

Abstract
Shared molecular genetic characteristics other than DNA and protein sequences can provide excellent sources of phylogenetic information, particularly if they are complex and rare and are consequently unlikely to have arisen by chance convergence. We have used two such characters, arising from changes in mitochondrial genetic code, to define a clade within the Platyhelminthes (flatworms), the Rhabditophora. We have sampled 10 distinct classes within the Rhabditophora and find that all have the codon AAA coding for the amino acid Asn rather than the usual Lys and AUA for Ile rather than the usual Met. We find no evidence to support claims that the codon UAA codes for Tyr in the Platyhelminthes rather than the standard stop codon. The Rhabditophora are a very diverse group comprising the majority of the free-living turbellarian taxa and the parasitic Neodermata. In contrast, three other classes of turbellarian flatworm, the Acoela, Nemertodermatida, and Catenulida, have the standard invertebrate assignments for these codons and so are convincingly excluded from the rhabditophoran clade. We have developed a rapid computerized method for analyzing genetic codes and demonstrate the wide phylogenetic distribution of the standard invertebrate code as well as confirming already known metazoan deviations from it (ascidian, vertebrate, echinoderm/hemichordate).
Monophyly of the flatworms (phylum Platyhelminthes) and the placement of putatively “primitive” flatworm groups have been challenged by molecular data on a number of occasions recently. The problematic taxa include the Acoelomorpha (Acoela and Nemertodermatida) and the Catenulida, both taxa considered to be basal flatworms by morphologists. Nonmonophyly of the Platyhelminthes and our understanding of their early origins and radiation have implications for understanding the early history of bilaterally symmetrical metazoans. Ehlers (1, 2) (Fig. 1) provided the first cladistic analysis founded on morphological data and placed Catenulida as the most basal group, followed by the Acoelomorpha (Fig. 1). In his ladder-like phylogeny of the flatworms, the earliest diverging taxa, the “Archoophora” (including Catenulida, Acoelomorpha, Macrostomida, and Polycladida) possess the (presumably plesiomorphic) spiralian cleavage of endolecithal eggs in early development. The remaining Turbellaria and the monophyletic obligate parasites known as the Neodermata constitute the Neoophora, being linked by an idiosyncratic derived form of spiral cleavage and ectolecithal eggs (3). A more recent morphological analysis by Rohde (4, 5) supported the monophyly of the Platyhelminthes but suggested Acoelomorpha might be the most basal taxon. In contrast the morphological–cladistic analyses by Haszprunar have shown platyhelminthes as polyphyletic (6, 7).
Figure 1.

Phylogeny of the Platyhelminthes based on a morphological analysis of Ehlers (2), indicating the major clades and the basal position of the Catenulida and the Acoelomorpha.
Various molecular analyses have given contradictory results for the placement of the Acoela, Nemertodermatida, and Catenulida relative to each other and to the Rhabditophora. In different analyses based on 18S rDNA sequences, all three groups have been found either within the Rhabditophora or basal to the Rhabditophora or even unrelated to the Rhabditophora, suggesting a polyphyletic phylum Platyhelminthes (supplementary Fig. 4 a–e; see www.pnas.org).
Most recently, a densely sampled analysis of the Platyhelminthes in the context of the Metazoa placed the Acoela as the most basal extant group of Bilateria, unrelated to the other flatworms; the Catenulida were found to be the sister group of the Rhabditophora, and the Nemertodermatida were nested within the Rhabditophora (8). In an attempt to throw further light on this bewildering diversity of phylogenetic positions of the Acoela, Nemertodermatida, and Catenulida, we have focused on an alternative source of molecular synapomorphy—changes in the mitochondrial genetic code.
Two Potential Flatworm Synapomorphies.
The rhabditophoran flatworm Fasciola (Trematoda: Digenea) has previously been shown to have two differences in its mitochondrial genetic code when compared with most other animals: the mitochondrial codon AUA codes for Ile rather than the normal Met and the mitochondrial codon AAA codes for Asn rather than Lys (9, 10).
We decided to investigate whether this unusual condition is conserved throughout the rhabditophoran flatworms and, if so, whether it also occurs in the Acoelomorpha and Catenulida. If all flatworm groups do share this unusual genetic code, it would provide a strong case for rejecting the studies indicating polyphyly of the Platyhelminthes (6–8). We have also looked for these code changes in two enigmatic taxa sometimes allied to the Platyhelminthes—Xenoturbella (11, 12) and the dicyemid mesozoan Dicyema misakiense (13). Studies of oogenesis (14) and Cox I and 18S rDNA data (15) have previously contradicted a platyhelminth affinity for Xenoturbella. 18S rDNA studies of Mesozoa were ambiguous because of long branch effects (13), but the presence of a lophotrochozoan type Hox gene in D. misakiense (16) has led to support for a derived (and hence potentially flatworm) position for this latter phylum rather than the basal position outside the triploblasts otherwise espoused (17).
Materials and Methods
For the purposes of this study and to use published gene sequences available on GenBank/European Molecular Biology Laboratory, we have concentrated on sequencing and aligning portions of the mitochondrial cytochrome c oxidase subunit I (Cox I) gene. In addition to the eight published platyhelminth sequences (seven parasitic Neodermata and a single turbellarian sequence), we have determined Cox I nucleotide sequences for 16 additional species representing a much greater diversity of flatworm taxa. A variety of PCR primer combinations were used to amplify fragments of the Cox I gene, and in total we had available sequences from 24 species of flatworm.
Previously published primers LCO1490 and HCO2198 (18) and newly designed primers (forward: CO15A2, CO15B and reverse: CO13A and CO13B) were used to amplify Cox I. Primer sequences are given in Table 1 with reference to their relative position against a published flatworm mitochondrial Cox I gene (S. mansoni, GenBank accession no. AF101196). Supplementary Table 3 (www.pnas.org) indicates the species sampled and the primer combinations successfully used, and the accession numbers and additional genes used for S. mansoni and Dicyema misakiense are shown.
Table 1.
Primers used to amplify cytochrome c oxidase I fragments, their source and position relative to a published sequence of cytochrome c oxidase subunit 1 from the digenean S. mansoni (AF101196)
| Primer | Sequence | Position on AF101196 |
|---|---|---|
| 5′ | ||
| COI5A2 | TAA TWG GTG GNT TYG GNA | 523–541 |
| COI5B | TTC TGR TTY TTY GGN CAY CC | 981–1000 |
| LCO1490 | GGT CAA CAA ATC ATA AAG ATA TT | 325–349 |
| 3′ | ||
| COI3A | TCA GGR TGN CCR AAR AAY CA | 984–1003 |
| COI3B | AAG TGT TGN GGR AAR AAN GT | 1551–1570 |
| HCO2198 | TAA ACT TCA GGG TGA CCA AAA AA | 984–1012 |
DNA Extraction, Amplification, and Sequencing.
All specimens had been fixed and stored in a minimum of five volumes of 95–100% ethanol. Genomic DNA was extracted as described in (4).
Partial Cox I fragments were amplified from each extract by using PCR Beads (Pharmacia), and the cycling conditions were generally hot start (95°C/5 min) followed by 30 cycles of 94°C/1 min, 50°C/1 min, and 72°C/1 min. At least two reactions were performed for each template. Amplified products were cleaned directly with Qiaquick PCR purification kit (Qiagen, Chatsworth, CA) or were run out on a 1% agarose gel, cut out, pooled, and purified by using Qiaquick gel extraction kit (Qiagen).
Gene fragments were directly sequenced by using standard reaction mixes and procedures on a 373 or 377 Applied Biosystems automated sequencer with the Big Dye Readymix reaction kit (Applied Biosystems, Perkin–Elmer). PCR fragments were sequenced by using the same PCR primers used for amplification. Both strands of the amplified DNA products were sequenced, and contiguous regions were assembled with sequencher ver. 3.0 (Gene Codes, Ann Arbor, MI).
Alignments.
Cox I nucleotide sequences from the flatworms, Xenoturbella, and a mesozoan, as well as a wide diversity of higher eukaryotes (fungi, diploblastic and triploblastic metazoa) were aligned to the amino acid alignments of Cox I proteins published by Castresana et al. (19). The nucleotide sequences were first translated into amino acids, which were then aligned to the Castresana data set by using clustalw. Aligning the original nucleotide sequences with their respective amino acid sequence matched up the nucleotide sequences with the protein alignments of Castresana et al. Sites judged to be of uncertain alignment were removed from the final alignment. The alignment file has been deposited with European Bioinformatics Institute and is available by anonymous file transfer protocol from ftp.ebi.ac.uk/pub/databases/embl/align under accession numbers DS43962 and DS43963.
Inferring Codon Usage by Alignment to Known Protein Sequences.
Our general approach for determining which amino acid each codon codes for follows the example of refs. 9 and 10. For example, if the AUA codon within a particular species consistently coincides with positions where most eukaryotes have the amino acid Ile, then we can have some confidence that AUA codes for Ile within this species.
However, this approach fails to take proper account of conservative substitutions. For example, Val is very similar to Ile in terms of amino acid side-chain properties and chemical structure. Thus, if we observe AUA aligned to Val, then we should add some weight to the prediction of AUA coding for Ile. In contrast, Gly is very different from Ile (and the two amino acids substitute rarely for each other), and so if we see AUA aligned with Gly, this correspondingly should reduce our confidence in the idea that AUA codes for Ile. We have accordingly developed a more sophisticated prediction method that takes into account amino acid substitution or exchange preferences. Unlike previous studies, the method is also automated, making a rapid and systematic analysis of large data sets practicable.
Amino acid exchange matrices provide estimates of the probability of one amino acid substituting for another based on an analysis of many thousands of aligned sequences. For this study, we used the blosum62 matrix, which provides logarithm of odds scores for amino acid substitutions (20). Considering each species, for each of the 64 codons in turn, we use this matrix to calculate a score representing the relative probability that the codon under consideration codes for each of the 20 possible amino acids. The score for each amino acid is an average of its blosum62 exchange values for the amino acids that we observe aligned to the codon. The amino acid with the highest score is proposed as that most likely coded for by the particular codon.
More formally, let the number of times that a particular codon, C, aligned to an amino acid, j, be njC.
We then define a score SiC for each amino acid, i, as:
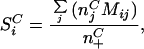 |
where Mij is the blosum62 exchange matrix value for the transition between amino acids i and j (including i = j) and n+C is the total number of amino acids aligned to the codon C. Note that we ignored positions where gaps were present in any of the protein sequences. SiC is essentially an averaged logarithm of odds score representing the likelihood that the codon C codes for the amino acid i and is roughly akin to a very simple profile specific to each codon (21). We predict the amino acid with the highest SiC to be that coded for by the codon C in a given species.
For example, there are a total of four positions aligned to the codon ATA in
D. japonica:
 |
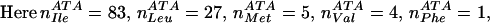 |
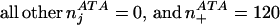 |
To calculate the probability that ATA codes for Ile, we need to consider the blosum62 exchange values for changing Ile→Ile (4), Leu→Ile (2), Met→Ile (1) Val→Ile (3) and Phe→Ile (0). Substituting these values into the above equation gives:
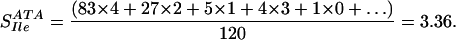 |
Similarly, for Val, we have exchange values Ile→Val (3), Leu→Val (1), Met→Val (1), Val→Val (4), and Phe→Val (−1), giving:
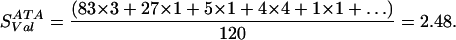 |
Repeating the calculation for the remaining 18 amino acids reveals that Val is the second highest scoring amino acid despite the fact that Val is observed infrequently. The blosum matrix is accounting for the high degree of similarity between the Ile and Val side chains.
SiC values are shown for the AUA and AAA codons in Fig. 2.
Figure 2.

Graphs showing SiC values of the 20 amino acids for the two codons AUA (black bars) and AAA (white bars) for 24 flatworms, a mesozoan and a xenoturbellid. The number of codons used to calculate SiC scores are shown in brackets on each graph in the order AUA/AAA.
Results and Discussion
Accuracy of the Method.
To evaluate the accuracy of our method, we tried predicting the amino acids coded for by codons closely related to the two codons of direct interest (AAA and AUA). AUU and AUC code for Ile, AUG for Met, AAU and AAC for Asn, and AAG for Lys, and there are no known deviations from these codings in any eukaryotic, bacterial, or organellar genome (22). We examined how often we were able to make correct predictions for these 6 codons from 53 eukaryotes. We find that our method works extremely reliably, at least for the Cox I gene, when these codons occur more than twice (Table 2).
Table 2.
Data showing that if the Cox I codons AUU, AUC, AUG, AAU, AAC, or AAG occur three or more times, our method allows us >95% chance of correctly predicting the amino acid they code for
| Occurrences | Observations | % correct |
|---|---|---|
| 1 | 49 | 65 |
| 2 | 29 | 86 |
| 3 | 27 | 96 |
| 4 | 25 | 96 |
| 5 | 137 | 100 |
AAA Codes for Asn, and AUA Codes for Ile in Rhabditophoran Mitochondria. Acoelomorpha and Catenulida Have the Standard Invertebrate Code.
We have extended previous findings of an unusual code in flatworms to cover the great diversity of rhabditophoran flatworms, including archoophoran and neoophoran Turbellaria, and the parasitic Trematoda, Monogenea, and Cestoda. This code is not shared by members of the Acoelomorpha or Catenulida (Fig. 2). Although there are relatively few AAA codons, the consistent pattern within the Acoelomorpha and Catenulida (AAA = Lys) and within the Rhabditophora (AAA = Asn) strengthens our interpretation. The result AAA = Ile in Temnocephala sp. and Taenia solium are almost certainly errors, only a single codon being sampled. From a practical perspective, our results mean that the mitochondrial genetic codes recorded on the National Center for Biotechnology Information for both trematodes and platyhelminthes need updating. There should be just one code for all rhabditophoran flatworms, including the trematoda but excluding the Acoelomorpha and Catenulida, which should be included in the invertebrate code.
AAA = Asn Is a Rhabditophoran Synapomorphy Supporting Their Monophyly.
We have shown that, in common with the echinoderms but unlike all other triploblasts so far studied, all of the rhabditophoran flatworms in our analyses have AAA coding for Asn and AUA coding for Ile. In Fig. 3a, we show that the observation of AAA = Asn in Rhabditophora is most parsimoniously interpreted as a synapomorphy gained in parallel with the echinoderms. This firmly links the disparate groups of Rhabditophora to the exclusion of the Acoelomorpha and Catenulida: the endolecithal “Archoophora” (Macrostomida and Polycladida) with the ectolecithal Neoophora (all others) and the free-living “Turbellaria” with the parasitic Neodermata.
Figure 3.

Mitochondrial genetic codes mapped onto a likely phylogeny of the Metazoa. The trees are rooted by the Fungi as an outgroup. (A) It is most parsimonious (three changes: 1, 2, 3) to assume that both the Echinodermata and Rhabditophora independently evolved AAA = Asn, which must therefore be synapomorphies of the groups. Assuming AAA primitively codes for Asn in the Metazoa requires four changes (1′, 2′, 3′, 4′). AAA is not seen in hemichordate mitochondria. (B) In ignorance of the position of the Acoelomorpha and Catenulida, it is equally parsimonious to presume that, in the Metazoa, AUA primitively coded for Ile [with changes to Met in Chordata (1′), Lophotrochozoa (2′) and Ecdysozoa (3′)] or that AUA changed to coding for Met in a basal metazoan (1) and reverted to Ile in Echinodermata/Hemichordata (2) and Rhabditophora (3). It is hence impossible to polarize the changes based on this knowledge. (C) Positioning the Acoelomorpha and/or Catenulida anywhere on the thickened line forces us to conclude that AUA = Ile in Rhabditophora is a synapomorphy (1, 2, 3 changes) rather than primitive (1′ 2′, 3′, 4′ changes). If Acoela are indeed basal (ref. 8), this also suggests the AUA = Ile in Echinodermata/Hemichordata is a synapomorphy.
AUA = Ile/Met Can Be Polarized (and Supports Rhabditophoran Monophyly) if We Make Reasonable Assumptions About the Position of the Basal Flatworms.
On the basis of the known distribution of the two states of this character and of our current ideas of animal phylogeny (Fig. 3b), the character AUA = Ile can be as parsimoniously interpreted as plesiomorphic or synapomorphic within the Platyhelminthes and Echinodermata (i.e., it cannot be polarized). This uncertainty disappears if we accept either the recently published position of the Acoela as the most basal triploblasts or the more traditional position for any one or more of the Acoela, Nemertodermatida, or Catenulida as the sister group of the Platyhelminthes (2). In any of these cases, the character state AUA = Ile, as seen in the Rhabditophora, is most parsimoniously interpreted as a derived character (Fig. 3c). In fact, if we add the basal flatworms to our basic tree, they inevitably turn AUA = Ile in the Rhabditophora into a derived character unless they are severally considered sister groups of the Chordata or Arthropoda (very unlikely) or closer sister groups of the Spiralia (e.g., Mollusca) than are the Rhabditophora (Fig. 3c).
Only if Acoela Are Basal Metazoa Can the Character AUA = Ile in Echinoderms and Hemichordates Be Considered a Synapomorphy.
Knowledge of the state of these characters in basal flatworms potentially has ramifications beyond strengthening our inference of a monophyletic Rhabditophora based on the apomorphy AAA = Asn: if (and only if) the acoels really are the most basal Metazoa (8), then it also allows us to polarize the AUA = Ile character seen in Echinodermata and Hemichordata into a synapomorphy linking these two groups. Other positions for the acoels on the protostome branch do not alleviate our ignorance of the polarization of the echinoderm/hemichordate AUA = Ile character state (Fig. 3c). This is an important result, as this character has been claimed as a synapomorphy supporting the sister-group relationship of these two phyla, and yet we can see this inference depends on the position of the Acoela (19).
We Cannot Further Refine the Positions of the Acoela, Nemertodermatida, and Catenulida.
As discussed, of the three supposedly basal flatworms—the Acoela, Nemertodermatida, and Catenulida—none shares the mitochondrial genetic code seen in the Rhabditophora. These three taxa have, instead, the standard, invertebrate mitochondrial genetic code. Our experiment aiming to test the relationship of the basal flatworms to the Rhabditophora has given support to the idea that the Rhabditophora are distinct from the Acoelomorpha and Catenulida contradicting results placing any of these groups within the Rhabditophora (5, 23–26) but consistent with the morphology-based results of Ehlers (1, 2). However, the pleisiomorphic states of these characters in these basal flatworms also means there is no way of refining their position further. Although it is clear that the three classes did not emerge from within the Rhabditophora, we cannot choose between the two other proposed positions for these taxa: the sister group(s) of the Rhabditophora or the most basal triploblasts.
No Evidence That UAA Codes for Tyrosine in Rhabditophora.
Based on a single observation in the Cox I gene of the planarian D. japonica, it has been suggested that, in Platyhelminthes, the codon UAA codes for Tyr rather than Stop, as seen in almost all other known genetic codes (10, 27). We can find no further evidence to support this idea. UAA does not occur at all among the 4,598 codons we have aligned from 18 rhabditophorans of broad phylogenetic distribution. In contrast, the other codons coding for Tyr, UAU, and UAC occur 166 and 14 times respectively. UAA is seen twice in the reported S. mansoni Cox I sequence but only at the 3′ extremity beyond any alignable amino acid residues, and the first of these almost certainly represents the termination codon as has been reported (28). The instance discovered by Bessho et al. (10) came from individual specimens of D. japonica, from which multiple distinct Cox I sequences were PCR amplified, suggesting heteroplasmy (more than one clone of mitochondrion). It is possible that this is a pseudogene or an idiosyncrasy of this species, but it seems not to be characteristic of the Rhabditophora.
The Changes Are Unlikely to Be Explained by mRNA Editing.
Our results come from sequences of mitochondrial DNA, and it is possible that, in a manner analogous to green plants, changes at the level of the DNA are not reflected in the RNA such that there is no true change in mitochondrial genetic code (22). In green plant mitochondria, the DNA triplet CGG is seen in conserved Trypotophan positions, whereas CGG usually codes for Arginine. This is explained by the evolution of molecular machinery that alters the CGG in the mRNA to UGG, the universal code for Trypotophan, maintaining the universal coding. mRNA editing can be ruled out at least in the case of S. mansoni AUA codons, where identical nucleotides are seen in genomic and cDNA clones (data not shown). cDNA sequences are not available for the Cox I genes of other flatworms, but the rarity of RNA editing means this is a very unlikely explanation of the differences we see between Rhabditophora and other Metazoa. Further evidence for the absence of mRNA editing is that the Fasciola tRNAlys anticodon is CUU (29). The usual tRNALys anticodon is UUU, which will pair with both AAG and AAA, whereas our inference that only AAG codes for Lys in Rhabditophora fits with the likely specificity of the anticodon CUU to AAG only.
Are Dicyemid Mesozoans or Xenoturbella spp. Derived Rhabditophoran Flatworms?
We also attempted to infer the mitochondrial genetic codes of two other taxa sometimes claimed to be derived from within the Platyhelminthes: the dicyemid Mesozoa—symbionts of cephalopod kidneys—and the genus Xenoturbella. We were able to use concatenated Cox I, II, and III sequences from the mesozoan Dicyema misakiense, and from this analysis it seems they have the canonical invertebrate mitochondrial genetic code rather than sharing either of the two rhabditophoran synapomorphic changes. Xenoturbella has recently been proposed, based on 18S rDNA and Cox I sequences and on embryological similarities, to be a secondarily derived mollusk rather than a turbellarian (12, 15). Our data support this view, in that Xenoturbella lacks both rhabditophoran genetic code synapomorphies, although this does not rule out derivation of either group from the Acoelomorpha or Catenulida.
Generality of Invertebrate Codes.
We have also assayed the generality of the standard invertebrate mitochondrial code in as many classes and phyla as we could extract from GenBank (see supplementary data, www.pnas.org). We ignore apparent deviations from the standard code if based on two or fewer observations of a codon (see Table 2) as well as apparent deviations when the wrongly identified amino acid is very similar to the likely correct one (Ile/Val/Leu and His/Gln). In none of the protostome clades we investigated (Insecta, Crustacea, Onychophora, Nematoda, oligochaete, polychaete and hirudinean Annelida, Pogonophora, Vestimentifera, cephalopod, polyplacophoran and gastropod Mollusca, Sipunculida, Echiura, and Bryozoa) were there any convincing deviations from the standard invertebrate code. We also found the nonstandard codes expected in Vertebrata, Hemichordata, Echinodermata, Urochordata, and Cnidaria.
Conclusions
Detecting two synapomorphic codon reassignments in the mitochondrial genetic code of the rhabditophoran Platyhelminthes has provided us with a valuable and compelling source of systematic information. Two taxa traditionally placed within the phylum Platyhelminthes, the Catenulida and the Acoelomorpha (Acoela + Nemertodermatida), do not share these characters, providing evidence for their separation from the Rhabditophora congruent with conclusions based on morphology. We point out, however, that the strength of our results lies, in part, in the density of sampling—many orders of Rhabditophora, several examples of the Catenulida and Acoelomorpha, and the greatest diversity yet of the Metazoa. Thanks to this dense sampling, we are able to make two important points. First, a codon reassignment in one member of a phylum cannot necessarily be extrapolated to the rest of the phylum (e.g., the Platyhelminthes) with potentially important consequences for users of translation tables. Second, only with dense enough sampling can these characters be polarized and hence used as true synapomorphies. We also note that even these apparently unlikely changes are subject to convergence, as both rhabditophoran anomalies are also seen in the Echinodermata.
Our efficient method for determining mitochondrial genetic codes complements other analytical techniques but seems unlikely to provide any further information regarding the phylogeny of the Metazoa, although as several phyla remain unexamined we hope we will be proved wrong. We end by agreeing with Hillis, who points out that “no one technique is a perfect solution for all phylogenetic problems, even though each provides us with a new perspective on evolution” (30).
Supplementary Material
Acknowledgments
We thank José Castresana (European Molecular Biology Laboratory, Heidelberg) for providing mitochondrial amino acid alignments and Ulf Jondelius for sending us the Xenoturbella Cox I sequence. R.B.R. thanks Jim Fickett and David Searls (SmithKline Beecham) for encouragement. D.T.J.L. and E.A.H. were funded by a Wellcome Senior Fellowship to D.T.J.L. (043965/Z/95/Z).
Footnotes
References
- 1.Ehlers U. Verh Naturwiss Ver Hamburg (NF) 1984;27:291–294. [Google Scholar]
- 2.Ehlers U. In: The Origins and Relationships of Lower Invertebrates. Conway Morris S, George J D, Gibson R, Platt H M, editors. Oxford: Clarendon; 1985. pp. 143–158. [Google Scholar]
- 3.Ellis C H, Fausto-Sterling A. In: Embryology. Constructing the Organism. Gilbert S F, Raunio A M, editors. Sunderland, MA: Sinauer; 1997. pp. 115–130. [Google Scholar]
- 4.Littlewood D T J, Rohde K, Bray R A, Herniou E A. Biol J Linn Soc. 1999;68:257–287. [Google Scholar]
- 5.Littlewood D T J, Rohde K, Clough K A. Biol J Linn Soc. 1999;66:75–114. [Google Scholar]
- 6.Haszprunar G. J Zool Syst Evol Res. 1996;34:41–48. [Google Scholar]
- 7.Haszprunar G. In: Origin and Evolutionary Radiation of the Mollusca. Taylor J, editor. Oxford: Oxford Univ. Press; 1996. pp. 1–28. [Google Scholar]
- 8.Ruiz Trillo I, Riutort M, Littlewood D T J, Herniou E A, Baguñà J. Science. 1999;283:1919–1923. doi: 10.1126/science.283.5409.1919. [DOI] [PubMed] [Google Scholar]
- 9.Ohama T, Osawa S, Watanabe K, Jukes T H. J Mol Evol. 1990;30:329–332. doi: 10.1007/BF02101887. [DOI] [PubMed] [Google Scholar]
- 10.Bessho Y, Ohama T, Osawa S. J Mol Evol. 1992;34:331–335. doi: 10.1007/BF00160240. [DOI] [PubMed] [Google Scholar]
- 11.Ehlers U, Sopott-Ehlers B. Verh Dtsch Zool Ges. 1997;90:68. [Google Scholar]
- 12.Lundin K. Zool Scripta. 1998;27:263–270. [Google Scholar]
- 13.Pawlowski J, Montoya-Burgos J L, Fahrni J F, Wüest J, Zaninetti L. Mol Biol Evol. 1996;13:1128–1132. doi: 10.1093/oxfordjournals.molbev.a025675. [DOI] [PubMed] [Google Scholar]
- 14.Israelsson O. Nature (London) 1997;390:32. [Google Scholar]
- 15.Norén M, Jondelius U. Nature (London) 1997;390:31–32. [Google Scholar]
- 16.Kobayashi M, Furuya H, Holland P W H. Nature (London) 1999;401:762. doi: 10.1038/44513. [DOI] [PubMed] [Google Scholar]
- 17.Meglitsch P A, Schram F R. Invertebrate Zoology. Oxford: Oxford Univ. Press; 1991. pp. 49–53. [Google Scholar]
- 18.Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. Mol Mar Biol Biotechnol. 1994;3:294–299. [PubMed] [Google Scholar]
- 19.Castresana J, Feldmaier-Fuchs G, Pääbo S. Proc Natl Acad Sci USA. 1998;95:3703–3707. doi: 10.1073/pnas.95.7.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henikoff S, Henikoff J G. Proc Natl Acad Sci USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durbin R, Eddy S, Krogh A, Mitchinson G. Biological Sequence Analysis. Cambridge U.K.: Cambridge Univ. Press; 1998. [Google Scholar]
- 22.Osawa S. Evolution of the Genetic Code. Oxford: Oxford Univ. Press; 1995. [Google Scholar]
- 23.Katayama T, Nishioka M, Yamamoto M. Zool Sci. 1996;13:747–756. doi: 10.2108/zsj.13.747. [DOI] [PubMed] [Google Scholar]
- 24.Carranza S, Baguñà J, Riutort M. Mol Biol Evol. 1997;14:485–497. doi: 10.1093/oxfordjournals.molbev.a025785. [DOI] [PubMed] [Google Scholar]
- 25.Jondelius U. Hydrobiologia. 1998;383:147–154. [Google Scholar]
- 26.Campos A, Cummings M P, Reyes J L, Laclette J P. Mol Phylogenet Evol. 1998;10:1–10. doi: 10.1006/mpev.1997.0483. [DOI] [PubMed] [Google Scholar]
- 27.Bessho Y, Ohama T, Osawa S. J Mol Evol. 1992;34:324–330. doi: 10.1007/BF00160239. [DOI] [PubMed] [Google Scholar]
- 28.Blair D, Le T H, Després L, McManus D P. Parasitology. 1999;119:303–313. doi: 10.1017/s0031182099004709. [DOI] [PubMed] [Google Scholar]
- 29.Garey J R, Wolstenholme D R. J Mol Evol. 1989;28:374–387. doi: 10.1007/BF02603072. [DOI] [PubMed] [Google Scholar]
- 30.Hillis D M. Proc Natl Acad Sci USA. 1999;96:9979–9981. doi: 10.1073/pnas.96.18.9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.