

Abstract
Chemokines and chemokine receptors play important roles in HIV-1 infection and tropism. CCR5 is the major macrophage-tropic coreceptor for HIV-1 whereas CXC chemokine receptor 4 (CXCR4) serves the counterpart function for T cell-tropic viruses. An outstanding biological mystery is why only R5-HIV-1 is initially detected in new seroconvertors who are exposed to R5 and X4 viruses. Indeed, X4 virus emerges in a minority of patients and only in the late stage of disease, suggesting that early negative selection against HIV-1–CXCR4 interaction may exist. Here, we report that the HIV-1 Tat protein, which is secreted from virus-infected cells, is a CXCR4-specific antagonist. Soluble Tat selectively inhibited the entry and replication of X4, but not R5, virus in peripheral blood mononuclear cells (PBMCs). We propose that one functional consequence of secreted Tat is to select against X4 viruses, thereby influencing the early in vivo course of HIV-1 disease.
Keywords: chemokine antagonist, viral coreceptor
Infection of cells by HIV-1 requires CD4 and a seven-transmembrane-domain chemokine receptor (1). Historically, HIV-1 isolates have been classified as either macrophage tropic (MT) and non-syncytium-inducing (NSI) or T cell tropic (TT) and syncytium-inducing (SI). Recent findings demonstrate that entry of MT/NSI HIV-1 into susceptible cells requires CC chemokine receptor 5 (CCR5), a receptor for the CC-chemokines RANTES, macrophage inflammatory protein (MIP-1α, and MIP-1β (2–4) whereas entry of TT/SI viruses requires CXC chemokine receptor 4 (CXCR4), the receptor for the CXC chemokine SDF-1. These results have prompted the nomenclature R5 and X4 to denote usage of CCR5 or CXCR4, respectively (5).
Individuals infected with a swarm of R5 and X4 viruses typically transmit successfully only the R5 form to recipients (6). In recipient seroconvertors, eventual emergence of X4-HIV-1 is surprisingly slow and generally not before 7 yr after infection (7). This delayed evolution of R5 to X4 virus is unexpected in view of HIV-1's exceptionally high rates of replication/mutation (8) and the fact that only one or two substitutions in the viral envelope protein sequence sufficiently convert R5 phenotype to X4 (9–11). Moreover, many more CD4+ T cells express CXCR4 than CCR5 in both peripheral blood and lymphoid tissues (12–14); and macrophages express amounts of CXCR4 sufficient for infection by X4 isolates although it remains controversial whether X4-tropic HIV-1 can infect macrophages in vivo (15). Findings on cellular and humoral immunity in infected individuals unsatisfactorily explain the 7-yr-long X4 virus-free duration. Collectively, these observations have prompted the suggestion by some of an early in vivo negative selection against X4 virus (16), which could explain delayed emergence.
In patients, the emergence of X4-HIV-1 correlates with rapidly progressive immunodeficiency and clinical deterioration. These in vivo events may be causally related because X4 strains are also more virulent in vitro. The host, as well as the virus, may benefit biologically by restricting emergence of virulent virus strains. Currently, the only known host factor able to selectively block X4-HIV-1 infection is SDF-1, the chemokine-ligand for CXCR4. Here we report the first viral factor with this property, the HIV-1 Tat protein, acting as a non-chemokine CXCR4-selective antagonist.
Materials and Methods
Competitive Ligand Binding.
For 125I-labeled chemokine binding to whole cells, 1 × 106 cells were incubated for 1 h at room temperature in buffer [HBSS plus 1% BSA and 0.1% sodium azide (pH 7.4)] containing 0.1 nM 125I-labeled chemokines (NEN) in the presence or absence of unlabeled chemokines or Tat. Specific activity for each chemokine used was 2200 Ci/mmol, as indicated by the manufacturer. Cells were centrifuged through Hanks' balanced salt solution (HBSS)/1% BSA containing 0.5 M NaCl, the supernatants were removed, and pellets were counted in a gamma counter. Each condition was tested in triplicate. Data are presented as unadjusted cell-associated counts. Where indicated, binding was competed with a 40-aa Tat peptide corresponding to residues 11 to 50. SDF-1α binding to CXCR4 used human PBMCs; RANTES or MIP-1β binding to CCR5 used HEK293 cells stably expressing transfected hCCR5 (17). Specific competition used an excess (100 nM) of unlabeled cognate chemokine.
Calcium Mobilization Assays.
Calcium-mobilization was measured by Fura-2 fluorescence as previously described (18).
Purification of Proteins.
1-Exon (1–72) and 2-exon () Tat or mutant derivatives were fused to the maltose-binding protein in Escherichia coli expression vector pMAL-c2. Recombinant proteins were purified according to the manufacturer's protocol (New England BioLabs). 1-Exon and 2-exon Tat fusion proteins were checked to be biologically active for cellular uptake and transactivation of an unintegrated HIV-long terminal repeat (LTR)-reporter.
Cell Culture, HIV-1 Infection, and Reverse Transcriptase (RT) Assays.
PBMCs were stimulated with phytohemagglutinin 3 days before infection and were maintained in medium containing IL-2. Infections were performed in duplicate. Cells (1 × 105) were infected with 100 tissue culture ID50 (TCID50) of X4-HIV-1 strain NL4-3 with the indicated concentrations of soluble proteins. Fresh proteins were replenished into culture medium every 4 days, and culture supernatants were sampled for RT activity as previously described (19). All HIV-1 infections were repeated a minimum of 3 times.
Immunodetection of Tat in Serum.
Poly(vinylidene difluoride) (PVDF) (Immobilon P, Millipore) membrane was activated with methanol for 30 s and washed four times (1 min each) with TN buffer [50 mM Tris⋅HCl (pH 8.0)/50 mM NaCl]. Membrane was placed on a sheet of Whatman 3M paper lightly soaked with TN buffer and positioned atop a dry pad of absorbent paper. Serum samples (0.5 ml) were mixed with an equal volume of 2× TN buffer. Samples were spotted 50 ml at a time onto PVDF membrane and allowed to slowly air dry. Membranes were then treated with blocking buffer [0.2% I-block reagent (Tropix)/1× PBS/0.1% Tween 20] followed by incubation with a mixture of two polyclonal rabbit anti-Tat sera (raised to Tat peptide residues 1–25 and 62–86). Parallel assays were performed in which 5 μg of maltose-binding protein (MBP)-Tat () protein was added to anti-Tat sera for 1 h at room temperature before incubation with filters. Washing and chemiluminescent detection were according to manufacturer's protocol (Tropix, Bedford, MA).
PCR Analyses.
Infected cells (1 × 106) were washed and resuspended into 100 ml of lysis buffer [10 mM Tris⋅HCl (pH 8.0)/0.5 mM EDTA/0.001% Triton X-100/0.0001% SDS). PCR was performed with HIV-1-specific primers and actin primers as detailed in the legends to Fig. 4 and Fig. 5. Each 100-μl PCR reaction contained 5 μl of cell lysate. Specific PCR products were confirmed by Southern hybridization to 32P-labeled HIV-1 LTR probe (Fig. 4).
Figure 4.

Tat inhibits X4-dependent infection at the step of viral entry into cells. HOS-CD4 cells stably expressing individual coreceptors (AIDS Reagent Repository, National Institute of Allergy and Infectious Diseases) were infected either with DNase I-treated NL4-3 (X4-tropic) or NLAD8 (ref. 30; NL4-3 modified to contain an R5-tropic envelope). At time of infection, affinity-purified MBP or MBPTat72 fusion protein was added separately to final concentrations of 100 ng/ml. Cells were harvested at indicated times (hours postinfection), washed, and resuspended into PCR lysis buffer [10 mM Tris⋅HCl (pH 8.0)/0.5 mM EDTA/0.001% Triton X-100/0.0001% SDS). PCR products from nef-R primer pair (5′-AGCTGTAGATCTTAGCCACTT-3′ and 5′-AGGCTCAGATCTGGTCTAA-3′) were visualized after hybridization with a 32P-labeled HIV-1 LTR probe. Control PCRs on the same samples used β-actin-specific primers (ethidium bromide-stained bands). (A) Schematics of the HIV-1 LTR and primers used. Virus-specific signal is 522 bp. (B) PCR analysis of NL4-3/NLAD8 infection of HOS-CXCR4/HOS-CCR5 (Left and Right, respectively). Tat inhibited NL4-3 entry into HOS-CXCR4 (lanes 4 and 6) but not NLAD8 entry into HOS-CCR5 (lanes 10 and 12). HIV-1 (arrow) points to the virus-specific 522-bp band. (C) MAGI analysis of R5- or X4-dependent viral entry into cells. RT-normalized virus stocks (3,000 cpm) of NL4-3 or NLAD8 were used to infect either U373-MAGI-CXCR4 or U373-MAGI-CCR5 cells in the presence of MBP (−Tat) or MBPTat72 (+Tat). Cells were processed for beta-galactosidase staining (31) 48 h after infection. Values are averages from three assays.
Figure 5.

Extracellular Tat selects against X4-tropism. (A) A two-step PCR protocol for detecting changes at residue 25 of the V3 loop. The region surrounding position 25 was first amplified by using primers (5′, GTAATTAATTGTACAAGACCCAAC and 3′, CTACTAACGTTACAATGAGCTTGTC). Changes at position 25 were queried by a further one-cycle PCR separately by using one of three codon-specific 32P-labeled inner primers (“labeling” primer). (B and C) Evolution at position 25 in V3 when X4-NL4-3-infected PBMCs were maintained with either MBPTat72 (B) or MBP (C). Cells were harvested at the indicated days postinfection and analyzed by PCR. K to Q change seen in the MBPTat72 samples is absent in the MBP samples. (D) Phenotypic analysis of MBPTat72-selected NL4-3 virus. RT-normalized (3,000 cpm) viral samples were collected at the indicated days after infection and assayed on U373-MAGI-CXCR4 or U373-MAGI-CCR5 cells. Assays were in triplicate, and average values are presented. Range of values varied by less than 20%. Proportion of NL4-3 virus that showed R5 phenotype at day 35 was calculated by using the formula [(U373-MAGI-CCR5 day 35 value) − (U373-MAGI-CCR5 day 0 value)]/[(U373-MAGI-CCR5 day 35 value) + (U373-MAGI-CXCR4 day 35 value)]. The proportion of R5-tropic NL4-3 at day 35 was (11 − 7)/[11 + 165] = 2%; and (29 − 7)/[129 + 29] = 14% for −Tat and +Tat samples, respectively.
RESULTS
HIV-1 Tat Binds CXCR4.
Tat is selectively secreted from HIV-1-infected cells (20, 21), suggesting potential extracellular functions (22, 23) in addition to its well-characterized nuclear transcriptional role. Fortuitously, we noted amino acid similarities between Tat and chemokines. When Tat was compared with 14 CC-chemokines (including I-309, RANTES, MIP-1α, MIP-1β, eotaxin, and others), several conserved elements, including a CC-motif (at Tat amino acids 30 and 31) and a high density of basic amino acids were revealed (data not shown; alignment comparisons available on request). Related features including a CXC-motif (at Tat amino acids 25 and 27) were also seen between Tat and CXC-chemokines (SDF-1, MIP-2α, MIP-2β, NAP-2, IL-8, GCP-2, and ENA-78; data not shown). Despite these similarities, the overall sequence identities between Tat and chemokines remain, at best, modest; and the three-dimensional structures are completely different.
Chemokines regulate leukocyte trafficking by activating specific G protein-coupled receptors. Many chemokine receptors, including CXCR4 (24) and CCR5 (2–4), have been characterized to function as cell entry (co)-factors for HIV. Ligands for these chemokine receptors can affect HIV-coreceptor activity (2–4, 25, 26). The weak sequence-relatedness between Tat and chemokines prompted us to ask whether Tat affects HIV coreceptor activity. We first checked whether Tat is an authentic ligand for chemokine receptor(s) by using a ligand-receptor competition assay (Fig. 1a). Cells expressing either CXCR4 or CCR5 were separately equilibrated with 125I-labeled ligands (SDF-1α, RANTES, or MIP-1β) with or without escalating amounts of Tat or 100 nM unlabeled chemokine. Tat competed efficiently (as effective as unlabeled SDF-1) for 125I-SDF-1a binding to CXCR4+ cells (Fig. 1a Left), but did not compete for binding of either 125I-RANTES (Fig. 1a Center) or 125I-MIP-1β to CCR5 (Fig. 1a Right). Consistent with this, protein affinity chromatography results showed direct binding of glutathione S-transferase (GST)-Tat () to CXCR4 without significantly detectable binding to CCR8, CCR5, or CCR4 (Fig. 1b). A similar binding specificity by Tat for CXCR4 protein was also verified in yeast two-hybrid assays (data not shown).
Figure 1.

Tat binds directly to CXCR4 and competes for 125I-SDF-1α binding to CXCR4. (a) Separate bindings of 125I-labeled SDF-1α (Left), RANTES (Center), or MIP-1β (Right) to cells were competed either with escalating concentrations (12.5 nM, 25 nM, and 100 nM) of a 40-aa Tat peptide corresponding to residues 11 to 50 or with an excess (100 nM) of unlabeled cognate chemokine (last bar in each panel). SDF-1α binding to CXCR4 was measured by using human PBMCs; RANTES or MIP-1β binding to CCR5 used CCR5/293 cells stably transfected with hCCR5. (b) GST-protein affinity chromatography shows direct binding of Tat to CXCR4. 35S-labeled chemokine receptors were translated in vitro from T7-generated transcripts in rabbit reticulocyte lysate and then incubated with 50 μl of glutathione-Sepharose beads saturated with either GST (lane 2) or GST-Tat (lane 3). Beads were washed twice (each time with 20 column volumes of buffer containing 100 mM NaCl). Proteins retained by either GST or GST-Tat were visualized by boiling washed beads in reducing SDS-sample buffer followed by PAGE and autoradiography.
Tat Antagonizes CXCR4-Function.
The physical interaction between Tat and CXCR4 (Fig. 1 and data not shown) directed us to consider functional significance (i.e., would extracellular Tat perturb SDF-1/CXCR4 signaling?). To ensure that any observed biological effect(s) emanated from Tat and not from endotoxins that might copurify in minute quantities with recombinant protein expressed in E. coli, we synthesized Tat and mutant polypeptides bearing alanine substitutions in the CC or CXC motifs, which were verified for purity by mass spectroscopy (J. Coligan, unpublished data). Stimulus-induced calcium flux occurred when PBMCs were exposed to SDF-1 (Fig. 2, panel 1, top recording); however, pretreatment with Tat peptide completely abolished this flux (Fig. 2, panel 1, bottom recording). Mutant Tat polypeptide changed in the CXC motif failed to affect SDF-1-signaling (Fig. 2; panel 1, middle recording). Collectively, these results show a functional interaction between Tat and CXCR4 that requires an intact CXC motif.
Figure 2.

Tat antagonizes signaling by CXCR4, but not CCR5, agonists. (Panel 1) Tat wild-type (wt) (residues 11–50), but not Tat-mCXC (cysteines at residues 25 and 27 changed to alanines), peptide-desensitized SDF-1-induced calcium mobilization. Tat peptides were added 50–100 s before SDF-1; measurements of calcium flux followed. Tat wt peptide did not affect RANTES signaling in PBMCs (panel 2) and monocytes (panel 3); nor MIP1α (panel 4) or MIP1β (panel 5) signaling in PBMCs. In each panel, the top tracing is the addition of chemokine alone (100 nM) whereas the bottom tracing(s) is pretreatment with Tat peptide(s) (200 nM), followed by addition of indicated chemokine (100 nM).
Specificity of Tat for CXCR4 was compared with effects on PBMCs, monocytes, or a CCR5-expressing HEK293 cell line (hCCR5/293; Fig. 2; panels 2–5) by CCR5 agonists, RANTES, MIP-1α, and MIP-1β. In contrast with SDF-1-signaling (Fig. 2, panel 1), Tat did not affect signaling by these CCR5 agonists (Fig. 2; panels 2–5; lower recordings).
Tat Inhibits CXCR4- but Not CCR5-Tropic Infection of Cells by HIV-1.
The above findings suggest that soluble Tat might selectively affect HIV-1 envelope–CXCR4 interaction. To address this issue, biologically active forms of 1-exon (Tat 72) and 2-exon (Tat 101) Tat proteins fused to MBP were expressed and purified. MBPTat72, MBPTat101, and MBP alone were tested at concentrations of 2 mg/ml and 20 ng/ml (Fig. 3 a and b) in X4-tropic infection of PBMCs. Compared with MBP alone, MBPTat101 at 20 ng/ml significantly inhibited infection; curiously, the same protein at 2 mg/ml slightly promoted infection (Fig. 3a). Opposing concentration-dependent findings have been described for several cytokines (27). A possible explanation for our findings here is that two separate Tat activities, a chemokine receptor-blocking role at low concentration and a dominant nuclear trans-activation function secondary to cellular uptake of protein at high concentration, coexist. Indeed, the 72 aa 1-exon Tat protein, which has been shown to be defective in its transcriptional activation of integrated HIV-1 provirus (19), inhibited virus replication >90% at both high (2 mg/ml) and low (20 ng/ml) concentrations (Fig. 3 a and b). MBPTat72 inhibition persisted at >50% even at 2 ng/ml (data not shown). We note that Tat and β-chemokines (MIP-1α, MIP-1β, and RANTES) are comparably sized small proteins, and that both inhibit HIV-1 at ng/ml concentrations (28). Hence, anti-HIV-1 potencies of Tat and β-chemokines are similar, with the latter being selective for R5- and the former for X4-HIV-1.
Figure 3.

Tat inhibits CXCR4-dependent infection of cells by HIV-1 NL4-3. Phytohemagglutinin-stimulated PBMCs were infected with 100 TCID50 of NL4-3 in the presence of indicated proteins at either 2 mg/ml (a) or 20 ng/ml (b). Fresh proteins were replenished into cultures at days 4, 8, and 12, and media supernatants were sampled for RT activity. Representative day 4 RT values (similar profiles were also seen on days 8 and 12; not shown) are presented; results were replicated three times. (c) Tat and SDF-1 synergistically inhibit NL4-3-infection of PBMCs. PBMCs with MBP alone, MBP + SDF-1, or MBP + SDF-1 + MBPTat72 were infected with NL4-3. SDF-1 concentration was 800 ng/ml; MBP or MBPTat72 was at 2 μg/ml. RT average values from three independent experiments are from day 8 after infection.
SDF-1 inhibits X4 virus infection of PBMCs (29). We wondered whether Tat potentiates this SDF-1 inhibition. At concentrations of 20 ng/ml, 200 ng/ml, and 800 ng/ml, SDF-1-treated cells showed 6-, 8.5-, and 10-fold inhibition of HIV-1 infection, respectively, when compared with mock-treated PBMCs (data not shown). When SDF-1 (800 ng/ml) was incubated with 1-exon Tat (2 mg/ml) or with the wild-type peptide used in Fig. 2 (data not shown), X4 infection of PBMCs was reduced by an additional 10-fold over that of SDF-1 alone (Fig. 3c). Because both Tat and SDF-1 can bind to CXCR4 (Figs. 1 and 2), we interpret these results as Tat contributing to further occupy CXCR4, which might otherwise be vacant despite 800 ng/ml of SDF-1. However, we cannot exclude that there could be additionally complex interactions between HIV-1 envelope/SDF-1/Tat with CD4 and CXCR4.
Tat Inhibits HIV-1 Infection at the Step of Entry.
The above findings suggest, but do not prove, that Tat inhibits entry of X4 virus into cells. To test this hypothesis directly, we measured virus entry by using PCR (Fig. 4 a and b) and multinuclear activation of a galactosidase indicator (MAGI)-based (Fig. 4c) assays. Tat's interference with CXCR4-dependent entry was checked by comparing infection of HOS-CXCR4 cells by X4-NL4-3 with HOS-CCR5 cells by R5-NLAD8 (30; Fig. 4b). Paired infections incubated with either MBP alone (Fig. 4b Left) or MBPTat72 (Fig. 4b Right) were analyzed by PCR at 0 (Fig. 4b; lanes 1, 2, 7, and 8), 3 (Fig. 4b; lanes 3, 4, 9, and 10), and 6 h (Fig. 4b; lanes 5, 6, 11, and 12) after virus inoculation. MBPTat72 was found to inhibit X4- (Fig. 4b, lanes 4 and 6) but not R5-dependent (Fig. 4b, lanes 10 and 12) virus infection. Similar results were achieved by using wild-type Tat peptide (Fig. 2; data not shown). We also checked Tat's CXCR4-specific activity by using the widely accepted MAGI-entry assay (31). Here, MBPTat72 inhibited X4-tropic NL4-3 infection of U373-MAGI-CXCR4 cells, but did not affect R5-tropic NLAD8 infection of U373-MAGI-CCR5 cells (Fig. 4c).
Soluble Tat Selects Against CXCR4-Tropic Env Residue.
We asked next whether Tat's selective effect at CXCR4 influences evolution of viral tropism. Several residues in the V3 loop of gp120 are known to influence tropism; changes at one or two of these positions can sufficiently alter coreceptor specificity (9, 10, 11). One corollary, amongst many, on coreceptor usage by HIV-1 is that the charge properties at positions 11 and 25 of the V3 loop are important. Positively charged residues (e.g., K or R) at 11 and 25 correlate with X4 use (10, 11) whereas neutral amino acids (frequently S) at 11 with another neutral (frequently Q) or a negatively charged (frequently D or E) residue at position 25 favor R5 use. X4-NL4-3 virus has an S (neutral residue, normally favoring R5 usage) at 11 and a K (basic residue, normally favoring X4 usage) at 25. In this configuration, should Tat antagonize X4 use, it might be expected to select for a change in the positive nature of K25 in NL4-3.
To test whether Tat would select for this change, we assayed parallel NL4-3-infected PBMCs with either soluble MBPTat72 or MBP alone. At days 3, 7, and 28, the identity of V3 amino acid 25 in the infected cells was checked by using a PCR-based mismatch assay (Fig. 5A). The assay used three different primers to identify K, E, or Q at position 25. Input NL4-3 has a K (basic); preservation of K during infection would be consistent with maintenance of X4-tropism. By contrast, a change to either E (acidic) or Q (neutral) would indicate selection against X4 use. Indeed, by day 28, MBPTat72 selected significantly for a K to Q change (Fig. 5B; compare lanes 1–3 with 7–9) whereas MBP maintained K25 (Fig. 5C; compare lanes 1–3 with 7–9). Viral tropism was also examined after 35 days of MBPTat72 selection (Fig. 5D). By using RT-normalized amounts of virus to infect U373-MAGI-CXCR4 or U373-MAGI-CCR5 cells, we found that, after subtracting for background, the R5 proportion of X4-tropic NL4-3 increased from 2% (−Tat selection) to 14% (+Tat selection; Fig. 5D). By contrast, MBPTat72 selection did not affect at all the phenotype of R5-tropic NLAD8. The distinct, although incomplete, phenotypic conversion in NL4-3 after 35 days of Tat selection agrees with findings in the literature that the charge of V3 residue 25 exerts an important, albeit only partial, contribution to tropism.
Detection of Soluble Tat in HIV-1 Seropositive Individuals.
If Tat functions in vivo as an X4 restriction factor, then circulating Tat protein in HIV-1-infected individuals perhaps could be verified. Meaningful in vivo measurements of Tat have been difficult for several reasons. First, existing Tat antibodies are weak in avidity and are poorly standardized from laboratory to laboratory (ref. 32; K.T.J., unpublished data). Quantitative values from different investigators are thus difficult to compare. Second, the relevant body compartment (blood, mucosa, lymph node?) to sample for Tat is not known. Third, Tat most likely is a locally acting cytokine that can be trapped by molecules such as heparin sulfates becoming sequestered on extracellular matrices. Recognizing these caveats, we attempted to measure Tat protein in 80 anonymous HIV-1+ patient sera from the National Institutes of Health Clinical Center.
More than 33% of the 80 sera had humoral reactivity to Tat (data not shown). We next checked for soluble Tat in these sera by using a mixture of two hyperimmune rabbit antibodies raised by us to synthetic Tat peptides. The antisera at 1:1000 dilution easily detected 0.1 ng of membrane-bound MBPTat101 (Fig. 6, top row, left). When HIV-1+ patient sera were individually spotted onto 1 × 1 cm2 PVDF filters and were reacted with rabbit anti-Tat followed with detections by chemiluminescence, 5 of 80 samples showed clear reactivity to anti-Tat (Fig. 6, rows 4 and 5). These signals were Tat specific because competitions with excess MBPTat101 protein eliminated detection (Fig. 6, rows 2 and 3). Extrapolating from Tat standards, the five positives approximated concentrations from 2 ng/ml to 40 ng/ml. These values could be underestimations because local concentrations of Tat in lymphoid tissues might be higher and Tat in vivo might be sequestered by endogenous anti-Tat and/or by glycosaminoglycans. Until more reliable methods can be developed to measure in vivo amounts of viral protein, the full biological implications of circulating Tat protein cannot be entirely determined.
Figure 6.
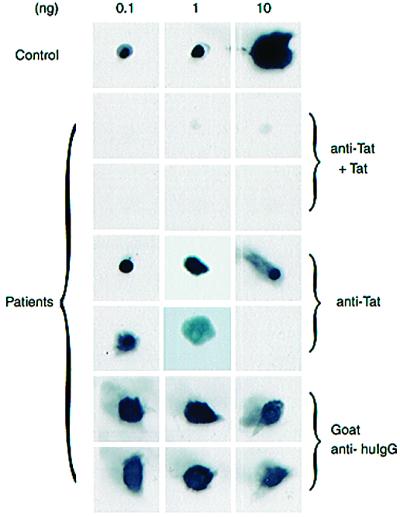
Examples of soluble Tat detected in HIV-1+ sera. Recombinant MBP-Tat protein (0.1 ng, 1 ng, or 10 ng) were spotted onto PVDF-squares (top row). Eighty anonymous patient sera in 0.5-ml aliquots were dot-spotted onto individual filters and were screened in two parallel sets by using rabbit anti-Tat polyclonal sera with (anti-Tat + Tat) or without (anti-Tat) competition with soluble MBP-Tat protein. Six examples, which include five anti-Tat reactive and one nonreactive (bottom rightmost square, anti-Tat) samples, are shown. The same six samples were reacted with goat anti-human IgG to verify for equivalence in spotting (last two rows; goat anti-huIgG).
Discussion
Current findings on neutralizing antibodies and cytotoxic T cell responses insufficiently explain the slow appearance of X4 viruses in HIV-1-infected individuals. The relatively late emergence of X4-HIV-1 (>7 yr) after infection has prompted several investigators to suggest the existence of an early negative selection against X4 virus (6, 16). Here, we propose the virally encoded Tat protein as an early selective factor against X4-HIV-l. We show that Tat binds CXCR4 (but not CCR5; Fig. 1), abolishes SDF-1/CXCR4 (but not β-chemokine/CCR5) signaling (Fig. 2), inhibits X4- (but not R5-) mediated viral infection/entry of cells (Figs. 3 and 4), and selects against X4-HIV-1 tropism in infections of PBMC (Fig. 5).
Why might HIV-1 encode a factor against X4 tropism? Chronologically, appearance of X4-HIV-1 in vivo coincides with a rapid decline of CD4+ T cells and onset of clinical immunodeficiency (29, 33, 34). From several perspectives X4-HIV-1 is considered more pathogenic than its R5 counterpart (34). Thus, if moderate (i.e., R5 virus) pathogenicity benefits HIV-1 for purposes of transmission and maintenance in host populations, then X4 antagonism through Tat may represent a means for the virus to achieve host-independent moderation of virulence.
Currently, we do not understand how selectivity for CXCR4 is achieved by Tat. Our experiments do suggest that Tat's CXC motif is important (Figs. 1 and 2). Additional mechanistic insights emerge from three recent reports characterizing X4-specific small molecule inhibitors (AMD3100, ref. 35; T22, ref. 36; and ALX40-4C, ref. 37). A common concept that emerges from these three diverse inhibitors is a principle that positively (basic) charged molecules show strong binding for CXCR4 (38). Thus, AMD3100 is highly cationic under physiological conditions (39); ALX40-4C is a nonapeptide of 9 arginines; and T22 is an 18-mer with 8 positively charged (lysine or arginine) amino acids. Interestingly, we note that the basic TAR RNA-binding domain of Tat has charge characteristics (40) equivalent to 9 consecutive arginines (i.e., ALX40-4C). Moreover, regions of Tat outside of its arginine-rich motif are also unusually biased in basic amino acids. For instance, clade B NL4-3 Tat has either an Arg or a Lys (total = 20) once every five amino acids. These residues are equally distributed between the first- (14 of 72) and second-coding (6 of 29) exon. Hence, one could view Tat's interaction with CXCR4 as being contributed in part through its CXC motif and in part through its high density of basic amino acids. Interestingly, CXCR4's extracellular surface is extremely acidic whereas CCR5's surface is more neutral to basic; these charge properties are also consistent with the Tat/coreceptor specificity described here. Accordingly, the antiviral potency of Tat [>90% inhibition of X4 viruses at 20 ng/ml (Fig. 3) and ≈50% inhibition at 2 ng/ml; data not shown] is similar to that of AMD3100 (35) and T22 (36) and is slightly superior to ALX40-4C (37).
Our hypothesis of selective X4-antagonism must be evaluated cautiously in view of recent success in vaccinating subhuman primates with Tat (41). Should it act in vivo to slow R5 to X4 transition, then a potential consequence of immunization with Tat might be to accelerate emergence of X4 virus when the vaccinee is subsequently infected with HIV-1. Whereas it is unclear what early emergence of X4 virus in humans might mean, extant observations suggest that this change would not benefit disease course (29, 33, 34). Balanced against this prediction are recent reports that immunization with Tat in macaques partially protected the host against subsequent challenge with a highly pathogenic simian-HIV (SHIV; refs. 42 and 43). However, because monkeys naturally do not use CXCR4 as a coreceptor for infection by simian immunodeficiency virus (SIV; ref. 44), it is unclear whether loss of Tat's CXCR4 antagonism holds the same implication for SHIV/macaques as for HIV-1/humans. Proposed and ongoing studies on vaccinating humans with Tat (41) should yield clarifying information.
Our findings here are somewhat at odds with two recent reports that suggested that Tat up-regulates both CXCR4 and CCR5 (45) or only CXCR4 (46) expression. In our experiments, we consistently have failed to observe either CXCR4 or CCR5 expression being modulated by Tat. However, we cannot exclude that cell surface antagonism of CXCR4 by Tat might lead to compensatory up-regulation of expression. Indeed, as noted in Fig. 3, several competing effects of Tat likely coexist with optimal suppression of X4 virus requiring a complex interaction of Tat + SDF-1 at CXCR4. In this regard, despite suppression from Tat + SDF-1, in a minority of cases X4-HIV-1 does eventually emerge in vivo. Possibly, this emergence is a consequence of progressive degradation of lymphoid architecture by R5 viruses leading to loss of SDF-1 production (16) coupled with the contribution to virus replication, at this stage, by Tat's induction of CXCR4 expression (45, 46).
Acknowledgments
We thank I. Quinto, T. Murakami, S. Smith, and B. Berkhout for critical readings; M. Van and L. Lin for preparation of manuscript; C. Lane for anonymous sera; M. Emerman for cells; J. Coligan for help with peptides; and AIDS Targeted Antiviral Program from the Office of the Director, National Institutes of Health for funding.
Abbreviations
- RANTES
regulated upon activation, normal T cell expressed and secreted
- SDF
stromal cell-derived factor
- CCR5
CC chemokine receptor 5
- CXCR4
CXC chemokine receptor 4
- PBMC
peripheral blood mononuclear cell
- MIP
macrophage inflammatory protein
- LTR
long terminal repeat
- RT
reverse transcriptase
- PVDF
poly(vinylidene difluoride)
- MBP
maltose-binding protein
- GST
glutathione S-transferase
- MAGI
multinuclear activation of a galactosidase indicator
Note Added in Proof.
After submission of this work, we noted that similar findings were reported by Ghezzi et al. (47).
References
- 1.Berger E A, Murphy P M, Farber J M. Annu Rev Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- 2.Alkhatib G, Combadiere C, Broder C C, Feng Y, Kennedy P E, Murphy P M, Berger E A. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 3.Deng H K, Unutmaz D, KewalRamani V N, Littman D R. Nature (London) 1996;381:661–666. [Google Scholar]
- 4.Dragic T, Litwin V, Allaway G P, Martin S R, Huang Y, Nagashima K A, Cayanan C, Maddon P J, Koup R A, Moore J P, Paxton W A. Nature (London) 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 5.Berger E A, Doms R W, Fenyo E M, Korber B T, Littman D R, Moore J P, Sattentau Q J, Schuitemaker H, Sodroski J, Weiss R A. Nature (London) 1998;391:240. doi: 10.1038/34571. (lett.). [DOI] [PubMed] [Google Scholar]
- 6.Zhu T, Mo H, Wang N, Nam D S, Cao Y, Koup R A, Ho D D. Science. 1993;261:1179–1181. doi: 10.1126/science.8356453. [DOI] [PubMed] [Google Scholar]
- 7.Connor R I, Sheridan K E, Ceradini D, Choe S, Landau N R. J Exp Med. 1997;185:621–628. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perelson A S, Neumann A U, Markowitz M, Leonard J M, Ho D D. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 9.Cocchi F, DeVico A L, Garzino-Demo A, Cara A, Gallo R C, Lusso P. Nat Med. 1996;11:1244–1247. doi: 10.1038/nm1196-1244. [DOI] [PubMed] [Google Scholar]
- 10.Chesebro B, Wehrly K, Nishio J, Perryman S. J Virol. 1996;70:9055–9059. doi: 10.1128/jvi.70.12.9055-9059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao L, Owen S M, Goldman I, Lal A A, Dejong J J, Goudsmit J, Lal R B. Virology. 1998;240:83–92. doi: 10.1006/viro.1997.8924. [DOI] [PubMed] [Google Scholar]
- 12.Berkowitz R D, Beckerman K P, Schall T J, McCune J M. J Immunol. 1998;161:3702–3710. [PubMed] [Google Scholar]
- 13.Kitchen S G, Zack J A. J Virol. 1997;71:6928–6934. doi: 10.1128/jvi.71.9.6928-6934.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dairaghi D J, Franz-Bacon K, Callas E, Cupp J, Schall T J, Tamraz S A, Boehme S A, Taylor N, Bacon K B. Blood. 1998;91:2905–2913. [PubMed] [Google Scholar]
- 15.Yi Y, Rana S, Turner J D, Gaddis N, Collman R G. J Virol. 1998;72:772–777. doi: 10.1128/jvi.72.1.772-777.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michael N L, Moore J P. Nat Med. 1999;5:740–741. doi: 10.1038/10462. [DOI] [PubMed] [Google Scholar]
- 17.Combadiere C, Ahuja S K, Tiffany H L, Murphy P M. J Leukocyte Biol. 1996;60:147–152. doi: 10.1002/jlb.60.1.147. [DOI] [PubMed] [Google Scholar]
- 18.Tiffany H L, Lautens L L, Gao J L, Pease J, Locati M, Combadiere C, Modi W, Bonner T I, Murphy P M. J Exp Med. 1997;186:165–170. doi: 10.1084/jem.186.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neuveut C, Jeang K T. J Virol. 1996;70:5572–5581. doi: 10.1128/jvi.70.8.5572-5581.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ensoli B, Barillari G, Salahuddin S Z, Gallo R C, Wong-Staal F. Nature (London) 1990;345:84–86. doi: 10.1038/345084a0. [DOI] [PubMed] [Google Scholar]
- 21.Westendorp M O, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin K M, Krammer P H. Nature (London) 1995;375:497–500. doi: 10.1038/375497a0. [DOI] [PubMed] [Google Scholar]
- 22.Albini A, Barillari G, Benelli R, Gallo R C, Ensoli B. Proc Natl Acad Sci USA. 1995;92:4838–4842. doi: 10.1073/pnas.92.11.4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trinh D P, Brown K M, Jeang K T. Biochem Biophys Res Commun. 1999;256:299–306. doi: 10.1006/bbrc.1999.0317. [DOI] [PubMed] [Google Scholar]
- 24.Feng Y, Broder C C, Kenedy P E, Berger E A. Science. 1996;272:872–877. [Google Scholar]
- 25.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath P D, Wu L, Mackay C R, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J. Cell. 1996;85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 26.Doranz B J, Rucker J, Yi Y, Smyth R J, Samson M, Peiper S C, Parmentier M, Collman R G, Doms R W. Cell. 1996;85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 27.Belardelli F, Gresser I. Immunol Today. 1996;17:369–372. doi: 10.1016/0167-5699(96)10027-X. [DOI] [PubMed] [Google Scholar]
- 28.Cocchi F, DeVico A L, Garzino-Demo A, Arya S K, Gallo G, Lusso P. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 29.van't Wout A B, Ran L J, Kuiken C L, Kootstra N A, Pals S T, Schuitemaker H. J Virol. 1998;72:488–496. doi: 10.1128/jvi.72.1.488-496.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith S M, Markham R B, Jeang K T. Proc Natl Acad Sci USA. 1996;93:7955–7960. doi: 10.1073/pnas.93.15.7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vodicka M A, Goh W, Wu L, Rogel M, Bartz S, Schweickart V, Raport C, Emerman M. Virology. 1997;233:193–198. doi: 10.1006/viro.1997.8606. [DOI] [PubMed] [Google Scholar]
- 32.Cohen S S, Li C, Ding L, Cao Y, Pardee A B, Shevach E M, Cohen D I. Proc Natl Acad Sci USA. 1999;96:10842–10847. doi: 10.1073/pnas.96.19.10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tersmette M, de Goede R E, Al B J, Winkel I N, Gruters R A, Cuypers H T, Huisman H G, Miedema F. J Virol. 1988;62:2026–2032. doi: 10.1128/jvi.62.6.2026-2032.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richman D D, Bozzette S A. J Infect Dis. 1994;169:968–974. doi: 10.1093/infdis/169.5.968. [DOI] [PubMed] [Google Scholar]
- 35.Schols D, Struyf S, Van Damme J, Este J A, Henson G, De Clercq E. J Exp Med. 1997;186:1383–1388. doi: 10.1084/jem.186.8.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murakami T, Nakajima T, Koyanagi Y, Tachibana K, Fujii N, Tamamura H, Yoshida N, Waki M, Matsumoto A, Yoshie O, Kishimoto T, Yamamoto N, Nagasawa T. J Exp Med. 1997;186:1389–1393. doi: 10.1084/jem.186.8.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doranz B J, Grovit-Ferbas K, Sharron M P, Mao S H, Goetz M B, Daar E S, Doms R W, O'Brien W A. J Exp Med. 1997;186:1395–1400. doi: 10.1084/jem.186.8.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berkhout, B. & Das, A. T., Emerging Infect. Dis.4, 335–336. [DOI] [PMC free article] [PubMed]
- 39.Este J A, Cabrera C, Clercq E D, Struyf S, Damme J V, Bridger G, Skerlj R T, Abrams M J, Henson G, Gutierrez A, Clotet B, Schols D. Mol Pharmacol. 1999;55:67–73. doi: 10.1124/mol.55.1.67. [DOI] [PubMed] [Google Scholar]
- 40.Jeang K T, Xiao H, Rich E A. J Biol Chem. 1999;274:28837–28840. doi: 10.1074/jbc.274.41.28837. [DOI] [PubMed] [Google Scholar]
- 41.Gallo R C. Proc Natl Acad Sci USA. 1999;96:8324–8326. doi: 10.1073/pnas.96.15.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cafaro A, Caputo A, Fracasso C, Maggiorella M T, Goletti D, Baroncelli S, Pace M, Sernicola L, Koanga-Mogtomo M L, Betti M, et al. Nat Med. 1999;5:643–650. doi: 10.1038/9488. [DOI] [PubMed] [Google Scholar]
- 43.Pauza C D, Trivedi P, Wallace M, Ruckwardt T J, Buanec H L, Lu W, Bernard B L, Burny A, Zagury D, Gallo R C. Proc Natl Acad Sci, USA. 2000;97:3515–3519. doi: 10.1073/pnas.070049797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Z, Zhou P, Ho D D, Landau N R, Marx P A. J Virol. 1997;71:2705–2714. doi: 10.1128/jvi.71.4.2705-2714.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang L, Bosch I, Hofmann W, Sodroski J, Pardee A B. J Virol. 1998;72:8952–8960. doi: 10.1128/jvi.72.11.8952-8960.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Secchiero P, Zella D, Capitani S, Gallo R C, Zauli G. J Immunol. 1999;162:2427–2431. [PubMed] [Google Scholar]
- 47.Ghezzi S, Noonan D M, Aluigi M G, Vallanti G, Cota M, Benelli R, Morini M, Reeves J D, Vicenzi E, Poli G, et al. Biochem Biophys Res Commun. 2000;270:992–996. doi: 10.1006/bbrc.2000.2523. [DOI] [PubMed] [Google Scholar]