dried gel was visualized by autoradiography.
Phosphoaminoacid analysis
Immunoprecipitated [32P]-labeled VP16 isolated from infected HeLa cells at 8 hpi was separated on a 10% SDS-PAGE gel and electrophoretically transferred to a PVDF membrane. The labeled species corresponding to VP16 was identified by autoradiography. The corresponding PVDF fragment was excised and digested in 6N hydrochloric acid for 1 hour at 100°C. The sample was dried and mixed with phosphoamino acid standards (2 μg each in 5 μL) and then spotted on a cellulose thin-layer electrophoresis plate. Following electrophoresis at 500V for 1 hour at pH 2.5, the plate was dried and sprayed with ninhydrin (0.5% in acetone), then developed at 70°C until the standards were clearly visible. The radiolabeled amino acids were visualized by autoradiography.
Mass spectrometry
Unlabelled VP16 was immunoprecipitated from 109 HeLa cells infected with HSV-1 strain KOS at 8 hpi using the method described above except that no [32P]-orthophosphate was added and 25 μL of LP1 was used for each sample. Protein eluted from the immunoprecipitate was separated by electrophoresis in a 10% polyacrylamide denaturing gel (SDS-PAGE). A gel slice corresponding to VP16 (detected by staining with Coomassie Blue) was washed in acetonitrile, dried and frozen. Following in-gel digestion by trypsin, chymotrypsin and endoproteinase AspN, sequence analysis of proteolytic fragments was performed by microcapillary reverse-phase HPLC nano-electrospray tandem mass spectrometry (μLC/MS/MS) on a Finnigan LCQ DECA XP Plus quadrupole ion trap mass spectrometer.
Gene expression assays
Steady-state levels of viral mRNAs were determined using quantitative reverse-transcriptase PCR assays (qRT-PCR). Total RNA was isolated from approximately 2 x 106 infected HeLa cells at 2 or 4 hours post-infection using TRI-Reagent (Molecular Research Center). cDNA was prepared using 1 μg total RNA and a randomly-primed reverse transcription system (Promega). Viral or cellular gene fragments (Table 1) were amplified using SYBR Green Core reagents and ABI Prism 7700 (Applied Biosystems). PCR conditions included 0.1–0.25 μM each primer, 2–3 mM MgCl2 and 0.75 U Taq DNA polymerase (Applied Biosystems) in 30 μL final volume. Relative levels of viral gene expression were obtained by the 2-ΔΔCt method (Livak and Schmittgen, 2001) using wild type infections as standard and 18S rRNA as endogenous control. Bars represent the average expression levels of two or three independent infections each measured in duplicate. Errors bars indicate the range of the biological triplicates.
Table 1.
Oligonucleotides used as primers in quantitative real-time PCR to detect viral immediate early (IE), late (L), or cellular (C) gene fragments. The orientations of the primers are indicated relative to the transcription unit (F for forward and R for reverse). PCR end points are defined relative to the transcription start site (+1) of each gene.
| Gene | Class | Region | Oligonucleotide sequence | PCR endpoints |
|---|---|---|---|---|
| ICP0 | IE | P | (F) 5′-GGCCGTGCATGCTAATGATA
(R) 5′-CTTATACCCCACGCCTTTCC |
−172 to −20 |
| ORF | (F) 5′-CTGTCGCCTTACGTGACCAA
(R) 5′-CCATGTTTCCCGTCTGGTC |
3022 to 3133 | ||
| ICP4 | IE | ORF | (F) 5′-GAAGTTGTGGACTGGGAAGG
(R) 5′-GTTGCCGTTTATTGCGTCTT |
4125 to 4257 |
| ICP22 | IE | ORF | (F) 5′-TTTGGGGAGTTTGACTGGAC
(R) 5′-CAGACACTTGCGGTCTTCTG |
1575 to 1712 |
| ICP27 | IE | P | (F) 5′-GTCCCGTTACCAAGACCAAC
(R) 5′-GCACAGACAAGGACCAATCA |
−235 to −102 |
| ORF | (F) 5′-TGCATCCTTCGTGTTTGTCATTCTGG
(R) 5′-GCCGTCAACTCGCAGACACGACTC |
1099 to 1249 | ||
| ICP47* | IE | ORF | (F) 5′-GTACGACCATCACCCGAGTC
(R) 5′-GACGGCACGCCTTTTAAGTA |
808 to 935 |
| VP16 | L | P | (F) 5′-CAGCCCGCTCCGCTTCTCG
(R) 5′-GCCGCCCCGTACCTCGTGAC |
−272 to −47 |
| U3 | C | P | (F) 5′-GCACCACACCAGGAGCAAAC | −295 to −71 |
| snRNA | (R) 5′-CGCTAGTTCCGATGCCATTAGG | |||
| 18S | C | (F) 5′-CCGCAGCTAGGAATAATGGA | 850 to 975 | |
| rRNA | (R) 5′-CGGTCCAAGAATTTCACCTC |
The ICP47 primer overlaps with the US11 transcript. At early times of infections US11 is not expressed and thus this PCR detects ICP47 gene expression levels.
Chemical crosslinking and immunoprecipitation assays
Chromatin immunoprecipitation assays (ChIPs) were performed as described previously (Herrera and Triezenberg, 2004). In brief, HeLa cells (approx. 3 x 107 cells) were infected with wild type HSV-1 (KOS strain) or SJO2 strain at a multiplicity of 5 pfu/cell. Two hours after infection, protein-DNA complexes were crosslinked by adding formaldehyde to the medium overlaying infected cells to a final concentration of 1%. Chromatin was prepared and sonicated to obtain DNA fragments with an average length of 300–400 basepairs. Immunoprecipitations were performed using specific antibodies directed against VP16 (C8-5), Oct-1 (#143, a gift from W. Herr, Cold Spring Harbor Laboratory) and histone H3 (ab1791, Abcam), using concentrations of approx. 50 μg/ml at 4°C overnight. Antigen-antibody complexes were precipitated using protein-G agarose beads (Upstate). The beads were washed extensively before protein-DNA complexes were eluted. Crosslinks were reversed and samples were subsequently digested with proteinase K (Boehringer). The DNA was purified using QIAquick PCR Purification Kit (QIAGEN) and resuspended in 75 μl TE pH 8.0. Quantitative polymerase chain reactions (qPCRs) were performed to detect specific viral or cellular gene fragments in the immunoprecipitated samples (Table 1). qPCRs were performed on a ABI Prism 7700 Sequence Detection System using SYBR Green Core Reagents (Applied Biosystems). Standard PCR conditions included 3 μL of template (IP or “No Ab” samples), 0.25 μM of each primer, 0.75 U Taq DNA polymerase (Applied Biosystems) and 3 mM MgCl2 in 30 μL final volume. The enrichment of a particular promoter over background levels in the IP reactions (Relative IP) were obtained by subtracting CT values of “No Ab” sample from Ct values of a particular IP and using the formula 2-ΔCt to calculate the fold enrichment. For ChIP assays using histone H3 antibodies, the presence of specific DNA in the IP sample is expressed relative to input (pre-IP) DNA. Viral DNAs in nuclear DNA preparations were measured by Q-PCR, normalized to cellular DNA fragments from the U3 and IFN-β promoters, and then normalized to wildtype (KOS) DNA levels. The figures show representative experiments, with error bars that represent the range of the technical duplicates.
Acknowledgments
The phosphorylation analysis and mutant virus construction was performed by SO, with mass spectrometric analysis by WL. Gene expression and ChIP assays were performed by FH and JD. Viral growth curves were performed by AH and SM. The research was supervised and the manuscript written by SJT. Oct-1 antibodies were a gift from Dr. Winship Herr, and LP1 monoclonal antibody against VP16 was a gift from Dr. Anthony Minson. This work was supported by NIH grant AI-27323 and by the MSU Department of Biochemistry and Molecular Biology. FH was supported in part by a dissertation completion fellowship from MSU. JD was supported in part by undergraduate research awards from the American Society for Microbiology.
References
- Ace CI, McKee TA, Ryan JM, Cameron JM, Preston CM. Construction and characterization of a herpes simplex virus type 1 mutant unable to transinduce immediate-early gene expression. J Virol. 1989;63:2260–2269. doi: 10.1128/jvi.63.5.2260-2269.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babb R, Huang CC, Aufiero DJ, Herr W. DNA recognition by the herpes simplex virus transactivator VP16: a novel DNA-binding structure. Mol Cell Biol. 2001;21:4700–12. doi: 10.1128/MCB.21.14.4700-4712.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SL, Pina B, Silverman N, Marcus GA, Agapite J, Regier JL, Triezenberg SJ, Guarente L. Genetic isolation of ADA2 - a potential transcriptional adaptor required for function of certain acidic activation domains. Cell. 1992;70:251–265. doi: 10.1016/0092-8674(92)90100-q. [DOI] [PubMed] [Google Scholar]
- Cress A, Triezenberg SJ. Nucleotide and deduced amino acid sequences of the gene encoding virion protein 16 of herpes simplex virus type 2. Gene. 1991a;103:235–8. doi: 10.1016/0378-1119(91)90278-j. [DOI] [PubMed] [Google Scholar]
- Cress WD, Triezenberg SJ. Critical structural elements of the VP16 transcriptional activation domain. Science. 1991b;251:87–90. doi: 10.1126/science.1846049. [DOI] [PubMed] [Google Scholar]
- Elliott G, Mouzakitis G, O’Hare P. VP16 interacts via its activation domain with VP22, a tegument protein of herpes simplex virus, and is relocated to a novel macromolecular assembly in coexpressing cells. J Virol. 1995;69:7932–7941. doi: 10.1128/jvi.69.12.7932-7941.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiman RN, Herr W. Viral mimicry: common mode of association with HCF by VP16 and the cellular protein LZIP. Genes Dev. 1997;11:3122–7. doi: 10.1101/gad.11.23.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerster T, Roeder RG. A herpesvirus trans-activating protein interacts with transcription factor OTF-1 and other cellular proteins. Proc Natl Acad Sci USA. 1988;85:6347–6351. doi: 10.1073/pnas.85.17.6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson W, Roizman B. Proteins specified by herpes simplex virus. X Staining and radiolabelling properties of B capsid and virion proteins in polyacrylamide gels. J Virol. 1974;13:155–165. doi: 10.1128/jvi.13.1.155-165.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves R, O’Hare P. Separation of requirements for protein-DNA complex assembly from those for functional activity in the herpes simplex virus regulatory protein Vmw65. J Virol. 1989;63:1641–1650. doi: 10.1128/jvi.63.4.1641-1650.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves RF, O’Hare P. Structural requirements in the herpes simplex virus type 1 transactivator Vmw65 for interaction with the cellular octamer-binding protein and target TAATGARAT sequences. J Virol. 1990;64:2716–2724. doi: 10.1128/jvi.64.6.2716-2724.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann JG, Sharff AJ, O’Hare P, Luisi B. Molecular shapes of transcription factors TFIIB and VP16 in solution: implications for recognition. Biochemistry. 2001;40:6267–74. doi: 10.1021/bi0028946. [DOI] [PubMed] [Google Scholar]
- Herr W, Cleary MA. The POU domain: versatility in transcriptional regulation by a flexible two-in-one DNA-binding domain. Genes Dev. 1995;9:1679–93. doi: 10.1101/gad.9.14.1679. [DOI] [PubMed] [Google Scholar]
- Herrera FJ, Triezenberg SJ. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J Virol. 2004;78:9689–96. doi: 10.1128/JVI.78.18.9689-9696.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer P, Highkin M. High level, stable production of recombinant proteins in mammalian cell culture using the herpesvirus VP16 transactivator. Biotechnology (N Y) 1993;11:1037–41. doi: 10.1038/nbt0993-1037. [DOI] [PubMed] [Google Scholar]
- Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–49. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan R, Schang L, Schaffer PA. Transactivation of herpes simplex virus type 1 immediate-early gene expression by virion-associated factors is blocked by an inhibitor of cyclin-dependent protein kinases. J Virol. 1999;73:8843–7. doi: 10.1128/jvi.73.10.8843-8847.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katan M, Haigh A, Verrijzer CP, Vandervliet PC, O’Hare P. Characterization of a cellular factor which interacts functionally with Oct-1 in the assembly of a multicomponent transcription complex. Nucleic Acids Res. 1990;18:6871–6880. doi: 10.1093/nar/18.23.6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knez J, Bilan PT, Capone JP. A single amino acid substitution in herpes simplex virus type 1 VP16 inhibits binding to the virion host shutoff protein and is incompatible with virus growth. J Virol. 2003;77:2892–902. doi: 10.1128/JVI.77.5.2892-2902.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristie TM, LeBowitz JH, Sharp PA. The octamer-binding proteins form multi-protein - DNA complexes with the HSV alpha TIF regulatory protein. EMBO J. 1989;8:4229–4238. doi: 10.1002/j.1460-2075.1989.tb08608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Boissiere S, Izeta A, Malcomber S, O’Hare P. Compartmentalization of VP16 in cells infected with recombinant herpes simplex virus expressing VP16-green fluorescent protein fusion proteins. J Virol. 2004;78:8002–14. doi: 10.1128/JVI.78.15.8002-8014.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai JS, Herr W. Interdigitated residues within a small region of VP16 interact with Oct-1, HCF, and DNA. Mol Cell Biol. 1997;17:3947–3954. doi: 10.1128/mcb.17.7.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam Q, Smibert CA, Koop KE, Lavery C, Capone JP, Weinheimer SP, Smiley JR. Herpes simplex virus VP16 rescues viral mRNA from destruction by the virion host shutoff function. EMBO J. 1996;15:2575–2581. [PMC free article] [PubMed] [Google Scholar]
- LaMarco KL, McKnight SL. Purification of a set of cellular polypeptides that bind to the purine-rich cis-regulatory element of herpes simplex virus immediate early genes. Genes Dev. 1989;3:1372–1383. doi: 10.1101/gad.3.9.1372. [DOI] [PubMed] [Google Scholar]
- Leighton IA, Dalby KN, Caudwell FB, Cohen PT, Cohen P. Comparison of the specificities of p70 S6 kinase and MAPKAP kinase-1 identifies a relatively specific substrate for p70 S6 kinase: the N-terminal kinase domain of MAPKAP kinase-1 is essential for peptide phosphorylation. FEBS Lett. 1995;375:289–93. doi: 10.1016/0014-5793(95)01170-j. [DOI] [PubMed] [Google Scholar]
- Lemaster S, Roizman B. Herpes simplex virus phosphoproteins. II Characterization of the virion protein kinase and of the polypeptides phosphorylated in the virion. J Virol. 1980;35:798–811. doi: 10.1128/jvi.35.3.798-811.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Gong W, Huang CC, Herr W, Cheng X. Crystal structure of the conserved core of the herpes simplex virus transcriptional regulatory protein VP16. Genes Dev. 1999;13:1692–703. doi: 10.1101/gad.13.13.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lu R, Misra V. Zhangfei: a second cellular protein interacts with herpes simplex virus accessory factor HCF in a manner similar to Luman and VP16. Nucleic Acids Res. 2000;28:2446–54. doi: 10.1093/nar/28.12.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Yang P, Padmakumar S, Misra V. The herpesvirus transactivator VP16 mimics a human basic domain leucine zipper protein, luman, in its interaction with HCF. J Virol. 1998;72:6291–7. doi: 10.1128/jvi.72.8.6291-6297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean C, Buckmaster A, Hancock D, Buchanan A, Fuller A, Minson AC. Monoclonal antibodies to three nonglycosylated antigens of herpes simplex virus type 2. J Gen Virol. 1982;63:297–305. doi: 10.1099/0022-1317-63-2-297. [DOI] [PubMed] [Google Scholar]
- Memedula S, Belmont AS. Sequential recruitment of HAT and SWI/SNF components to condensed chromatin by VP16. Curr Biol. 2003;13:241–6. doi: 10.1016/s0960-9822(03)00048-4. [DOI] [PubMed] [Google Scholar]
- Mittler G, Stuhler T, Santolin L, Uhlmann T, Kremmer E, Lottspeich F, Berti L, Meisterernst M. A novel docking site on Mediator is critical for activation by VP16 in mammalian cells. EMBO J. 2003;22:6494–504. doi: 10.1093/emboj/cdg619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriuchi H, Moriuchi M, Pichyangkura R, Triezenberg SJ, Straus SE, Cohen JI. Hydrophobic cluster analysis predicts an amino-terminal domain of varicella-zoster virus open reading frame 10 required for transcriptional activation. Proc Natl Acad Sci U S A. 1995;92:9333–7. doi: 10.1073/pnas.92.20.9333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison EE, Stevenson AJ, Wang YF, Meredith DM. Differences in the intracellular localization and fate of herpes simplex virus tegument proteins early in the infection of Vero cells. J Gen Virol. 1998;79 ( Pt 10):2517–28. doi: 10.1099/0022-1317-79-10-2517. [DOI] [PubMed] [Google Scholar]
- Morrison EE, Wang YF, Meredith DM. Phosphorylation of structural components promotes dissociation of the herpes simplex virus type 1 tegument. J Virol. 1998;72:7108–14. doi: 10.1128/jvi.72.9.7108-7114.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss H. Properties of the herpes simplex virus type 2 trans-inducing factor Vmw65 in wild-type and mutant viruses. J Gen Virol. 1989;70:1579–1585. doi: 10.1099/0022-1317-70-6-1579. [DOI] [PubMed] [Google Scholar]
- Neely KE, Hassan AH, Wallberg AE, Steger DJ, Cairns BR, Wright APH, Workman JL. Activation domain-mediated targeting of the SWI/SNF complex to promoters stimulates transcription from nucleosome aarrays. Mol Cell. 1999;4:649–655. doi: 10.1016/s1097-2765(00)80216-6. [DOI] [PubMed] [Google Scholar]
- Nogueira ML, Wang VE, Tantin D, Sharp PA, Kristie TM. Herpes simplex virus infections are arrested in Oct-1-deficient cells. Proc Natl Acad Sci U S A. 2004;101:1473–8. doi: 10.1073/pnas.0307300101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hare P. The virion transactivator of herpes simplex virus. Sem Virol. 1993;4:145–155. [Google Scholar]
- O’Hare P, Goding CR. Herpes simplex virus regulatory elements and the immunoglobulin octamer bind a common factor and are both targets for virion transactivation. Cell. 1988;52:435–445. doi: 10.1016/s0092-8674(88)80036-9. [DOI] [PubMed] [Google Scholar]
- O’Reilly D, Hanscombe O, O’Hare P. A single serine residue at position 375 of VP16 is critical for complex assembly with Oct-1 and HCF and is a target of phosphorylation by casein kinase II. EMBO J. 1997;16:2420–30. doi: 10.1093/emboj/16.9.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon APW, Roizman B. The phenotype in vitro and in infected cells of herpes simplex virus 1 alpha-trans-inducing factor (VP16) carrying temperature-sensitive mutations introduced by substitution of cysteines. J Virol. 1995;69:7658–7667. doi: 10.1128/jvi.69.12.7658-7667.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regier JL, Shen F, Triezenberg SJ. Pattern of aromatic and hydrophobic amino acids critical for one of two subdomains of the VP16 transcriptional activator. Proc Natl Acad Sci USA. 1993;90:883–887. doi: 10.1073/pnas.90.3.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–44. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schang LM, Bantly A, Knockaert M, Shaheen F, Meijer L, Malim MH, Gray NS, Schaffer PA. Pharmacological cyclin-dependent kinase inhibitors inhibit replication of wild-type and drug-resistant strains of herpes simplex virus and human immunodeficiency virus type 1 by targeting cellular, not viral, proteins. J Virol. 2002;76:7874–82. doi: 10.1128/JVI.76.15.7874-7882.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schang LM, Rosenberg A, Schaffer PA. Transcription of herpes simplex virus immediate-early and early genes is inhibited by roscovitine, an inhibitor specific for cellular cyclin-dependent kinases. J Virol. 1999;73:2161–72. doi: 10.1128/jvi.73.3.2161-2172.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P, Knez J, Capone JP. Amino acid substitutions in the herpes simplex virus transactivator VP16 uncouple direct protein-protein interaction and DNA binding from complex assembly and transactivation. J Biol Chem. 1995;270:29030–29037. doi: 10.1074/jbc.270.48.29030. [DOI] [PubMed] [Google Scholar]
- Simmen KA, Newell A, Robinson M, Mills JS, Canning G, Handa R, Parkes K, Borkakoti N, Jupp R. Protein interactions in the herpes simplex virus type 1 VP16 induced complex: VP16 peptide inhibition and mutational anlysis of the host cell factor requirements. J Virol. 1997;71:3886–3894. doi: 10.1128/jvi.71.5.3886-3894.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smibert CA, Popova B, Xiao P, Capone JP, Smiley JR. Herpes simplex virus VP16 forms a complex with the virion host shutoff protein vhs. J Virol. 1994;68:2339–2346. doi: 10.1128/jvi.68.4.2339-2346.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley JR, Duncan J. Truncation on the C-terminal acidic transcriptional activation domain of herpes simplex virus VP16 produces a phenotype similar to that of the in1814 linker insertion mutation. J Virol. 1997;71:6191–6193. doi: 10.1128/jvi.71.8.6191-6193.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand SS, Leib DA. Role of the VP16-binding domain of vhs in viral growth, host shutoff activity, and pathogenesis. J Virol. 2004;78:13562–72. doi: 10.1128/JVI.78.24.13562-13572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand SS, Vanheyningen TK, Leib DA. The virion host shutoff protein of herpes simplex virus type 1 has RNA degradation activity in primary neurons. J Virol. 2004;78:8400–3. doi: 10.1128/JVI.78.15.8400-8403.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan SM, Horn PJ, Olson VA, Koop AH, Nu W, Ebright RH, Triezenberg SJ. Mutational analysis of a transcriptional activation region of the VP16 protein of herpes simplex virus. Nucleic Acids Res. 1998;26:4487–4496. doi: 10.1093/nar/26.19.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal-Singer R, Pichyangkura R, Chung E, Lasner TM, Randazzo BP, Trojanowski JQ, Fraser NW, Triezenberg SJ. The transcriptional activation domain of VP16 is required for efficient infection and establishment of latency by HSV-1 in the murine peripheral and central nervous systems. Virology. 1999;259:20–33. doi: 10.1006/viro.1999.9756. [DOI] [PubMed] [Google Scholar]
- Triezenberg SJ. Structure and function of transcriptional activation domains. Curr Opin Genetics Dev. 1995;5:190–196. doi: 10.1016/0959-437x(95)80007-7. [DOI] [PubMed] [Google Scholar]
- Triezenberg SJ, Kingsbury RC, McKnight SL. Functional dissection of VP16, the trans-activator of herpes simplex virus immediate early gene expression. Genes Dev. 1988;2:718–729. doi: 10.1101/gad.2.6.718. [DOI] [PubMed] [Google Scholar]
- Triezenberg SJ, LaMarco KL, McKnight SL. Evidence of DNA:protein interactions that mediate HSV-1 immediate early gene activation by VP16. Genes Dev. 1988;2:730–743. doi: 10.1101/gad.2.6.730. [DOI] [PubMed] [Google Scholar]
- Utley RT, Ikeda K, Grant PA, Cote J, Steger DJ, Eberharter A, John S, Workman JL. Transcriptional activators direct histone acetyltransferase complexes to nucleosomes. Nature. 1998;394:498–502. doi: 10.1038/28886. [DOI] [PubMed] [Google Scholar]
- Walker S, Greaves R, O’Hare P. Transcriptional activation by the acidic domain of Vmw65 requires the integrity of the domain and involves additional determinants distinct from those necessary for TFIIB binding. Mol Cell Biol. 1993;13:5233–5244. doi: 10.1128/mcb.13.9.5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker S, Hayes S, O’Hare P. Site-specific conformational alteration of the Oct-1 POU domain-DNA complex as the basis for differential recognition by Vmw65 (VP16) Cell. 1994;79:841–52. doi: 10.1016/0092-8674(94)90073-6. [DOI] [PubMed] [Google Scholar]
- Wang L, Grossman SR, Kieff E. Epstein-Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc Natl Acad Sci U S A. 2000;97:430–5. doi: 10.1073/pnas.97.1.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinheimer SP, Boyd BA, Durham SK, Resnick JL, O’Boyle DR. Deletion of the VP16 open reading frame of herpes simplex virus type 1. J Virol. 1992;66:258–269. doi: 10.1128/jvi.66.1.258-269.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werstuck G, Capone JP. Mutational analysis of the herpes simplex virus trans-inducing factor Vmw65. Gene. 1989;75:213–224. doi: 10.1016/0378-1119(89)90267-9. [DOI] [PubMed] [Google Scholar]
- Wu TJ, Monokian G, Mark DF, Wobbe CR. Transcriptional activation by herpes simplex virus type 1 VP16 in vitro and its inhibition by oligopeptides. Mol Cell Biol. 1994;14:3484–3493. doi: 10.1128/mcb.14.5.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao P, Capone JP. A cellular factor binds to the herpes simplex virus type-1 transactivator Vmw65 and is required for Vmw65-dependent protein-DNA complex assembly with Oct-1. Mol Cell Biol. 1990;10:4974–4977. doi: 10.1128/mcb.10.9.4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, DeBeaumont R, Zhou S, Naar AM. The activator-recruited cofactor/Mediator coactivator subunit ARC92 is a functionally important target of the VP16 transcriptional activator. Proc Natl Acad Sci U S A. 2004;101:2339–44. doi: 10.1073/pnas.0308676100. [DOI] [PMC free article] [PubMed] [Google Scholar]