

Gene silencing mediated by RNA interference (RNAi) has been observed in a wide range of eukaryotes including fungi, nematodes, insects, plants and, more recently, mammalian cells. RNAi has been technologically developed to become a powerful tool to assess the function of genes. RNAi is induced by the presence of double stranded RNA (dsRNA) whose sequence corresponds to a portion of the gene being targeted for silencing. The RNAi mechanism includes the degradation of dsRNA into short interfering RNAs (siRNAs) by DICER and the degradation of the targeted RNA by RISC [for recent reviews see (1–3)].
One major barrier to the experimental use of RNAi for targeted gene silencing in mammalian systems was the non-specific gene silencing triggered by long dsRNAs through the interferon response pathway [for review see (4)]. This difficulty was overcome when Tuschl and colleagues discovered that siRNAs (21 bp), normally generated from long dsRNA during RNAi, can induce a specific and potent RNAi effect in cultured mammalian cells (5). Initially, siRNAs corresponding to the targeted genes were chemically synthesized in vitro and then were transfected into cultured mammalian cells resulting in specific gene silencing. Soon thereafter, several different plasmids were designed to produce siRNAs in vivo from RNA transcripts generated by host cell RNA polymerases [(6–10); reviewed in (11)]. These plasmids contain a promoter and terminator from genes transcribed by RNA polymerase III (such as the RNase P H1 RNA or U6 snRNA genes) so as to direct the expression of short RNAs with defined ends. Two different designs have been used for these siRNA-expressing vectors. In one strategy, the sense and antisense strands are expressed as two independent transcripts that anneal within the cell to form structures resembling in vitro synthesized siRNAs (7,8). In the second approach, the sense and antisense strands are expressed as a part of a single transcript separated by a short intervening loop. Therefore, the sense and antisense strand form a hairpin structure that after intracellular processing produces siRNAs (6,9,10).
Since sequence complementary between the siRNA and the targeted gene transcript is critical for efficient RNAi (6,10,12), it becomes important to ensure that the siRNA expression plasmids contain no mutations introduced during the cloning process. However, siRNA vectors that express sense and antisense separated by an intervening loop will have hairpin secondary structures in the DNA that can interfere with standard sequencing reactions. Here we describe the use of digestion by restriction enzymes to relieve the secondary structure barriers to allow efficient DNA sequencing of inserts cloned into siRNA vectors.
We employed the pSUPER vector (6) to express siRNAs with sequence homology to the mammalian transcriptional coactivators BRG1 or p300 by cloning annealed oligonucleotides into the Bgl II and Hind III sites of pSUPER as described (6). The Bgl II site was not restored after cloning. The sequence of the inserted double-stranded oligonucleotide for the BRG1 siRNA is indicated by the capital letters in Figure 1A.
Figure 1.
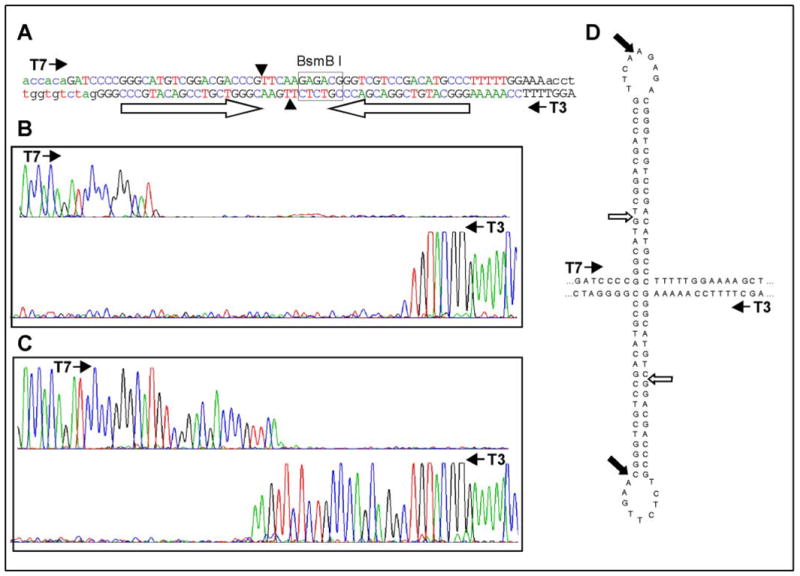
Sequence analysis of the pSUPER-BRG1 siRNA expression plasmid. (A) Capital letters indicate the sequence of the DNA insert and lower case letters indicate flanking sequence from the pSUPER vector. Letters are colored and aligned to match the sequence chromatograms shown below. Open arrows mark the inverted repeat sequences. A BsmB I recognition site (boxed) directs cleavage at sites indicated by arrowheads within the intervening “loop” region. Filled arrows indicate the direction of sequencing reactions using T7 and T3 primers. (B) DNA sequencing chromatograms of the undigested pSUPER-BRG1 plasmid using T7 and T3 primers. (C) DNA sequencing chromatograms of the pSUPER-BRG1 plasmid after digestion with BsmB I. (D) DNA secondary structure predicted to occur within the siRNA encoding region due to sequence complementarity of the inverted repeats. Positions of premature termination of sequencing reactions are indicated with open arrows. Solid arrows indicate run-off termination of sequencing reactions at the template ends following digestion with BsmB I.
The DNA sequences of the inserted fragments were analyzed using the Big Dye™ Terminator Cycle Sequencing Kit (Applied Biosystems, Inc., Foster City, CA) on a capillary-based ABI PRISM® 3100 Genetic Analyzer (Applied Biosystems, Inc.). Sequencing reactions on the two strands were initiated using primers corresponding to the T7 and T3 RNA polymerase promoters (5′-TAATACGACTCACTATAGGG-3′and 5′-ATTAACCCTCACTAAAGGGA-3′, respectively). Problematic sequencing data for both the BRG1 and p300 siRNA plasmids were consistently returned for sequencing reactions from either direction. These reads were typically accurate (Phred values >20) until encountering the inserted DNA oligonucleotides but then prematurely terminated about 7–9 nt into the siRNA encoding region (Figure 1B). A likely explanation for the premature termination of the sequencing reactions is the presence of strong secondary structure in the DNA insert (Figure 1D) that might inhibit the process of the DNA polymerase during DNA sequencing. Attempts to overcome the premature termination by modifying the standard sequencing reactions, including addition of DMSO and increased annealing temperature, were unsuccessful (data not shown).
In an attempt to remove the impeding secondary structure, the siRNA expression plasmids were cleaved with restriction endonucleases that recognize sequences near the loop between the inverted repeats. For example, pSUPER-BRG1 was digested with BsmB I, which recognizes the asymmetric sequence 5′-CGTCTC-3′ and cleaves to leave a four-nucleotide 5′ overhang. The recognition site for BsmB I was fortuitously created near the loop end of the second repeat of the siRNA insert, and the cleavage sites fall within the loop between the inverted repeats (Figure 1A). Initial digestion for one hour under standard conditions resulted in inadequate cleavage of the plasmid, perhaps due to interference by the DNA secondary structure. Overnight restriction digestion was performed and the resulting DNA subjected to DNA sequencing from both directions. Figure 1C shows that using this strategy the DNA sequencing reactions extended past the site of premature termination to the end of the DNA template at the site of BsmB1 cleavage. Since the BsmB I cleavage site is in the loop region, the sequences of the two inverted repeats were determined from separate reactions using T7 and T3 primers respectively. The sequences obtained with T3 and T7 primers contain 4 nt of overlap due to the asymmetry in the cleavage by the restriction endonuclease (Figure 1C), ensuring complete sequencing coverage of the insert DNA.
Similar sequencing difficulties were encountered with the undigested siRNA expression plasmid for the p300 transcriptional coactivator. In this case, a Bgl II site had been created at the junction of the loop region with one arm of the inverted repeat. Following digestion with Bgl II, sequencing reactions proceed to the ends of the linearized templates, providing a 4 nt overlap in the sequencing products using the T3 and T7 primers (data not shown). This result shows that the sequencing difficulty and the applicability of the solution described herein are not unique to the pSUPER-BRG1 plasmid.
To test whether the release of superhelical torsion of the plasmid pSUPER-BRG1 might be sufficient to obtain successful DNA sequencing, the plasmid was digested using either Hind III and EcoR I in separate reactions. These enzymes recognize unique restriction sites flanking the DNA insert or HI RNA promoter, respectively, and therefore digestion with either restriction enzyme resulted in a linear DNA. Nonetheless, the sequencing results following digestion with either Hind III or EcoR I resembled that of the undigested plasmid, prematurely terminating early in the inverted repeats (data not shown). We conclude that releasing superhelical torsion in the plasmid is not sufficient to overcome the structural barrier presented to the DNA polymerase in DNA sequencing reactions under these conditions.
Our results indicate that siRNA-encoding DNA, with its inherent secondary structure, can be effectively sequenced following restriction digestion to disrupt the secondary structure and separate the inverted repeats. In our example with pSUPER-BRG1, a unique and fortuitous BsmBI restriction site was formed as a consequence of the sequence chosen for its homology to our target gene. In the case of pSUPER-p300, a unique Bgl II site was formed at the junction of the loop and one arm of the inverted repeat. Other restriction sites might be similarly engineered within or adjacent to the loop connecting the inverted repeats. Users of this or similar siRNA expression vectors can incorporate this feature as one parameter in designing siRNA cassettes. The restriction sites should be either inherently asymmetric (as for BsmB I in our pSUPER-BRG1 plasmid) or asymmetrically placed (as for Bgl II in our pSUPER-p300 plasmid) to avoid the potential complications of additional secondary structure inherent in symmetric restriction sites. Moreover, the restriction enzyme chosen should cleave to leave 5′ extended ends, so that the DNA sequences coming from opposite directions will overlap.
Several advantages might favor the use of plasmids to generate siRNA over in vitro chemical synthesis including cost, stability of DNA over RNA, and the possibility of generating stable cell lines expressing a particular siRNA. Since RNAi requires sequence specificity between siRNAs and the targeted RNA, the accurate determination of the DNA sequences cloned into siRNA-producing vectors represents an important step in altering the expression of selected genes using RNAi technology. Although we have demonstrated the value of this tactic using the pSUPER expression vector, the sequencing problem (and thus this solution) are associated with the nature of the inverted-repeat inserts and not to specific components of the vector itself. Thus, this method should be widely applicable for many plasmid or virally based siRNA expression systems.
Acknowledgments
We thank Dr. R. W. Henry, Dr. Amy Hark, and Dean Shooltz for comments on the manuscript. This work was supported by NIH grant AI27323 to SJT and by funds from the Department of Biochemistry and Molecular Biology, MSU. Address correspondence to Dr. Steven J. Triezenberg, Department of Biochemistry and Molecular Biology, Michigan State University, East Lansing, MI 48824-1319, USA. E-mail: triezenb@msu.edu.
References
- 1.Zamore PD. Ancient pathways programmed by small RNAs. Science. 2002;296:1265–1269. doi: 10.1126/science.1072457. [DOI] [PubMed] [Google Scholar]
- 2.Hannon GJ. RNA interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 3.McManus MT, Sharp PA. Gene silencing in mammals by small interfering RNAs. Nat Rev Genet. 2002;3:737–747. doi: 10.1038/nrg908. [DOI] [PubMed] [Google Scholar]
- 4.Kumar M, Carmichael GG. Antisense RNA: function and fate of duplex RNA in cells of higher eukaryotes. Microbiol Mol Biol Rev. 1998;62:1415–1434. doi: 10.1128/mmbr.62.4.1415-1434.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 6.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 7.Miyagishi M, Taira K. U6 promoter-driven siRNAs with four uridine 3′ overhangs efficiently suppress targeted gene expression in mammalian cells. Nat Biotechnol. 2002;20:497–500. doi: 10.1038/nbt0502-497. [DOI] [PubMed] [Google Scholar]
- 8.Lee NS, Dohjima T, Bauer G, Li H, Li MJ, Ehsani A, Salvaterra P, Rossi J. Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat Biotechnol. 2002;20:500–505. doi: 10.1038/nbt0502-500. [DOI] [PubMed] [Google Scholar]
- 9.Paul CP, Good PD, Winer I, Engelke DR. Effective expression of small interfering RNA in human cells. Nat Biotechnol. 2002;20:505–508. doi: 10.1038/nbt0502-505. [DOI] [PubMed] [Google Scholar]
- 10.Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002;16:948–958. doi: 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuschl T. Expanding small RNA interference. Nat Biotechnol. 2002;20:446–448. doi: 10.1038/nbt0502-446. [DOI] [PubMed] [Google Scholar]
- 12.Elbashir SM, Martinez J, Patkaniowska A, Lendeckel W, Tuschl T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 2001;20:6877–6888. doi: 10.1093/emboj/20.23.6877. [DOI] [PMC free article] [PubMed] [Google Scholar]