

Abstract
In the yeast Saccharomyces cerevisiae, meiotic recombination is initiated by double-strand DNA breaks (DSBs). Meiotic DSBs occur at relatively high frequencies in some genomic regions (hotspots) and relatively low frequencies in others (coldspots). We used DNA microarrays to estimate variation in the level of nearby meiotic DSBs for all 6,200 yeast genes. Hotspots were nonrandomly associated with regions of high G + C base composition and certain transcriptional profiles. Coldspots were nonrandomly associated with the centromeres and telomeres.
Hotspots are genomic regions with unusually high levels of recombination (1). From studies in yeast, several generalizations concerning hotspots can be made. First, most hotspots are intergenic rather than intragenic (2). Second, genetically defined hotspots are associated with local double-strand DNA breaks (DSBs) (1). Third, DSBs usually occur in DNase I-sensitive regions (3). Fourth, activity of the HIS4 hotspot in Saccharomyces cerevisiae requires the binding of transcription factors in the hotspot region (4–6), but does not require high levels of meiosis-specific transcription (7). Hotspots that require transcription factor binding are called α hotspots (8). Fifth, certain DNA sequences result in hotspots (β hotspots) that do not require the binding of known transcription factors (9).
Meiotic DSB formation requires Spo11p, a topoisomerase II-related protein that is transiently covalently attached to the 5′ ends of the DNA fragments (10, 11). In wild-type yeast cells, the Spo11p is removed to allow subsequent steps in recombination. In strains with the rad50S mutation, however, Spo11p stays covalently attached to the broken DNA ends (11).
Coldspots in yeast have received less attention than hotspots. Lambie and Roeder (12) showed that the centromere of chromosome III reduced crossing-over and gene conversion of nearby markers, and Baudat and Nicolas (13) noted a lack of DSB sites near the centromere. Several researchers have found a relative lack of DSB sites in rad50S strains near the telomeres (13, 14).
Although observations concerning individual hotspots and coldspots have given clues as to the mechanism of recombination initiation, our ability to predict hotspots and coldspots from DNA sequence information is very limited. A complementary approach is to map hotspots and coldspots globally and to determine whether they share common DNA sequences and/or structural elements. Such mapping studies have been performed to map DSB sites to single-gene resolution on chromosome III (13) and to the resolution of a pulsed-field gel on chromosomes I, III, and VI (14, 15). By using DNA samples enriched for meiosis-specific DSBs as hybridization probes on DNA microarrays, we extended these analyses to measure the global distribution of DSBs at single-gene resolution.
Materials and Methods
Yeast Strains.
FX4, FX6, and QFY105 are diploid rad50S strains that have been described (4). These strains (used in the microarray analysis) are isogenic except for changes introduced by transformation. The strain MJL2688 (MATa/MATα ura3/ura3 lys2/lys2 ho∷LYS2/ho∷LYS2 arg4-nsp,bgl/arg4-nsp,bgl SPO11–3XHA-His6∷KanMX4/SPO11–3XHA-His6∷KanMX4 rad50-K181∷URA3/rad50-K181∷URA3) is isogenic with SK1 (11).
Purification of DSB-Enriched DNA.
Cells were sporulated for 24 h (16). Genomic DNA was isolated by using a nonproteolyzing protocol (11). The DNA was sheared to a size of 2–3 kb. A portion of this DNA was used as a hybridization probe (total genomic DNA). From the remaining sample, we isolated DNA fragments covalently attached to proteins by using a glass fiber filter (11). This DNA was the DSB-enriched hybridization probe.
Microarray Analysis.
Both genomic and DSB-enriched DNA samples were used as templates for a two-step random PCR amplification. About 25 ng of each DNA sample was used for two 8-min extensions with 2.7 mM Round A primer [5′-GTTTCCCAGTCACGATCNNNNNNNNN (Integrated DNA Technologies, Coralville, IA), N being a mixture of all four nucleotides with 60% A + T and 40% G + C] at 37°C with 267 units/ml Sequenase 2.0 (Amersham Pharmacia). DNA was denatured at 94°C for 1.5 min, and cooled to 10°C, and Sequenase 2.0 was added between extensions. The resulting products were used as templates for 15 rounds of PCR using Taq polymerase (Perkin–Elmer) and 10 mM Round B primer (5′-GTTTCCCAGTCACGATC). This DNA then was labeled by 20 additional rounds of PCR including fluorescently labeled Cy3-dUTP or Cy5-dUTP (Amersham Pharmacia). These DNA samples were mixed and used as probes for DNA microarrays (17). Microarray images were acquired by using a GenePix 4000A scanner (Axon Instruments, Foster City, CA). For image analysis, genepix pro 3.0 software was used (Axon Instruments).
Analysis of Hotspots Using Nylon Filters.
DSB-enriched DNA was isolated from the SK1-derivative MJL2688 by chromatin immunoprecipitation (18) with the following alterations: (i) formaldehyde was omitted, (ii) the lysis buffer contained 1 M NaCl, and (iii) sonication resulted in DNA fragments of 1 kb. Hemagglutinin (HA)-tagged Spo11p was precipitated with anti-HA antibody 12CA5 (Roche Molecular Biochemicals) and Immobilized Protein G (Pierce). Immunoprecipitates were washed five times with lysis buffer with 1 M NaCl, three times with lysis buffer with 0.5 M NaCl, followed by two washes as described (18). DSB-enriched DNA and total genomic DNA (25 ng) were labeled by the incorporation of 33P-dCTP by using the High Prime DNA Labeling system (Roche Molecular Biochemicals).
Genes were amplified individually from strain S288C, mostly using primers purchased from Research Genetics, Huntsville, AL. About 2 ng of each PCR product was spotted on Zeta-Probe GT membranes (Bio-Rad) (19). Hybridization methods were as described (20), except that 5× Denhardt's solution replaced skim milk. Filters were analyzed on a BAS2000 PhosphorImager system after being exposed for 40–70 h and quantitated by using image gauge 3.3 software (Fuji Medical Systems, Stamford, CT). For each filter, the average background was subtracted from the measured intensity of all members of the array. The data were normalized by using the ratio of the median signal intensities of the array members measured from each filter. The normalized data for each gene were used to calculate the ratio of (Spo11p-enriched DNA)/(genomic DNA).
Data Analysis.
Most of the data analysis was done by using GCG programs. The microarray data generated in our study has the website address: http://derisilab.ucsf.edu/hotspots/. Statistical analyses were done with the instat 1.12 program for Macintosh.
Results and Discussion
Experimental Rationale.
The mapping of hotspots and coldspots is based on the assumptions that: (i) Spo11p-catalyzed meiosis-specific DSBs are the initiating lesions for most recombination events in yeast, and (ii) DNA fragments from a rad50S strain (induced to undergo meiosis) that are covalently attached to proteins will be enriched for the Spo11p-associated DNA sequences adjacent to hotspots and deficient for the DNA sequences adjacent to coldspots.
Microarray Mapping of Hotspots and Coldspots.
Two closely related rad50S yeast strains were used for the microarray analysis, FX4 and FX6 (4). FX4 has a small insertion upstream of HIS4 that elevates the already high level of recombination at this locus (5), and FX6 has the same insertion upstream of ARG4.
We prepared two samples of meiotic DNA, one containing total genomic DNA (probe 1, P1) and one enriched for DNA fragments covalently bound to proteins, including Spo11p (probe 2, P2) (11); we will refer to the P2 sample as DSB-enriched. Southern analysis showed that P2 was enriched at least 100-fold for DNA fragments resulting from DSB formation (Fig. 1). P1 and P2 were labeled with different fluorescently tagged nucleotides (Cy3-dUTP and Cy5-dUTP), mixed, and hybridized to DNA microarrays containing the 6,200 ORFs of the genome (17). Most DSBs occur within the intergenic regions (3, 13); because the probe DNA is derived from DNA fragments 2–3 kb in size and the intergenic regions in yeast average 500 bp (21), P2 DNA should contain portions of ORFs that border the intergenic regions.
Figure 1.

Characterization of DSB-enriched probe. (a) Map of the HIS4 recombination hotspot in FX4. An insertion of telomeric DNA with three Rap1 binding sites results in high levels of meiosis-specific DSBs at the position indicated by the arrow (4). The position of the probe (pDN42; 4) used in b is shown by the vertical line adjacent to the map. (b) Southern analysis of FX4 meiotic DNA samples used in the microarray experiments. Lanes 1 and 2 contain BglII-treated DSB-enriched DNA (P2) isolated by using glass fiber filters (11), and BglII-treated total genomic DNA (P1), respectively. Lane 3 contains HindIII-treated λ DNA. Arrows indicate the position of the intact 2.9-kb HIS4-BIK1 BglII fragment and the 1.9-kb fragment resulting from the meiosis-specific DSB.
Dual-wavelength images of microarrays were obtained by scanning at a resolution of 10 microns/pixel. The relative hybridization signal at each spot was determined by calculating the ratio of mean pixel intensity within a spot for both wavelengths, each wavelength measuring the hybridization of one of the two probes. The hybridization signals were normalized to a value of approximately 1 over all 6,200 ORFs. Thus, a ratio of hybridization of 1 represents an ORF with an average level of nearby DSBs. Seven ORF-containing microarrays were analyzed, four with FX4 and three with FX6. In four of these experiments (two with FX4 and two with FX6 DNA), P1 was labeled with the Cy5-dUTP (red fluorophore) and P2 was labeled with the Cy3-dUTP (green fluorophore); in the other three experiments, the labeling scheme was reversed. This procedure controls for the possibility of DNA sequences that preferentially label with one type of fluorophore.
Fig. 2 is a graph of the frequency distribution of all genomic ORFs with various P2/P1 hybridization ratios (log of the median P2/P1 ratio). The frequency distribution is asymmetric, with approximately one-eighth of the ORFs located in a shoulder of high P2/P1 ratios. We classified an ORF as “hot” if it ranked in the top 12.5% in at least five of the seven microarrays and “cold” if it ranked in the bottom 12.5% in at least five of seven experiments. A binomial expansion calculation predicts that these criteria will result in only three false-positive hot or cold ORFs. Thus, for our analysis, we operationally define hot and cold ORFs as those that reproducibly have hybridization ratios in the top and bottom eighths, respectively. This classification scheme is conservative, although somewhat arbitrary, and emphasizes the hottest and coldest ORFs in the genome.
Figure 2.

Hotspot activity of yeast ORFs. To estimate DSB formation adjacent to each ORF, we measured the ratio of hybridization to a DSB-enriched probe (P2) to a total genomic probe (P1) in seven microarrays. The log of the median hybridization ratio for each of the 6,200 ORFs is graphed; ratios were grouped into bins of 0.015 log units. An arrow marks the separation between the ORFs with hybridization ratios ranked in the top eighth from those in the bottom seven-eighths.
By these criteria, we detected 303 hot ORFs, clustered into 177 hotspots (Table 1). Hot ORFs were considered to represent a single hotspot if they were adjacent or if they were separated by a single nonhot ORF. We found 49 cold ORFs, clustered into 40 coldspots (Table 2). The positions of the hotspots and coldspots along the chromosomes are shown in Fig. 3.
Table 1.
Recombination activity of hotspot ORFs
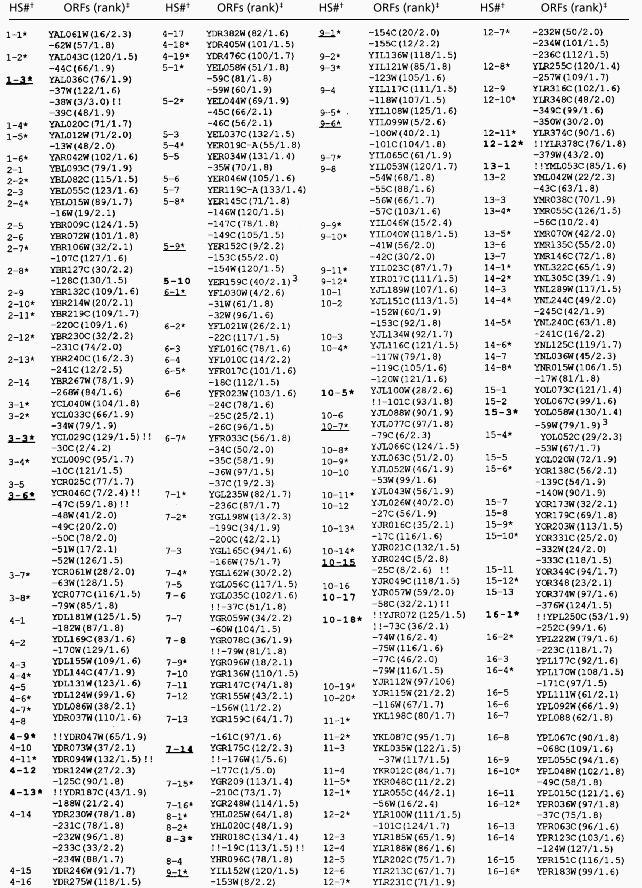 |
† Hotspots (HS) represent one or more ORFs ranked in the top 12.5% in five or more of the seven microarrays. Hotspots are numbered consecutively from the left end of the chromosome. Boldface indicates that a DSB has been mapped by Southern analysis; an asterisk indicates that the center of the hotspot is located within 2.5 kb of a peak of %(G+C) base composition that is at least 3% above the average %(G+C) for the chromosome. Underlining indicates that the hotspot has at least one ORF ranked among the 12 “hottest” ORFs in the genome.
‡ The designation of the hotspot ORFs is based on that used in the Stanford Genome Database. The ranking of each ORF (1 representing the “hottest”) is shown in parentheses as the first number. The procedure used to determine the ranking is described in the text. The second number in parentheses, separated by a slash from the ranking, is the median ratio of hybridization (DSB-enriched probe/genomic probe). Double exclamation points indicate the intergenic location of mapped DSBs. For example, the DSB for HS1-3 mapped between YAL038W and YAL039C. Exclamation points preceding the hotspot ORF indicate that the DSB was between the ORF and the next ORF to the left. Some hotspots had more than one DSB.
DSBs were located within the YER157W and YOL059W ORFs.
Table 2.
Recombination activity of coldspot ORFs
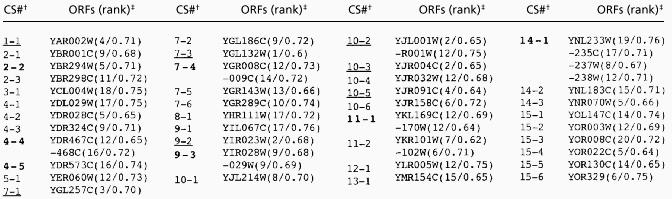 |
† Coldspots (CS) represent one or more ORFs ranked in the bottom 12.5% in five or more of the seven microarrays. Boldface indicates that Southern analysis was performed; no DSBs were detected for any coldspots examined. In addition, none of the coldspots was located within 2.5 kb of a GC peak. Underlining indicates that the coldspot has at least one ORF ranked among the seven “coldest” ORFs in the genome.
‡ Coldspots were ranked in the same manner described in Table 1 with one exception. After assignment of the median ranking for each coldspot ORF, the rankings were reordered with the coldest ORF ranked 1. As in Table 1. the number after the ranking is the median ratio of hybridization (DBS-enriched probe/genomic probe).
Figure 3.

Chromosomal map positions of the hotspots (red) and coldspots (green). Ovals indicate centromeres. Blue lines represent peaks of G + C base composition that are 3% higher than the average for the chromosome.
The relative strengths of the hot ORFs were determined by the following procedure: (i) within each microarray data set, each ORF was assigned a ranking based on the P2/P1 hybridization ratio, (ii) the median ranking of each ORF within the seven data sets was determined, and (iii) the 303 hot ORFs were reordered by their median ranking and renumbered with the highest ranking assigned a value of 1. These values are shown in Table 1 (first number in parentheses); because some hotspots share the same ranking, the values extend from 1 to 134. An analogous method was used to rank the coldspots (Table 2). In Tables 1 and 2, we also show the median ratio of P2/P1 hybridization (second number in parentheses). We chose to use the median ranking, instead of the median ratio of hybridization, to compare hotspots and coldspots because the rank positions in different microarrays varied less than the ratios of hybridization.
The highest ranked ORFs (YGR176W and YGR177C) had ratios of hybridization (P2/P1) of about five. The weakest hot ORFs had ratios of hybridization about 1.4-fold above the average. As described above, the strains FX4 and FX6 were isogenic except for small recombination-stimulating insertions located between BIK1 and HIS4 in FX4 and ARG4 and DED81 in FX6. ARG4 (YHR018C) in FX4 had a hotspot ranking of 134, and a hybridization ratio of 1.4 (Table 1); in FX6, the ARG4 ranking was 2, with a hybridization ratio of 5.5. No significant difference was observed for the HIS4/BIK1 hotspot in FX4 compared with FX6 (rankings of 4 and 2, and hybridization ratios of 3.2 and 4.2, respectively), possibly because of the very high level of hotspot activity in the wild-type (FX6) strain. We also examined two microarrays (one using FX4- and one using FX6-derived probes) that had both intergenic regions and ORFs. The results obtained were consistent with those seen with the ORF-only arrays. For example, 76% of the top 25 ranked hotspots from the ORF-only arrays were next to one of the intergenic regions ranked in the top 5% in the two ORF + intergenic microarrays.
Several arguments suggest that the microarray analysis reliably identifies hotspots and coldspots. First, all of the regions previously shown to have strong hotspot activity by genetic or physical criteria are represented among the 177 microarray-identified hotspots including: HIS4, ARG4, DED81, CYS3, HIS2, ARE1/IMG1, and CDC19 (1). Second, most of the hotspots identified by the microarray analysis that we examined by Southern analysis had one or more nearby strong meiosis-specific DSBs; none of the seven coldspots examined had detectable DSBs (data not shown). The hotspots with mapped DSBs are shown in boldface in Table 1. Twenty of the 22 mapped DSB sites were located within intergenic regions.
For chromosome III, we compared the microarray mapping to other mapping procedures. Baudat and Nicolas (13) did a detailed mapping of DSB sites on chromosome III. In Fig. 4, we compare the “heat” of chromosome III ORFs (showed as log of the median P2/P1 hybridization ratios) as determined in our study (Fig. 4a) with the data from the Baudat and Nicolas analysis (Fig. 4b). Although there are quantitative differences in the patterns, both methods identify similar hot and cold regions on chromosome III. Some of the differences are likely to reflect strain-specific differences in hotspot activity. For example, the strain used in our study (related to S288c) has a very high level of hotspot activity at the HIS4 locus (5) relative to the SK1 derivative examined by Baudat and Nicolas.
Figure 4.

Comparison of different methods of mapping hotspots and coldspots on chromosome III. (a) Summary of hybridization ratios using microarrays for all chromosome III ORFs. These data were based on yeast strains FX4 and FX6, except for the HIS4 locus for which only the FX6 data were used. Each ORF was assigned the median hybridization ratio of the seven microarrays and we graphed the log function of this value. The 182 ORFs represented in the analysis (left to right on the x axis) are (inclusive Stanford Genome Database numbering system): YCL076W-YCL073C, YCL069W-YCL016C, YCL014W-YCL001W, YCR001W-YCR020C, YCR020C-A, YCR021C-YCR024C, YCR024C-A, YCR025C-YCR029C, YCR029C-A, YCR030C-YCR077C, YCR079W-YCR097WA, and YCR097WB-YCR107W. The hotspot labels correspond to those listed in Table 1. (b) Summary of data of Baudat and Nicolas (13). These researchers monitored DSBs in an SK1 derivative by Southern analysis. (c) Analysis of hotspots in an SK1 derivative using DNA from immunoprecipitated Spo11p/hemagglutinin-DNA complexes as a probe of nylon filters containing chromosome III ORFs. The y axis shows the log of the average normalized ratio of hybridization (DSB-enriched probe/total genomic DNA probe). Gaps on the x axis reflect ORFs that were not present on the nylon filters. (d) Analysis of hotspots in FX6 using macroarrays. In this experiment, the probe was prepared by the method used in a.
We also used a different method to examine chromosome III hotspots. PCR-amplified DNAs representing ORFs from chromosome III were spotted onto a nylon filter. Probe DNA was isolated from a rad50S SK1 derivative strain containing an epitope-tagged version of Spo11p. An antibody directed against the epitope was used to purify the Spo11p-associated DNA and that DNA sample was used as a hybridization probe. The resulting profile (Fig. 4c) of hybridization mimics that observed by Baudat and Nicolas. We also used the nylon filter array with an FX6-derived probe (isolated with the glass fiber filter method) (Fig. 4d). To obtain sufficient quantities of the hybridization probe for the microarray analysis and for the analysis shown in Fig. 4d, the DNA samples were amplified by PCR. We repeated the analysis using nylon filters with hybridization probes that had not been amplified. The patterns were similar (data not shown).
Distribution of Hotspots and Coldspots.
All of the chromosomes have at least one hotspot (Fig. 3). There was a significant correlation between chromosome size and the number of hotspots [parametric (Pearson) linear correlation P = 0.008]; there was also a significant correlation (P = 0.03) between chromosome size and the number of coldspots.
Large chromosomes in yeast have fewer crossovers per kb than small chromosomes (22). Although larger chromosomes have more hotspots than smaller chromosomes, the density of hotspots on the smaller chromosomes is significantly greater than on the larger chromosomes (P = 0.03). In addition, the hotspots on the smaller chromosomes are significantly “hotter” than those on the larger chromosomes (linear regression analysis, P = 0.05). The simplest interpretation of these data are that both hotspot strength and hotspot density contribute to the high levels of crossovers observed for smaller chromosomes.
The average spacing between hotspots was 54 kb (median value of 36 kb). The average distance between hotspots for intervals that include the centromere was 117 kb (median value of 94 kb). The average interhotspot distance excluding the intervals containing the centromeres was 48 kb, with a median distance of 32 kb. The interhotspot distances for the centromere-containing intervals exceed those for the intervals that do not contain the centromere (P < 0.0001, Mann–Whitney nonparametric comparison), consistent with other studies indicating suppression of meiotic recombination near the centromere (12, 13).
The average distance from the telomere to the proximal hotspot was 103 kb. If the 177 hotspots were placed randomly along the chromosome, we would expect 14 located within 30 kb of the telomeres. None were observed, a significant departure from this expectation (P < 0.001 by χ2 analysis). These results support previous observations indicating a lack of DSBs near the telomeres of rad50S yeast strains (13, 14). The distribution of coldspots is strikingly different from that observed for hotspots (Fig. 3). If the 40 coldspots were randomly distributed, we expect three within 30 kb of the telomeres and two within 30 kb of the centromeres. We observed 10 telomere-associated coldspots and eight centromere-associated coldspots, a significant departure from the random distribution (P = 0.003 by χ2 analysis).
Correlation Between Peaks of G + C Base Composition and Hotspots.
Sharp and Lloyd (23) noted that chromosome III had broad peaks of high G + C base composition located in the middle of the left and right arms. They pointed out that three of the four known hotspots were located within these regions. Similar patterns of variation in base composition have been detected in most yeast chromosomes by researchers using sliding windows of either 5 kb (19) or 30 kb (21). To determine whether hotspots or coldspots were correlated with peaks of G + C, we monitored base composition along all of the yeast chromosomes in 5- and 30-kb sliding windows, moving the windows in steps of 1 kb. As described below, there was a striking correlation between hotspots and peaks of high G + C obtained with the 5-kb window; the correlation was much less striking with the 30-kb window (data not shown). We found no obvious correlation between hotspot locations and peaks of gene density analyzed with a 30-kb window (24).
When scanned with a 5-kb window, chromosome III had eight G + C peaks in which the peak G + C content was at least 3% above the average (38.7%) for the chromosome. The midpoints of seven of the eight hotspots on chromosome III were within 2.5 kb of one of the eight peaks. This correlation is depicted in Fig. 5. In Fig. 3, the vertical blue lines drawn below each chromosome show peaks of G + C that are at least 3% above the average for the chromosome.
Figure 5.

Correlation of hotspots with peaks of high G + C content on chromosome III. (Upper) A scan of base composition at 1-kb intervals (5-kb sliding window). The marked peaks have a G + C base composition ≥ 3% above the average for chromosome III (38.7%). In addition, we show the log values of the median hybridization ratio (DSB-enriched probe/total genomic DNA). (Lower) The ORFs (two lines of black rectangles) and the positions of a number of structural chromosomal elements are indicated.
Several statistical tests were done to assess the significance of this correlation. A total of 221 peaks of G + C were observed. If the 177 hotspots were distributed at random with respect to the peaks, the expected number within 2.5 kb of a peak is 18. We observed 99 peak-associated hotspots, a deviation from this expectation (P < 0.001 by χ2 analysis). We also calculated the G + C content in a 5-kb window centered at the midpoint of each hotspot. In 162 of 177 hotspots, the G + C content exceeded the average G + C content of the chromosome (P < 0.001 by χ2 analysis). We also found an association between the ranking of the hotspots (Table 1) and their G + C content by linear regression analysis (P < 0.002). In addition, the average rankings for the peak-associated hotspots and the nonpeak-associated hotspots were different (P < 0.05).
Of 22 mapped DSBs, 14 (13 intergenic and one intragenic) were associated with high G + C peaks (Table 1). The observed G + C content of these intergenic regions was 38.7%. The average G + C content of yeast genomic intergenic regions is about 34% (21). The average G + C content for the DNA sequences flanking the intergenic regions in a 5-kb window was 43.3%, higher than the 40.2% G + C observed for the average yeast ORF. Thus, the high G + C content associated with many of the hotspots reflects higher than average G + C content in both the intergenic region and flanking ORFs.
Relationship Between Hotspots and Known Repetitive Chromosomal Elements.
We looked for associations between hotspots and repetitive DNA elements. One repetitive sequence is the ARS (autonomously replicating sequence) element; a subset of ARS elements are chromosomal replication origins. Active origins have been mapped only on chromosomes III (25) and VI (26, 27). There are 15 hotspots located on these two chromosomes that have a combined total length of 585 kb. A total of 75/585 or 0.128 of the sequences on these two chromosomes are within 2.5 kb of a hotspot. There are 17 active origins on the two chromosomes. If these are located randomly with respect to the hotspots, we expect to find 0.128 × 17 or two origins within 2.5 kb of a hotspot; we find one. Thus, there is no significant association between the location of replication origins and hotspots.
By similar arguments, we failed to find any association between hotspots and other repetitive DNA elements including the tRNA genes, small nuclear RNA genes, Ty elements (Ty1–5), and the long terminal repeat elements derived from the Ty elements (σ, δ, and τ). Our calculations were based on the numbers of repetitive DNA elements reported by Kim et al. (28) and the positions indicated in the Stanford Genome Database. In addition, no associations between repetitive elements and coldspots were found, except for the centromere/telomere associations described above.
Relationship Between Hotspots and Transcription.
α Hotspots require the binding of transcription factors (8). Consequently, we looked for associations between hotspots and various parameters of gene expression. We mapped 20 DSBs within intergenic regions of the hotspots (Table 1). Intergenic regions can be located between the 5′ ends of two genes (class 1), between the 3′ ends of two genes (class 2), or between the 5′ end of one gene and the 3′ end of the other (class 3). The expected ratio of these classes is 1:1:2 if the transcriptional orientation of the flanking genes is random; this ratio is approximately that observed if all intergenic regions are analyzed (1,342, class 1; 1,427, class 2; 2,621, class 3). In contrast, of the 20 DSB-associated intergenic regions, we found 13 class 1, six class 3, and only one class 2. One explanation of this departure from expectation (P = 0.0001 by Fisher exact test) is that it reflects the requirement of transcription factor binding for hotspot activity. Alternatively, the negative superhelical stress resulting from divergent transcription may promote hotspot activity.
It is likely that some transcription factors are more proficient at stimulating recombination than others. For example, activity of the HIS4 hotspot requires the binding of Bas1p, Bas2p, and Rap1p, but not the binding of Gcn4p (6). If other hotspots behave similarly, one might find conserved DNA sequences reflecting conserved transcription factor binding sites at hotspots. We compared putative transcription factor binding sites from our 20 mapped DSB sites with 31 random intergenic regions. There was a significant enrichment for Pho4p (P = 0.05) and Gcr1p (P = 0.02) binding sites. Because we examined the intergenic regions for the binding sites of 35 different factors, however, the enrichment for Pho4p and/or Gcr1p binding sites at hotspots might represent a false-positive. Using the meme program of GCG, we also looked for other conserved DNA sequence motifs specific to the 20 intergenic DSB sites. None were found. Blumenthal-Perry et al. (29) reported a motif associated with some recombination hotspots. We found no striking correlation between this motif and the hotspots identified in our study, suggesting that this motif might be associated with a subset of hotspots.
Several interesting correlations were observed between hotspot ORFs and gene functions. A total of 3,725 of the yeast ORFs have been assigned one or more functions (Munich Information Center for Protein Sequences database), and 181 of the hotspot ORFs are within this group. There was a very significant (P < 0.0001) over-representation of the hotspot ORFs in the metabolism functional class (81 of the 181 classified ORFs), particularly involving amino acid biosynthetic genes. Of the 25 hotspots with the highest rankings, 19 had one or more ORFs classified in the metabolism functional class. In addition, there was an over-representation (P = 0.03) of hotspot ORFs classified in the functional category of ionic homeostasis. There was an over-representation of coldspot ORFs in the categories of transport facilitation and intracellular transport (P = 0.007). One interpretation of these correlations is that certain categories of genes are associated with a particular chromatin structure that is favorable (hotspots) or unfavorable (coldspots) for initiating meiotic recombination.
Using a database that examined mRNA levels for most of the yeast genes in vegetative cells (30), we found that hotspot ORFs were expressed at higher levels than average ORFs. The average ORF has a median transcript level of 0.8/cell (30). Of the 250 hotspot ORFs in the database, 155 were expressed at higher levels than this value and 95 were expressed at lower values. This effect, at least in part, might be related to the correlation between hotspots and genes that function in metabolic pathways, because such genes are likely to be abundantly expressed in vegetative cells.
Microarrays have been used to examine transcription profiles for many conditions of cell growth including sporulation (31). There is an association between hotspot ORFs and genes whose expression is repressed in meiosis. This association is most significant at times 5 h and 11.5 h after induction of sporulation (P = 0.001 and P = 0.003, respectively, by contingency χ2 test). This association is not a useful predictor of hotspot activity, however, because most hotspot ORFs show no meiosis-specific reduction in gene expression and most ORFs that show such a reduction are not hot ORFs.
Caveats Concerning Our Hotspot and Coldspot Mapping.
Our mapping of hotspots and coldspots has several caveats. First, the method used to prepare P2 DNA enriches for any protein covalently bound to meiotic DNA (for example, other topoisomerases), in addition to Spo11p. It is unclear whether DSBs made by other proteins contribute to meiotic recombination. A related issue is that the difference in the level of hybridization between a hotspot ORF and a nonhotspot ORF in the microarray experiments (often less than a factor of 2) is smaller than expected by direct measurement of DSBs using standard Southern analysis (13). Although this difference may be an artifact of the PCR amplification required to prepare the probe, an alternative possibility is that the microarray analysis detects nonlocalized DSBs more sensitively than standard Southern analysis. Another caveat is that we restricted our analysis to DNA samples prepared from rad50S strains. Although the level of DSBs measured in rad50S strains often correlates well with the frequency of recombination measured by genetic techniques (3), a recent study has shown that DNA sequences near the telomeres of two chromosomes have lower levels of DSBs in rad50S strains than in wild-type strains (V. Borde, A. Goldman, and M.L., unpublished data). Finally, our analysis was restricted to one specific genetic background. As discussed above, the same locus (for example, HIS4 on chromosome III) can have very different levels of recombination in different genetic backgrounds.
Summary and Conclusions.
No single distinctive feature is shared among all of the hotspots. The strongest correlation is the association with peaks of high G + C. One interpretation of this association is that DSBs are dictated, at least partly, by chromosome structure. Cohesins are bound at AT-rich regions located at approximately 15-kb intervals along chromosome III (19); some subset of these sites may be used to structure the DNA loops (averaging 20 kb) (32) of meiotic yeast chromosomes, and it has been suggested that DSB formation may be related to DNA loop structure (33). Because the average distance between hotspots measured in our study (54 kb) exceeds the average size of the DNA loops, all DNA loops do not represent hotspots. An alternative possibility is that the GC-rich regions bind protein(s) that result in a chromatin structure that is open to the recombination machinery. A related possibility is that the GC-rich regions are open to the recombination machinery by default, access to other chromosomal regions being blocked by cohesins. The association between hotspots and GC-rich regions is consistent with the finding that GC-rich Escherichia coli plasmid sequences often act as hotspots in yeast (20, 34).
One interpretation of the correlations of hotspots with genes of specific functions and specific expression patterns is that most hotspots require the binding of transcription factors to open the chromatin to permit access for the recombination machinery. We suggest that certain transcription factors are preferred by the recombination machinery. The repression of transcription observed for genes located within some of the hotspots could reflect competitive interactions between the transcription and recombination machinery for access to the intergenic region.
We found, as had others previously, that many of the coldspots are nonrandomly associated with telomeres and centromeres. Borde et al. (35) showed that the lack of DSBs in cold regions does not reflect an absence of nuclease hypersensitive sites, but is likely to reflect some aspect of higher-order chromosome structure.
In conclusion, the position of hotspots may be regulated at two levels: the approximate position regulated by some feature of chromosome structure related to GC-richness, and a more precise position regulated by chromatin openness controlled by transcription factors. Although we (and others) have begun to define operational rules that may help in predicting hotspots, more mechanistic studies will be critical in refining these rules. In addition, studies in other organisms (such as Schizosaccharomyces pombe) eventually will reveal to what extent these rules are organism-specific.
Acknowledgments
We thank M. Dominska, L. Stefanovich, P. Greenwell, N. Perabo, and K. Werner for technical assistance and S. Whitfield for photographic assistance. We thank S. Keeney for hemagglutinin-tagged Spo11p and G. S. Roeder, C. Newlon, and S. Keeney for useful suggestions. We thank J. Sekelsky, Y. Blat, and Jing Zhu for assistance in data analysis. The research was supported by National Institutes of Health Grants GM24110 (T.D.P.) and HG00983 (P.O.B.) and by the Howard Hughes Medical Institute. P.O.B. is an Associate Investigator of the Howard Hughes Medical Institute. J.L.G. was supported by a training grant to the Lineberger Cancer Center (T32 CA09156) and by American Cancer Society Grant 5–39833); J.D.R. was supported by a Sandler grant.
Abbreviations
- DSB
double-strand DNA break
- P1
probe 1
- P2
probe 2
Footnotes
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on April 27, 1999.
References
- 1.Lichten M, Goldman A S. Annu Rev Genet. 1995;29:423–444. doi: 10.1146/annurev.ge.29.120195.002231. [DOI] [PubMed] [Google Scholar]
- 2.Petes T D, Malone R E, Symington L S. In: The Molecular and Cellular Biology of the Yeast Saccharomyces. Broach J, Jones E W, Pringle J R, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1991. pp. 407–521. [Google Scholar]
- 3.Wu T C, Lichten M. Science. 1994;263:515–518. doi: 10.1126/science.8290959. [DOI] [PubMed] [Google Scholar]
- 4.Fan Q-Q, Xu F, Petes T D. Mol Cell Biol. 1995;15:1679–1688. doi: 10.1128/mcb.15.3.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White M A, Wierdl M, Detloff P, Petes T D. Proc Natl Acad Sci USA. 1991;88:9755–9759. doi: 10.1073/pnas.88.21.9755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White M A, Dominska M, Petes T D. Proc Natl Acad Sci USA. 1993;90:6621–6625. doi: 10.1073/pnas.90.14.6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White M A, Detloff P, Strand M, Petes T D. Curr Genet. 1992;21:109–116. doi: 10.1007/BF00318468. [DOI] [PubMed] [Google Scholar]
- 8.Kirkpatrick D T, Fan Q-Q, Petes T D. Genetics. 1999;152:101–115. doi: 10.1093/genetics/152.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirkpatrick D T, Wang Y-H, Dominska M, Griffith J D, Petes T D. Mol Cell Biol. 1999;19:7661–7671. doi: 10.1128/mcb.19.11.7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergerat A, de Massy B, Gadelle D, Varoutas P C, Nicolas A, Forterre P. Nature (London) 1997;386:414–417. doi: 10.1038/386414a0. [DOI] [PubMed] [Google Scholar]
- 11.Keeney S, Giroux C N, Kleckner N. Cell. 1997;88:375–384. doi: 10.1016/s0092-8674(00)81876-0. [DOI] [PubMed] [Google Scholar]
- 12.Lambie E J, Roeder G S. Cell. 1988;52:863–873. doi: 10.1016/0092-8674(88)90428-x. [DOI] [PubMed] [Google Scholar]
- 13.Baudat F, Nicolas A. Proc Natl Acad Sci USA. 1997;94:5213–5218. doi: 10.1073/pnas.94.10.5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein S, Zenvirth D, Dror V, Barton A B, Kaback D B, Simchen G. Chromosoma. 1996;105:276–284. doi: 10.1007/BF02524645. [DOI] [PubMed] [Google Scholar]
- 15.Zenvirth D, Arbel T, Sherman A, Goldway M, Klein S, Simchen G. EMBO J. 1992;11:3441–3447. doi: 10.1002/j.1460-2075.1992.tb05423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goyon C, Lichten M. Mol Cell Biol. 1993;13:373–382. doi: 10.1128/mcb.13.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeRisi J L, Iyer V R, Brown P O. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 18.Strahl-Bolsinger S, Hecht A, Luo K, Grunstein M. Genes Dev. 1997;11:83–93. doi: 10.1101/gad.11.1.83. [DOI] [PubMed] [Google Scholar]
- 19.Blat Y, Kleckner N. Cell. 1999;98:249–259. doi: 10.1016/s0092-8674(00)81019-3. [DOI] [PubMed] [Google Scholar]
- 20.Wu T C, Lichten M. Genetics. 1995;140:55–66. doi: 10.1093/genetics/140.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dujon B. Trends Genet. 1996;12:263–270. doi: 10.1016/0168-9525(96)10027-5. [DOI] [PubMed] [Google Scholar]
- 22.Kaback D B, Steensma H Y, de Jonge P. Proc Natl Acad Sci USA. 1989;86:3694–3698. doi: 10.1073/pnas.86.10.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharp P M, Lloyd A T. Nucleic Acids Res. 1993;21:179–183. doi: 10.1093/nar/21.2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goffeau A, Aert R, Agostini-Carbone L, Ahmed A, Aigle M, Alberghina L, Albermann K, Albers M, Aldea M, Alexandraki D, et al. Nature (London) 1997;387:3–105. [Google Scholar]
- 25.Newlon C S, Collins I, Dershowitz A, Deshpande A M, Greenfeder S A, Ong L Y, Theis J F. Cold Spring Harbor Symp Quant Biol. 1993;58:415–423. doi: 10.1101/sqb.1993.058.01.048. [DOI] [PubMed] [Google Scholar]
- 26.Yamashita M, Hori Y, Shinomiya T, Obuse C, Tsurimoto T, Yoshikawa H, Shirahige K. Genes Cells. 1997;2:655–665. doi: 10.1046/j.1365-2443.1997.1530351.x. [DOI] [PubMed] [Google Scholar]
- 27.Friedman K L, Brewer B J, Fangman W L. Genes Cells. 1997;2:667–678. doi: 10.1046/j.1365-2443.1997.1520350.x. [DOI] [PubMed] [Google Scholar]
- 28.Kim J M, Vanguri S, Boeke J D, Gabriel A, Voytas D F. Genome Res. 1998;8:464–478. doi: 10.1101/gr.8.5.464. [DOI] [PubMed] [Google Scholar]
- 29.Blumenthal-Perry, A., Zenvirth, D., Klein, S., Onn, I. & Simchen, G. (2000) EMBO J., in press. [DOI] [PMC free article] [PubMed]
- 30.Holstege F C, Jennings E G, Wyrick J J, Lee T I, Hengartner C J, Green M R, Golub T R, Lander E S, Young R A. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- 31.Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown P O, Herskowitz I. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 32.Moens P B, Pearlman R E. BioEssays. 1988;9:151–153. doi: 10.1002/bies.950090503. [DOI] [PubMed] [Google Scholar]
- 33.Zickler D, Kleckner N. Annu Rev Genet. 1999;33:603–754. doi: 10.1146/annurev.genet.33.1.603. [DOI] [PubMed] [Google Scholar]
- 34.Stapleton A, Petes T D. Genetics. 1991;127:39–51. doi: 10.1093/genetics/127.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borde V, Wu T C, Lichten M. Mol Cell Biol. 1999;19:4832–4842. doi: 10.1128/mcb.19.7.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]