

Abstract
The metabolism of dietary essential amino acids by the gut has a direct effect on their systemic availability and potentially limits growth. We demonstrate that, in neonatal pigs bearing portal and arterial catheters and fed a diet containing 23% protein [high protein (HP) diet], more than half the intake of essential amino acids is metabolized by the portal-drained viscera (PDV). Intraduodenal or i.v. infusions of [U-13C]-lysine were used to measure the appearance across and the use of the tracer by the PDV. In HP-fed pigs, lysine use by the PDV was derived almost entirely from the arterial input. In these animals, the small amount of dietary lysine used in first pass was oxidized almost entirely. Even so, intestinal lysine oxidation (24 μmol/kg per h) accounted for one-third of whole-body lysine oxidation (77 μmol/kg per h). Total lysine use by the PDV was not affected by low protein (LP) feeding (HP, 213 μmol/kg per h; LP,186 μmol/kg per h). In LP-fed pigs, the use of lysine by the PDV accounted for more than 75% of its intake. In contrast to HP feeding, both dietary and arterial lysines were used by the PDV of LP-fed pigs in nearly equal amounts. Intestinal lysine oxidation was suppressed completely. We conclude that the PDV are key organs with respect to amino acid metabolism and that the intestines use a disproportionately large amount of the dietary supply of amino acids during protein restriction.
The portal-drained viscera (PDV; the intestines, pancreas, spleen, and stomach) contribute between 20% and 35% of whole-body energy expenditure and protein synthesis, even though they contribute less than 6% of body weight (1–3). Dietary amino acid use by the intestine could have a substantial effect on their systemic availability and thereby regulate whole-body protein deposition. Studies in a number of mammalian species show that dietary essential amino acids (EAA) are directly used by the intestines for protein synthesis and other biosynthetic pathways (4–6).
Futhermore, under high protein (HP) feeding conditions, intestinal energy production is derived largely from the oxidation of glutamate, glutamine, and aspartate (7–9). However, the oxidation of these substrates accounts for neither the total CO2 production nor the production of alanine and ammonia by the PDV. The failure to account for the nitrogen outflow from the metabolism of glutamate, glutamine, and aspartate has led us to conclude that other amino acids, possibly including EAA, are oxidized in the PDV. There is evidence that leucine and methionine are oxidized by the enterocytes (10, 11), but we know of no detailed in vivo investigations of intestinal catabolism of other EAA. In this context, the oxidation of lysine would be of particular consequence, because this amino acid is nutritionally first limiting in cereal-based diets and milk (12).
Investigations carried out 25 years ago were unable to find evidence for the presence of enzymes required for lysine catabolism in enterocytes (13, 14), and it is generally held that lysine catabolism occurs primarily in the liver (15–17). Whether lysine is catabolized by the intact intestine in vivo has not been investigated. Quantification of intestinal lysine oxidation was one main objective of the studies described in this report.
A unique feature of intestinal metabolism is that the mucosal cells receive substrates directly both from the diet and from the mesenteric circulation. Although both sources are used by the PDV (6, 11, 18), their relative contributions have received relatively little systematic study. The determination of the relative rates of arterial and dietary first-pass lysine metabolism in the PDV was the second objective of this study.
Given that most current evidence suggests that the total use of EAA by the intestinal tissues could be substantial, the degree to which it is sensitive to dietary protein intake is a critical question. On one hand, it might be hypothesized that the high rate of intestinal protein turnover enables a rapid down-regulation of the rate of intestinal amino acid use when dietary protein intake is restricted. The effect would be to maximize the systemic availability of amino acids. On the other hand, it is conceivable that, under protein-limiting conditions, the metabolic needs of the mucosa are satisfied preferentially by first-pass use, thereby resulting in a disproportionately low systemic availability of EAA. There is evidence for both possibilities (4, 19, 20). The investigation of the response of intestinal lysine use to a low protein (LP) intake formed the third objective of the present experiments.
Methods
Animals.
The Baylor College of Medicine Animal Protocol Review Committee approved the study. Housing and care of the animals conformed to the United States Department of Agriculture guidelines. The study involved 17 4-week-old female crossbred piglets (Large White × Hampshire × Duroc) purchased from the Texas Department of Criminal Justice (Huntsville, TX). The pigs were received at the laboratory when they were 2 weeks old. For the next week, they were fed a liquid milk replacer (Litterlife; Merrick, Union, WI) at a rate of 50 g of dry matter/kg per day. The composition (per kg dry matter) of the milk replacer was 500 g of lactose, 100 g of fat, and 250 g of protein.
Study Design.
At a postnatal age of 3 weeks, the piglets were fasted overnight and, by using aseptic technique, catheters were implanted into the stomach, the duodenum, the portal and jugular veins, and the common carotid artery. An ultrasonic blood flow probe (Transonics, Ithaca, NY) was placed around the portal vein. After surgery, the piglets received complete i.v. nutrition for 24–36 h. The pigs then received either regular Litterlife (HP diet) or a diet that contained only 40% of the protein present in Litterlife (LP diet). The protein intake of the LP animals was set deliberately at a rate that would maintain body nitrogen equilibrium. The diets were given at the same rate (50 g/kg per day) and were made isocaloric by adding lactose (Sigma) and corn oil in the same ratio in both the control (HP) diet and the LP diet. The pigs resumed full feed intake on day 3 after surgery, and the tracer infusions were started after a further 4 days. At the postnatal age of 28 days, whole-body CO2 production was measured with an infusion of [1-13C]bicarbonate. The [U-13C]lysine infusion protocols were carried out on postnatal days 29–31.
Infusion Protocol.
After an overnight fast, the pigs consumed a meal that supplied one-seventh of the preceding daily intake. This meal served to restore intestinal motility. Immediately thereafter, a continuous gastric infusion of diet was started. The infusion provided feed at a rate that provided 1/14 of the preceding daily intake each hour. Arterial and portal blood samples were drawn after 90, 105, and 120 min. On day 1, [1-13C]bicarbonate (99%; Cambridge Isotope Laboratories, Andover, MA) was infused into the jugular catheter at a rate of 10 μmol/kg per h. Arterial and portal blood samples (1 ml) were taken at 15-min intervals from 75 to 120 min of infusion. On days 2–4, the animals received a constant infusion of [U-13C]lysine (97%; Cambridge Isotope Laboratories) providing approximately 7 μmol/kg per h via either the duodenal or the jugular catheter. The order of the i.v. and enteral infusions was randomized. The infusion commenced after 2 h of feeding and continued for 4 h. During the last hour of the tracer infusion, four arterial and portal blood samples were drawn at 15-min intervals.
Sample Analysis.
Small aliquots (0.2 ml) were taken for determination of blood gases (Chiron), glucose, and lactate (Yellow Springs Instruments). An aliquot of whole blood (0.2 ml) was mixed with an equal volume of an aqueous solution of methionine sulfone (0.5 mmol/liter) and immediately frozen in liquid nitrogen. This aliquot was used for the measurement of amino acid concentrations by reverse-phase high performance liquid chromatography of their phenylisothiocyanate derivatives (PicoTag; Waters). An aliquot of whole blood (1.0 ml) was placed in a 10-ml Vacutainer (Becton Dickinson), and 0.5 ml of perchloric acid (10% wt/wt) was added. The Vacutainer was placed on ice. Room air (8 ml), filtered through soda lime (Sodasorb; Grace Container Products, Lexington, MA), was injected into the Vacutainer, removed into a gas tight syringe, and transferred to a second Vacutainer. The isotopic enrichment of the carbon dioxide in the gas sample was measured on a continuous flow gas isotope ratio mass spectrometer (ANCA; Europa Instruments, Crewe, U.K.). The blood/perchloric acid mixture was centrifuged at 3,000 × g for 10 min, and the supernatant stored at −20°C. Subsequently, the supernatant was thawed and brought to pH ≥ 4 with KOH (4 mol/liter). After centrifugation and acidification, the amino acids in the supernatant were bound to a 1-ml column of Dowex 50 Wx8 (H+ form), eluted with 3 ml of 3 mol/liter NH4OH, and dried under vacuum.
Mass spectrometric analysis of lysine was conducted with the tri-fluoro-acetyl-methylester derivative (21, 22). The isotopic enrichment was measured on a gas chromatograph (Hewlett–Packard) connected to a combustion oven (850OC) and an isotope ratio mass spectrometer (Europa Instruments). The atom percentage 13C-enrichment was converted to mole percentage of [13C]lysine enrichment, after accounting for the 2-fold dilution of carbon in the derivative and the measured 13C abundance (97%) of the tracer lysine.
Calculations.
A schematic model of the different fluxes through the PDV is shown in Fig. 1. The equations used to obtain the results are detailed in the Appendix.
Figure 1.

Schematic representation of the metabolic fate of enteral and systemic lysine in the PDV. A, dietary intake of lysine; B, arterial flux of lysine through the PDV; C, portal outflow of lysine; D, unidirectional, first-pass uptake of dietary lysine by the PDV; E, dietary lysine that is not metabolized by the PDV in first pass; F, unidirectional uptake of arterial lysine by the PDV; G, arterial lysine that is not metabolized by the PDV; H, recycled lysine derived from proteolysis or reabsorption of previously synthesized and secreted endogenous proteins but not derived from the diet; and I, lysine that is oxidized by the PDV.
Statistics.
The data are presented as the mean values for samples taken over the last hour of the tracer study ± SEM. Balances were tested against zero by one-tailed t tests. Fractional balances are the means of the ratios. Differences between the balances of the animals fed the HP or LP diet were tested by two-tailed t tests. A value of P < 0.05 was taken as significant.
Results
The rate of weight gain of pigs fed the LP diet (24 ± 2 g/kg per day) was significantly (P < 0.001) lower than that of pigs fed the HP diet (45 ± 2 g/kg per day). Portal blood flow (PBF), whole-body CO2 production (on average 59 ± 3 mmol/kg per h), and CO2 production by the PDV (7.3 ± 0.9 mmol/kg per h) were not affected by the diet.
Intestinal Lysine Metabolism.
The net portal balances of the EAA over the last hour of the study are shown in Table 1, in which the EAA concentrations and balances obtained on different days within a diet group are pooled for presentation. Within a diet group, we did not find statistically significant grouped differences in either the amino acid concentration or the PBF during the intraduodenal and i.v. tracer infusions. However, we found measurable random, day-to-day variability in the amino acid balances in a given pig measured on separate days.
Table 1.
Intake, arterial and portal concentrations, and net portal mass balances of essential amino acids in piglets receiving either HP or LP diets continuously
| EAA | HP-fed piglets (n =
9)
|
LP-fed piglets (n = 8)
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Intake | Concentration;
μmol/Liter
|
Portal
balance
|
Intake | Concentration;
μmol/Liter
|
Portal balance
|
|||||
| Arterial | Portal | μmol/kg per h | Percentage of intake | Arterial | Portal | μmol/kg per h | Percentage of intake | |||
| Thr | 934 | 918 ± 83 | 954 ± 85 | 152 ± 36 | 16 ± 4 | 374 | 456 ± 56 | 448 ± 61 | −31 ± 44* | −1 ± 18§ |
| Val | 765 | 572 ± 49 | 638 ± 48 | 315 ± 31 | 41 ± 4 | 306 | 277 ± 21 | 299 ± 23 | 100 ± 26* | 31 ± 10 |
| Iso | 780 | 299 ± 19 | 348 ± 22 | 218 ± 18 | 28 ± 2 | 312 | 187 ± 14 | 202 ± 14 | 58 ± 11* | 19 ± 4† |
| Leu | 748 | 406 ± 18 | 484 ± 22 | 350 ± 33 | 47 ± 4 | 299 | 237 ± 19 | 267 ± 21 | 119 ± 30* | 40 ± 10 |
| Phe | 254 | 110 ± 5 | 131 ± 5 | 94 ± 9 | 37 ± 4 | 102 | 52 ± 7 | 60 ± 7 | 30 ± 4* | 28 ± 4 |
| Lys | 518 | 616 ± 23 | 678 ± 26 | 277 ± 23 | 54 ± 4 | 207 | 385 ± 29 | 394 ± 28 | 33 ± 12* | 16 ± 6‡ |
Values shown are means ± SEM.
A significant difference between HP- and LP-fed piglets at P < 0.001.
A significant difference between HP- and LP-fed piglets at P < 0.05.
A significant difference between HP- and LP-fed piglets at P < 0.01.
No significant difference from zero.
In the HP group, the net portal balance of the EAA was 35% of the intake, although the fractional portal appearance, which ranged from 16% of intake for threonine to 54% of intake for lysine, varied markedly among the amino acids. In the LP group, the fractional portal appearance of all of the dietary EAA was lower, significantly so for lysine (−70%), isoleucine (−32%), and threonine (−100%).
Table 2 summarizes the tracer results during the intraduodenal and i.v. [U-13C]lysine infusions during HP or LP feeding. The fractional use of arterial lysine by the PDV (8% of arterial flux) was not affected by diet, although, because of the lower arterial lysine concentration, the absolute use of arterial lysine by the PDV was significantly lower in the LP group. The total lysine use by the PDV (Table 3) was not significantly different between the two feeding groups. However, after correction for the use of the enteral tracer that reentered the PDV via the arterial circulation, it seemed that during HP feeding there was little first-pass use of dietary lysine, whereas during LP feeding, 43% of the dietary lysine was used by the intestine in first pass. The diet-related difference in the distribution of lysine use by the PDV was highly significant.
Table 2.
The doses, arterial and portal enrichments, and portal blood flows in piglets receiving either an HP or an LP diet, after an i.v. or intraduodenal infusion of [U-13C]lysine
| Tracer experiment | [U-13C]lysine infusion rate, μmol/(kg per h) | Arterial enrichment, MPE | Portal enrichment, MPE | Portal blood flow, liter/(kg per h) |
|---|---|---|---|---|
| HP (i.v., n = 7) | 5.29 ± 0.46 | 0.772 ± 0.076 | 0.634 ± 0.057 | 4.7 ± 0.7 |
| LP (i.v., n = 6) | 6.94 ± 0.10 | 1.174 ± 0.034 | 1.069 ± 0.023 | 4.6 ± 0.4 |
| HP (i.d., n = 6) | 5.34 ± 0.54 | 0.668 ± 0.032 | 0.719 ± 0.032 | 4.9 ± 0.8 |
| LP (i.d., n = 6) | 6.94 ± 0.10 | 0.974 ± 0.022 | 1.093 ± 0.027 | 4.5 ± 0.5 |
Values shown are means ± SEM. i.d., intraduodenal; MPE, mole percent excess.
Table 3.
The lysine intake, first-pass, and systemic use of lysine by the PDV in piglets receiving either an HP or an LP diet
| Diet | Lysine intake, μmol/kg per h | Arterial use, μmol/kg per h | First-pass use, μmol/kg per h | Recycling, μmol/kg per h | Total lysine use, μmol/kg per h |
|---|---|---|---|---|---|
| HP | 518 | 230 ± 40 | −15 ± 63 | 2 ± 59* | 213 ± 32 |
| LP | 207 | 126 ± 25† | 91 ± 36§ | 31 ± 27* | 186 ± 29 |
Values shown are means ± SEM.
No significant difference from zero.
Significant difference between HP- and LP-fed piglets at P < 0.05.
Significant difference between HP- and LP-fed piglets at P < 0.001.
During HP feeding, the intestine oxidized dietary lysine at a rate of 24 ± 6 μmol/kg per h. However, the PDV apparently did not oxidize systemic lysine. In the HP group, lysine oxidation by tissues other than the PDV was 53 ± 15 μmol/kg per h. Thus, total body lysine oxidation was 77 ± 12 μmol/kg per h, of which 31% represented intestinal oxidation of dietary lysine. In the LP-fed pigs, whole-body lysine oxidation fell to 18 ± 5 μmol/kg per h, and lysine oxidation by the PDV fell to 0. The amount of lysine in the portal vein that derived either from intracellular proteolysis or from redigested, previously secreted enteral proteins, so-called “recycled lysine,” was very low and not different between the two groups.
Distribution of Whole-Body Lysine Metabolism.
The distribution of lysine use between the PDV, the liver, and the remainder of the body is shown in Table 4. Neither the whole-body lysine flux nor the whole nonoxidative lysine disposal (an estimate of protein synthesis) was affected significantly by LP feeding. In the HP group, the first-pass splanchnic extraction (119 μmol/kg per h) accounted for 22% of the lysine intake, whereas in the LP group, the first-pass splanchnic lysine extraction (134 μmol/kg per h) was the equivalent of 70% of the lysine intake. However, as indicated above, LP feeding was associated with an increase in the first-pass use of dietary lysine by the gut, and the calculated first-pass use of lysine by the liver fell by 61% in the LP group.
Table 4.
Effect of protein intake on the distribution of lysine metabolism (μmol/kg per h) among the PDV, liver, and peripheral tissues
| Diet | Whole-body
metabolism
|
First-pass metabolism
|
Hepatic lysine oxidation | ||||
|---|---|---|---|---|---|---|---|
| Flux | Oxidation | NOLD | Total | Intestine | Liver | ||
| HP | 799 | 77 | 722 | 119 | −15 | 134 | 55 |
| LP | 712 | 18 | 694 | 143 | 91 | 53 | 18 |
| PSEM | 26 | 8 | 18 | 33 | 49 | 33 | 10 |
| Diet effect | NS | P < 0.05 | NS | NS | <0.05 | <0.05 | <0.01 |
| NOLD
|
Body protein degradation | Lysine balance
|
|||||
|---|---|---|---|---|---|---|---|
| PDV | Liver | Periphery | Body | PDV | Periphery + liver | ||
| HP | 191 | 79 | 452 | 281 | 441 | 213 | 228 |
| LP | 217 | 35 | 492 | 505 | 189 | 186 | 3 |
| PSEM | 30 | 14 | 24 | 28 | 22 | 30 | 25 |
| Diet effect | NS | =0.07 | NS | <0.01 | <0.001 | NS | <0.001 |
NOLD, nonoxidative lysine disposal; PSEM, pooled standard error of the mean; NS, no statistically significant difference between pigs fed HP or LP diets.
When whole-body lysine kinetics were adjusted both for lysine oxidation in the gut and liver and for the estimated contributions of protein turnover in the PDV and liver, LP feeding had no significant effect on nonoxidative lysine disposal by the peripheral tissues but was associated with 80% increase in peripheral protein degradation. Whole-body lysine balance, calculated from the difference between nonoxidative disposal and whole-body protein degradation, was 57% lower in the LP group. Moreover, LP feeding was associated with a switch from a strongly positive lysine balance in the liver and periphery (149 ± 32 μmol/kg per h) to a value not significantly different from zero (3 ± 17 μmol/kg per h). Thus, in the HP group, lysine disposal within the PDV accounted for 48% of whole-body lysine balance, whereas in the LP group, whole-body lysine balance was accounted for entirely by net (presumably secretory) lysine use by the tissues of the PDV.
Discussion
In our previous work (7, 24), we showed in pigs receiving an HP diet that, although the oxidation of glutamate, aspartate, glutamine, and glucose by the PDV was considerable, their catabolism did not account for all of the production of alanine and ammonia by the intestine. This observation led us to hypothesize that EAA, possibly including lysine, were also oxidized by the intestinal tissues. In fact, despite the reports that no lysine catabolic enzymes are present within enterocytes (13, 14), the present results indicate that the intestinal oxidation of lysine in piglets receiving an HP diet accounts for approximately 30% of the total-body lysine oxidation. Surprisingly, lysine taken up by the PDV from the mesenteric artery is apparently not oxidized, suggesting that lysine metabolism is compartmentalized in these tissues. This observation adds further data to prior observations that mucosal protein synthesis (25, 26), mucosal metabolism of glutamate (27), and other aspects of mucosal amino acid metabolism (7) are also compartmentalized.
Several possibilities could explain the observation with regard to compartmentalization of lysine catabolism in the intestinal tissues. First, it is possible that only the upper villus cells, which are more likely to metabolize dietary amino acids, are capable of oxidizing lysine. This idea receives support from earlier studies of differences in the use of luminal and systemic amino acids for protein synthesis in different regions of the villus (18). Second, one could argue that the intracellular trafficking of amino acids depends strongly on whether amino acids enter the enterocyte across the apical or basolateral membrane. This possibility is supported by previous studies of mucosal glutamate metabolism (27, 28). A third explanation is that microorganisms within the intestines oxidize dietary lysine. We think that this explanation is the least likely. First, it is highly probable that a free amino acid directly infused into the duodenum would be absorbed at a rate exceeding that of bacterial lysine use, because recent studies (29, 30) indicate that even intact proteins are rapidly and almost completely digested and absorbed by the distal region of the jejunum. Second, although direct oxidation of lysine by microorganisms within the duodenum or upper part of the jejunum is possible, such a mechanism does not easily explain why the direct oxidation of enteral lysine was suppressed completely during protein restriction. Thus, it seems that the mucosa, with its many different types of cells, is responsible for the oxidation of dietary lysine during HP feeding. It should be added that, from a nutritional point of view, it is of little importance whether lysine is oxidized by the mucosa or by intraluminal microorganisms, but the cellular site of lysine oxidation is of critical mechanistic importance.
The second aim of the study was to determine whether lysine used by the PDV was derived from the diet or from the mesenteric artery. The answer to this question is of regulatory significance, because if the use of systemic amino acids is dominant, then intestinal amino acid use is potentially responsive to endocrine influences. If, on the other hand, direct, first-pass use is the more important, then it is possible that it is regulated locally. In pigs receiving an HP diet, the former seemed to apply, because we found that little dietary lysine was used by the intestine in first pass and the large majority of this lysine was oxidized. Hence, all of the lysine used by the PDV for protein synthesis was obtained from the arterial circulation. This finding is quite different from our earlier conclusion, which stressed the importance of first-pass EAA metabolism (5). However, although the present data are similar with regard to net lysine use by the PDV, the new data on the use of arterial lysine obtained in the present work reveal that we previously had underestimated the use of dietary amino acids that had recycled to the PDV in the arterial circulation. As a result we overestimated first-pass uptake of the dietary amino acids.
At face value, the lack of statistically significant first-pass use of lysine during HP feeding is incompatible with the observation of significant intestinal oxidation of dietary lysine. However, we believe that the apparent paradox has a technical basis. It is clear that the estimate of first-pass lysine use has a large variance; thus, it is not possible to detect a small, biologically significant rate of use at the statistical level. The large variance probably results from random variation in the portal balance between study days and the fact that the calculation of the first-pass use of lysine involves four independent measurements, with an inevitable propagation of errors. The sensitivity and specificity of the measurement of lysine oxidation are much higher, largely because it is a direct measurement.
Perhaps the most striking observations to emerge from the study were (i) that the total lysine use by the PDV was largely unaffected by an inadequate protein intake, a observation that indicates a high obligatory visceral need for lysine, and (ii) that, after a prolonged period of LP feeding, the PDV switched from an exclusive systemic source to a combination of dietary and systemic lysine as the source of protein synthesis.
The mechanisms that underlie the second observation are unclear. With regard to the use of arterial lysine by the PDV, it is of interest to note that the fractional extraction of the arterial lysine was unaffected by the prior level of protein intake. Thus, the use of arterial lysine by the PDV was primarily a function of systemic lysine flow through the PDV, itself a function of the arterial lysine concentration. Indeed, a post hoc investigation of the between animal variations in the visceral use of arterial lysine and the arterial lysine concentration revealed a highly significant (r = 0.8, P < 0.001) and linear relationship, spanning a range in lysine concentrations of between 100 and 700 μM.
The mechanistic basis of the alteration in the use of dietary lysine is even less clear. The observation that, during the HP intake, very little dietary lysine was metabolized in first pass might indicate that, at high luminal concentrations, the major amino acid absorptive pathway is paracellular, as has been suggested by Pappenheimer (31). If this is the mechanism then the lack of metabolism might indicate that dietary lysine was not available for metabolism by the cells of the mucosa. It is also possible that the switch in lysine use reflected differential expression of the amino acid transporters in the brush border and basolateral membranes, with a consequent change in the intracellular trafficking of lysine. Certainly, several amino acid transporters have been identified on both membranes, and the recent identification of two intestinal amino acid transporter genes opens up new possibilities for research into the mechanistic aspects of the different use rates (32, 33).
The third aim of the study was to examine the scale of the lysine metabolic response of the PDV to protein restriction and to investigate the extent to which intestinal lysine metabolism influenced its systemic availability. It is critical to emphasize that in the present study, the LP intake was set deliberately at a level that would maintain body nitrogen equilibrium. LP feeding was associated with an increase in the fractional use of all of the EAA by the PDV, indicating that these tissues used a disproportionately high amount of EAA during protein restriction. Indeed, in the LP group the use of threonine by the PDV was equal to the intake of the amino acid. In other words, the restricted peripheral tissue growth of the animals receiving the LP diet was primarily a reflection of the substantial suppression of the net absorption of two amino acids, lysine and threonine.
The observations with regard to first-pass lysine use seem to contradict those of Hoerr et al. (4) in young men adapted to protein-restricted diets. However, this earlier study was based on measurements of first-pass splanchnic, rather than intestinal, extraction. In fact, when we examined the first-pass splanchnic extraction of lysine, we found that protein restriction was not associated with a significant increase, but that this lack of difference concealed the fact that under LP feeding conditions the contribution of the intestinal tissue increased and that of the liver decreased.
The reason for the disproportional uptake of EAA by the intestine is unclear, although it may be speculated that a large quantity of amino acids is needed to maintain the integrity and function of the gut, in order for it to serve as a barrier for bacterial insults, to produce large amounts of immunoglobulins and mucins within the mucosa, and to be able to digest and absorb the diet. The consequence is that the intestinal use of some specific amino acids is so high that, although overall nitrogen balance is close to zero, visceral growth and metabolism are well maintained whereas the peripheral tissues are probably losing protein. This phenomenon has been observed in previous body compositional studies in young pigs fed protein-restricted diets (19, 34).
Finally, we should add the caveat that the tracer studies were of relatively short duration; thus, it is possible that the processes of digestion and reabsorption of previously synthesized intestinal proteins that were secreted in response to the start of the feeding were not yet apparent. This possibility might explain why virtually no lysine not derived directly from the diet appeared in the portal vein, whereas Metges et al. (23) found a significant contribution of microbially derived lysine to plasma lysine flux in human adults after a 24-h tracer protocol.
In conclusion, we have shown that during protein restriction accompanied by a normal energy intake, the PDV maintain a high rate of metabolism and continue to use a disproportionately large amount of EAA. The results also show that the source of lysine used by these tissues is sensitive to protein status, in as much as it is almost exclusively systemic under generous feeding conditions, but switches to mixed dietary and systemic lysine under protein restriction. Furthermore, although lysine is catabolized by the intestines and accounts for 31% of whole-body lysine oxidation in HP-fed piglets, it is strongly suppressed when protein becomes the limiting nutrient. Unfortunately, the present study does not allow us to discriminate between microbial or intestinal oxidation, nor does it allow us to determine which cells are responsible for lysine catabolism. However, taken together, the results highlight the critical importance of the gut in actively regulating EAA flow to the body as a whole and underscore the necessity for consideration of bioavailability of EAA in calculations of nutritional recommendations.
Acknowledgments
We thank Ms. L. Loddeke for her editorial help. This work is a publication of the U.S. Department of Agriculture/Agricultural Research Service, Children's Nutrition Research Center, Department of Pediatrics, Baylor College of Medicine and Texas Children's Hospital, Houston, TX. This work was supported in part by federal funds from U.S. Department of Agriculture, Agricultural Research Service Cooperative Agreement 58-6250-6001 and Cooperative State Research, Education, and Extension Service Grant 98-35206. The Sophia Foundation of Scientific Research, the Nutricia Research Foundation and the Royal Netherlands Academy of Arts and Sciences (Ter Meulen Fund) supported J.B.v.G.
Abbreviations
- PDV
portal-drained viscera
- EAA
essential amino acids
- LP
low protein
- HP
high protein
- PBF
portal blood flow
Appendix
Lysine Use Calculations
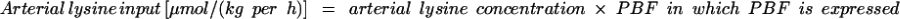 |
1 |
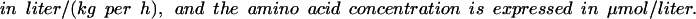 |
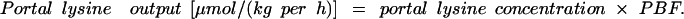 |
2 |
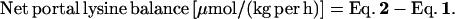 |
3 |
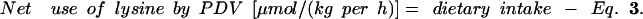 |
4 |
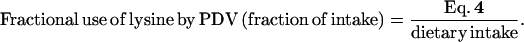 |
5 |
Substituting the amino acid concentration with the tracer concentration will give the tracer kinetics.
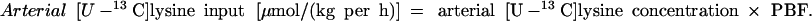 |
6 |
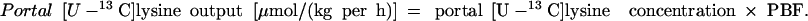 |
7 |
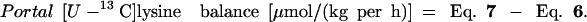 |
8 |
The percentage of arterial lysine that is used by the PDV is measured during the i.v. tracer study and is calculated as follows.
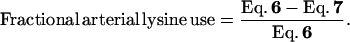 |
9 |
The amount of arterial lysine that is used by the PDV is calculated with the following equation.
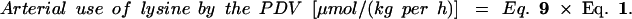 |
10 |
The amount of dietary lysine used on first pass by the PDV has to be corrected by the amount of dietary lysine that appears in the portal vein (and thus is not metabolized by the PDV) and then reenters the PDV but now from the arterial site. This lysine will be used by the PDV in the same proportion as the i.v. administered tracer (Eq. 9). Thus, during the enteral tracer infusion, the equation is
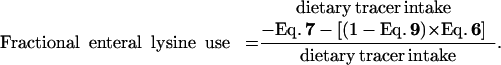 |
11 |
Thus, the amount of dietary lysine that is used by the PDV is calculated by
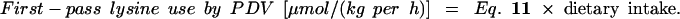 |
12 |
As stated in Calculations, some of the lysine appearing in the portal vein is derived neither from the diet nor from the artery. This so-called recycled lysine is derived from proteolysis, either intracellular or luminal, or from bacterial production. It was calculated as follows, with results from both the i.v. and the enteral tracer studies.
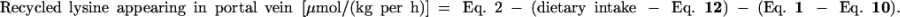 |
13 |
Lysine Oxidation by the PDV.
Lysine oxidation across the PDV was calculated as follows.
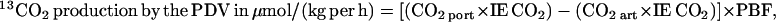 |
14 |
where CO2 art is the CO2 concentration in mmol/liter in arterial blood, CO2 port is the CO2 concentration in arterial blood, and IE is isotopic enrichment (atom/atom excess).
The lysine oxidation rate is calculated by dividing the 13CO2 production rate by the 13C input, either derived from dietary lysine or from arterial lysine.
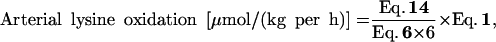 |
15 |
where the denominator is multiplied by a factor of 6 to account for the six carbon atoms that are labeled in the tracer.
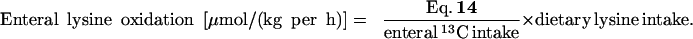 |
16 |
The enteral 13C intake was calculated by multiplying the tracer infusion rate by 6 and by the enrichment of the individual carbon atoms, 0.97.
Whole-Body Lysine Kinetics
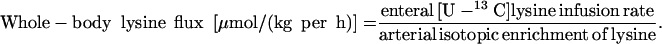 |
17 |
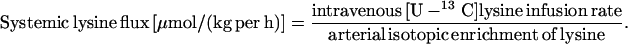 |
18 |
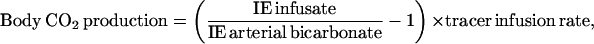 |
19 |
where IE infusate is the enrichment of H13CO3− in the infusate in mole percentage of excess, IE arterial bicarbonate is the bicarbonate enrichment in arterial blood in mole percentage of excess, and tracer infusion rate is the rate of [1-13C]bicarbonate in μmol/(kg per h) during the i.v. bicarbonate experiment during experimental day 1.
Hepatic lysine oxidation is calculated as follows.
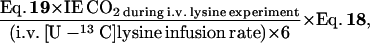 |
20 |
assuming that all non-PDV lysine oxidation is hepatic lysine oxidation. IE CO2 during i.v. lysine experiment represents the isotopic enrichment of arterial bicarbonate during the i.v. [U-13C]lysine tracer experiment.
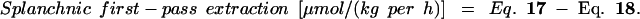 |
21 |
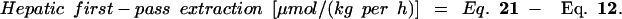 |
22 |
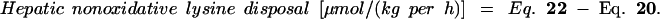 |
23 |
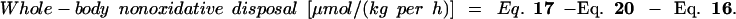 |
24 |
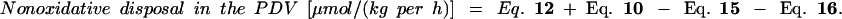 |
25 |
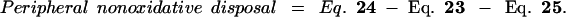 |
26 |
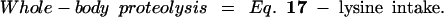 |
27 |
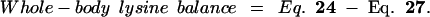 |
28 |
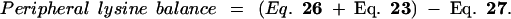 |
29 |
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.200371497.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.200371497
References
- 1.McNurlan M A, Garlick P J. Biochem J. 1980;186:381–383. doi: 10.1042/bj1860381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burrin D G, Ferrell C L, Britton R A, Bauer M. Br J Nutr. 1990;64:439–448. doi: 10.1079/bjn19900044. [DOI] [PubMed] [Google Scholar]
- 3.Lobley G E, Milne V, Lovie J M, Reeds P J, Pennie K. Br J Nutr. 1980;43:491–502. doi: 10.1079/bjn19800116. [DOI] [PubMed] [Google Scholar]
- 4.Hoerr R A, Matthews D E, Bier D M, Young V R. Am J Physiol. 1993;264:E567–E575. doi: 10.1152/ajpendo.1993.264.4.E567. [DOI] [PubMed] [Google Scholar]
- 5.Stoll B, Henry J, Reeds P J, Yu H, Jahoor F, Burrin D G. J Nutr. 1998;128:606–614. doi: 10.1093/jn/128.3.606. [DOI] [PubMed] [Google Scholar]
- 6.MacRae J C, Bruce L A, Brown D S, Calder A G. Am J Physiol. 1997;273:G1200–G1207. doi: 10.1152/ajpgi.1997.273.6.G1200. [DOI] [PubMed] [Google Scholar]
- 7.Stoll B, Burrin D G, Henry J, Yu H, Jahoor F, Reeds P J. Am J Physiol. 1999;277:E168–E175. doi: 10.1152/ajpendo.1999.277.1.E168. [DOI] [PubMed] [Google Scholar]
- 8.Windmueller H G, Spaeth A E. J Biol Chem. 1978;253:69–76. [PubMed] [Google Scholar]
- 9.Battezzati A, Brillon D J, Matthews D E. Am J Physiol. 1995;269:E269–E276. doi: 10.1152/ajpendo.1995.269.2.E269. [DOI] [PubMed] [Google Scholar]
- 10.Benevenga N J, Radcliff B C, Egan A R. Aust J Biol Sci. 1983;36:475–485. doi: 10.1071/bi9830475. [DOI] [PubMed] [Google Scholar]
- 11.Yu Y -M, Burke J F, Vogt J A, Chambers L, Young V R. Am J Physiol. 1992;262:E687–E694. doi: 10.1152/ajpendo.1992.262.5.E687. [DOI] [PubMed] [Google Scholar]
- 12.Davis T A, Nguyen H V, Costa D P, Reeds P J. Comp Biochem Physiol B Biochem Mol Biol. 1995;110:633–639. doi: 10.1016/0305-0491(94)00162-n. [DOI] [PubMed] [Google Scholar]
- 13.Chu S H, Hegsted D M. J Nutr. 1976;106:1089–1096. doi: 10.1093/jn/106.8.1089. [DOI] [PubMed] [Google Scholar]
- 14.Hutzler J, Dancis J. Biochim Biophys Acta. 1975;377:42–51. doi: 10.1016/0005-2744(75)90284-3. [DOI] [PubMed] [Google Scholar]
- 15.Fellows F C, Lewis M H. Biochem J. 1973;136:329–334. doi: 10.1042/bj1360329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hutzler J, Dancis J. Biochim Biophys Acta. 1968;158:62–69. doi: 10.1016/0304-4165(68)90072-x. [DOI] [PubMed] [Google Scholar]
- 17.Hutzler J, Dancis J. Biochim Biophys Acta. 1970;206:205–214. doi: 10.1016/0005-2744(70)90104-x. [DOI] [PubMed] [Google Scholar]
- 18.Alpers D H. J Clin Invest. 1972;51:167–173. doi: 10.1172/JCI106788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebner S, Schoknecht P, Reeds P, Burrin D. Am J Physiol. 1994;266:R1736–R1743. doi: 10.1152/ajpregu.1994.266.6.R1736. [DOI] [PubMed] [Google Scholar]
- 20.Dudley M A, Wykes L, Dudley A W, Jr, Fiorotto M, Burrin D G, Rosenberger J, Jahoor F, Reeds P J. J Nutr. 1997;127:687–693. doi: 10.1093/jn/127.5.687. [DOI] [PubMed] [Google Scholar]
- 21.Stalling D L, Gehrke C W, Zumwalt R W. Biochem Biophys Res Commun. 1968;23:616–622. doi: 10.1016/0006-291x(68)90523-8. [DOI] [PubMed] [Google Scholar]
- 22.Matthews D E, Ben-Galim E, Bier D M. Anal Chem. 1979;51:80–84. doi: 10.1021/ac50037a028. [DOI] [PubMed] [Google Scholar]
- 23.Metges C C, El-Khoury A E, Henneman L, Petzke K J, Grant I, Bedri S, Pereira P P, Ajami A M, Fuller M F, Young V R. Am J Physiol. 1999;277:E597–E607. doi: 10.1152/ajpendo.1999.277.4.E597. [DOI] [PubMed] [Google Scholar]
- 24.Reeds P J, Burrin D G, Jahoor F, Wykes L, Henry J, Frazer E M. Am J Physiol. 1996;270:E413–E418. doi: 10.1152/ajpendo.1996.270.3.E413. [DOI] [PubMed] [Google Scholar]
- 25.Bouteloup-Demange C, Boirie Y, Dechelotte P, Gachon P, Beaufrere B. Am J Physiol. 1998;274:E541–E546. doi: 10.1152/ajpendo.1998.274.3.E541. [DOI] [PubMed] [Google Scholar]
- 26.Stoll B, Burrin D G, Henry J F, Jahoor F, Reeds P J. Am J Physiol. 1999;276:G49–G57. doi: 10.1152/ajpgi.1999.276.1.G49. [DOI] [PubMed] [Google Scholar]
- 27.Reeds P J, Burrin D G, Stoll B, Jahoor F, Wykes L, Henry J, Frazer M E. Am J Physiol. 1997;273:E408–E425. doi: 10.1152/ajpendo.1997.273.2.E408. [DOI] [PubMed] [Google Scholar]
- 28.Murphy J M, Murch S J, Ball R O. J Nutr. 1996;126:878–886. doi: 10.1093/jn/126.4.878. [DOI] [PubMed] [Google Scholar]
- 29.Gaudichon C, Mahe S, Luengo C, Laurent C, Meaugeais P, Krempf M, Tome D. Eur J Clin Nutr. 1996;50:261–268. [PubMed] [Google Scholar]
- 30.Shulman R J, Gannon N, Reeds P J. Am J Clin Nutr. 1995;62:969–972. doi: 10.1093/ajcn/62.5.969. [DOI] [PubMed] [Google Scholar]
- 31.Pappenheimer J R. Am J Physiol. 1993;265:G409–G417. doi: 10.1152/ajpgi.1993.265.3.G409. [DOI] [PubMed] [Google Scholar]
- 32.Yao S Y, Muzyka W R, Elliott J F, Cheeseman C I, Young J D. Biochem J. 1998;330:745–752. doi: 10.1042/bj3300745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lauteala T, Horelli-Kuitunen N, Closs E, Savontaus M I, Lukkarinen M, Simell O, Cunningham J, Palotie A, Aula P. Hum Genet. 1997;100:80–83. doi: 10.1007/s004390050469. [DOI] [PubMed] [Google Scholar]
- 34.Seve B, Reeds P J, Fuller M F, Cadenhead A, Hay S M. Reprod Nutr Dev. 1986;26:849–861. doi: 10.1051/rnd:19860509. [DOI] [PubMed] [Google Scholar]