

Abstract
The miniaturization process applied to rubredoxins generated a class of peptide-based metalloprotein models, named METP (miniaturized electron transfer protein). The crystal structure of Desulfovibrio vulgaris rubredoxin was selected as a template for the construction of a tetrahedral (Sγ-Cys)4 iron-binding site. Analysis of the structure showed that a sphere of 17 Å in diameter, centered on the metal, circumscribes two unconnected approximately C2 symmetry related β-hairpins, each containing the -Cys-(Aaa)2-Cys- sequence. These observations provided a starting point for the design of an undecapeptide, which self assembles in the presence of tetrahedrally coordinating metal ions. The METP peptide was synthesized in good yield by standard methodologies. Successful assembly of the METP peptide with Co(II), Zn(II), Fe(II/III), in the expected 2:1 stoichiometry, was proven by UV-visible and circular dichroism spectroscopies. UV-visible analysis of the metal complexes indicated the four Cys ligands tetrahedrally arrange around the metal ion, as designed. Circular dichroism measurements on both the free and metal-bound forms revealed that the metal coordination drives the peptide chain to fold into a turned conformation. NMR characterization of the Zn(II)-METP complex fully supported the structure of the designed model. These results prove that METP reproduces the main features of rubredoxin.
Miniaturized proteins are peptide-based synthetic models of natural macromolecular systems. They contain a minimum set of constituents necessary for an accurate reconstruction of defined structures and for a fine-tuned reproduction of defined functions (1, 2). Their intermediate size between low molecular weight compounds and protein mutants makes miniaturized proteins suitable for structure–function relationship studies: they are simple enough to avoid ambiguities of interpretation associated with large proteins, and they provide sufficient size and chemical diversity to allow the construction of active sites.
The miniaturization process can be rationally organized, once a structural knowledge of the parent natural system to be miniaturized is available. It is necessary to define (i) the type and number of constituents to be assembled, (ii) the structure to be reconstructed, and (iii) the function to be reproduced. These aspects are strictly related. A larger number of constituents might be necessary for a more precise three-dimensional reconstruction and for a more specialized function.
Metalloproteins are well suited to miniaturization. The metal center represents a pivotal point where spheres of variable diameters that circumscribe part of the protein are centered: the larger the diameter of the sphere, the larger the number of constituents included in the model. Comparative studies, in terms of structure and function, of a set of protein models, defined by spheres of variable diameters, will allow better understanding of how the protein matrix modulates the unique features of the metal center, including electronic, redox, and catalytic properties (3–5). These properties are imprinted by the primary coordination shell—the composition, the number, and the geometry of ligands (6). They are also regulated by the secondary coordination shell—the immediate surroundings of the primary coordination sphere—and they are finely modulated by long-range electrostatic factors (6).
For a sphere of a given diameter around the metal center, the constituents therein circumscribed are in general unconnected parts. Several strategies can be conceived to obtain a miniaturized model. Two or more parts could spontaneously associate to give folded noncovalent self-assembled oligomers, or two or more parts could be covalently connected to give folded monomers (7–10). We described the latter strategy in the design of mimochromes, covalent helix-heme-helix sandwiches (11–14), and we described the former in the design of DF1, a small four-helix bundle protein with an oxo-di-iron site in its interior (15, 16). Mimochromes and DF1 have a common characteristic: they are both C2 symmetric dimers, intended to reproduce the quasi-symmetrical structure of a metalloprotein. The use of C2 symmetry is particularly advantageous because it simplifies the design and reduces the size of the molecules to be synthesized and may simplify their structural characterization. However, the use of a C2 symmetry axis to construct a symmetric dimer self assembled around metal ions leaves open the problem of possible diastereomeric forms. Λ and Δ isomers of Co(III)-mimochrome I were unpredicted but experimentally observed (12). DF1 was instead designed to self assemble as a trans antiparallel dimer, thus inhibiting the formation of a second diastereomeric form. Further, the design of noncovalent asymmetric dimers is complicated by homodimer formation.
This paper describes a design concept as applied to Rds (rubredoxins), the simplest iron–sulfur proteins. Rds are small proteins with a Fe(II)/Fe(III) high spin ion in a tetrahedral (Sγ-Cys)4 site, which seem to participate in electron transfer (17). They display high similarity in both their three-dimensional structures and metal-binding sequence, constituted by two pairs of Cys residues spaced as [-Cys-(Aaa)2-Cys-(Aaa)n-Cys-(Aaa)2-Cys-] (18–22).
Several investigations are being carried out on both natural Rds (17–23) and their mutants (24–32) and on model systems (33–39) to identify the factors tuning the properties of the active site.
We report here the design, synthesis, and characterization of a prototype miniaturized Rd named METP (miniaturized electron transfer protein). The coordination properties toward metal ions other than iron (zinc and cobalt) have also been examined to prove the versatility of METP in reproducing the structure and properties of other proteins with tetrahedral metal-binding sites.
Materials and Methods
Molecular Modeling.
A Silicon Graphics Indigo 2 workstation with the insight II/discover program package (Biosym Technologies, San Diego) was used. The consistent valence force field (CVFF) with suitable parameters for the Fe(II)-(Sγ-Cys)4 moiety (28) and with a distance-dependent dielectric constant was used for energy minimization, performed with the Newton–Raphson algorithm until convergence. The modeling of METP was based on the crystal structure of Desulfovibrio vulgaris Rd (ref. 19; PDB ID code 8RXN).
Peptide Synthesis and Purification.
METP peptide was synthesized by using a MilliGen peptide synthesizer by standard Fmoc/tert-butyl (But) strategy on a NovaSyn PR500 resin (Novabiochem, Switzerland). Cleavage from the resin and sidechain deprotection was achieved with ethanedithiol/H2O/triisopropylsilane/trifluoroacetic acid 0.25:0.25:0.1:9.5 (vol/vol/vol/vol) for 2 h. Extraction and precipitation with ether gave the crude material (yield 80%). It was reduced by 3-h incubation in aqueous solution at pH 8 and 10-fold excess DTT. Purification by preparative HPLC gave the desired pure product, as assayed by analytical HPLC and matrix-assisted laser desorption ionization time-of-flight spectrometry [molecular weight: calculated 1,151.4 atomic mass units (amu); obs 1152 ± 1 amu].
Metal-Binding Experiments.
All manipulations were performed under strictly anaerobic conditions. Both peptide and metal stock solutions were freshly prepared. The concentration of stock METP peptide solutions was determined spectrophotometrically by using ɛ230 = 1.52 (±0.01) × 103M−1⋅cm−1, as calculated from experimentally determined Lambert and Beer's law.
The Co(II) and Zn(II) complexes were prepared by addition of CoCl2 or ZnCl2, in 10% molar excess, to an aqueous solution of the reduced METP peptide at pH ≈4; the pH was then raised to 7.5 by addition of phosphate buffer (10 mM final buffer concentration).
Fe(II) complex was obtained anaerobically by using (NH4)2Fe(SO4)2. A 0.15 M stock solution of (NH4)2Fe(SO4)2 was prepared in 5 mM H2SO4 to minimize autooxidation. A 10% excess amount of (NH4)2Fe(SO4)2 was added to a 0.822 mM aqueous solution of METP peptide. The pH was then raised to 8.0 by addition of Tris buffer (10 mM final buffer concentration).
The Fe(III) complex was prepared in CH3OH. A 10% excess amount of FeCl3 was added to a 1.19 mM solution of METP peptide in CH3OH. The addition of N,N-diisopropylethylamine, 1.5 equivalents with respect to thiol groups, gave a deep red violet solution.
The pH dependence of the Co(II)-METP complex formation was determined by titrating NaOH into an aqueous solution of reduced METP peptide at 1.00 mM concentration and of CoCl2 in 10% molar excess.
The binding affinity of Co(II) to METP peptide was measured by spectrophotometric titration of CoNO3 into the reduced peptide at 0.754 mM concentration (10 mM phosphate buffer, pH 7.5). The observed absorbance intensities were fit [kaleidagraph, Synergy Software (Reading, PA); see Supplementary Material, www.pnas.org] to a binding isotherm (39–41) to estimate the dissociation constant KdissCo and the stoichiometry of the interaction n, according to the reaction:
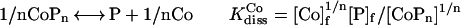 |
1 |
where [Co]f, [P]f, [CoPn] are the concentrations of free Co2+, free peptide (in monomeric units) and complex, respectively.
The affinity of Zn(II) was estimated by competition experiments on the Co(II) complex. Aliquots of ZnCl2 aqueous solution (19 mM) were titrated into a solution containing the reduced peptide at 0.571 mM concentration and a 3-fold molar excess of Co(II), at pH 7.5. The bleaching of the absorption spectrum was monitored. The relative affinity of the interaction was obtained by fitting the data to the equilibrium (40, 41):
 |
2 |
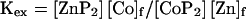 |
where [ZnP2], [CoP2], [Co]f, [Zn]f are the concentrations of Zn(II) complex, Co(II) complex, free Co2+, and free Zn2+, respectively. KdissZn was determined from KdissCo and Kex: KdissZn = KdissCo/(Kex)(1/2).
UV-Visible (UV-vis) Spectroscopy.
UV-vis spectra were recorded on a Perkin–Elmer Lambda 7 UV Spectrophotometer by using quartz cells of 1 cm path length. Wavelength scans were performed at room temperature, from 200 to 700 nm, with a 60 nm⋅min−1 scan speed. Anaerobic spectra were recorded in rubber-sealed cuvettes under nitrogen.
Circular Dichroism (CD) Spectroscopy.
CD spectra were obtained at room temperature on a Jasco (Easton, MD) J-715 dichrograph. Data were collected at 0.2-nm intervals with a 20 nm⋅min−1 scan speed, a 2-nm bandwidth, and a 16-s response, from 260 to 190 nm, and at 0.5-nm intervals with a 200 nm⋅min−1 scan speed, a 2-nm bandwidth, and a 2-s response from 250 to 800 nm; circular quartz cells of 0.01-cm and 1-cm path length were used for the far UV and near UV-vis regions, respectively. Peptide concentrations were in the range 0.20–1.2 mM. CD intensities in the far UV and near UV-vis regions are expressed as mean residue ellipticities (degree cm2⋅dmol−1⋅residue−1) and total molar ellipticities, respectively.
NMR Spectroscopy.
NMR sample was prepared directly into an NMR tube by addition of NaOD (D2O solution, 1.5 equivalents) to a CD3OH solution of reduced METP peptide (1.2 mM) and ZnCl2 (CD3OH solution, 1.1 equivalents). The NMR tube was flushed with nitrogen and sealed. Tetramethylsilane was used as internal reference.
1H NMR one-dimensional and two-dimensional spectra were collected at 298 K on a Varian Unity 400 spectrometer, operating at 400 MHz, and processed with felix software (Biosym Technologies, San Diego/Micron Separations). Assignment of the 1H NMR signals was accomplished by using a combination of scalar and dipolar correlation two-dimensional experiments (42). A total correlation spectroscopy experiment was carried out by using a 70 ms MLEV-17 spin lock. Nuclear Overhauser effect spectroscopy experiments were acquired at 200 and 300 ms, whereas rotating-frame Overhauser effect spectroscopy spectra were collected in a 200–120 ms range of mixing time.
Results and Discussion
Design.
The three-dimensional structure of Rd (19) was used as a template for the design of METP. We observed that a sphere of 17 Å in diameter, centered on the metal ion, circumscribes part of the molecule encompassing two quite similar type 3 β-hairpins (43): Val5-Glu12 and Val38-Pro45. Both contain two metal ion-coordinating Cys residues at position i and i+3 of type I-αRS α-turn (44, 45), and their backbone atoms are approximately C2 symmetry related. This pseudo C2 symmetry axis is almost coincident with one of the C2 symmetry axes of the coordination tetrahedron (Fig. 1). It passes through the metal ion and halfway between Cys9(6) and Cys42(39) Sγ atoms. The rms deviation was of 0.34 Å when overlapping the backbone atoms of Val5-Tyr11, Val38-Ala44 and the sidechains of Cys6, Cys9, Cys39, Cys42 with the backbone atoms of Val38-Ala44, Val5-Tyr11 and the side chains of Cys39, Cys42, Cys6, Cys9 of a copy of the molecule, respectively.
Figure 1.

Stereo view of Val5-Glu12, Val39-Pro45 from Rd (19) as ribbon drawing. C2 symmetry axis relating backbone atoms is indicated as an arrow.
These observations led us to conceive a β-hairpin peptide with two properly spaced Cys residues that could dimerize symmetrically (via a C2 axis) in the presence of a tetrahedrally coordinating metal ion to give a miniaturized model for Rd, METP. We began modeling a pentapeptide sequence, Cys-(Aaa)2-Cys-Aaa, in an α-turn conformation, as observed in several proteins (44). The χ1 angles of both Cys residues were set as found for Cys6(39) (χ1 = 180°) and Cys9(43) (χ1 = 75°) in Rd. In this particular conformation, the Sγ atoms are properly oriented to bind metal ions in a tetrahedral geometry (Fig. 2B). A dimeric structure can be obtained by use of a C2 axis. In the coordination tetrahedron so far defined, there are three C2 axes: two are productive in terms of symmetric dimer construction, and one is unproductive. The unproductive C2 axis (the a axis in Fig. 2B) is the one that passes through the metal ion and the edge of the coordination tetrahedron formed by the Sγ atoms of the two intrachain Cys residues. This axis would generate a copy of the peptide that reproduces Cys1 on Cys4 and vice versa. The productive C2 axes (axes b and c in Fig. 2 A and C) are those that pass through the metal ion and the edge of the coordination tetrahedron formed by the Sγ atoms of two interchain Cys residues. In the tetrahedral symmetry, these two axes are related by a symmetry plane. The dimers obtained from these axes should be Λ and Δ enantiomers (Fig. 2 A and C), but they are actually diastereomers because of the chirality of the amino acids. The configuration around the metal ion in Rd is Λ.
Figure 2.

Symmetric dimer reconstruction from Cys-(Aaa)2-Cys-Aaa pentapeptide by the use of productive C2 symmetry axes. (A) Λ isomer obtained through b axis; (B) unproductive a symmetry axis; (C) Δ isomer obtained through c axis.
Much attention was paid to the design to overcome the problem of two diastereomeric forms of METP. Either of the two possible diastereomers should contain interchain interactions that stabilize one isomer (positive design) and destabilize the other (negative design) (2). Two pentapeptide sequences, Cys-(Aaa)2-Cys-Aaa, with an α-turn conformation, may provide the four Cys residues in the appropriate orientation for metal ion binding, but they are too small to give a sufficient number of either stabilizing or destabilizing interchain interactions. A reasonable compromise between the smallest molecular size and the largest number of interactions was reached at an undecapeptide level. About 50% of the residues (the -Cys-(Aaa)2-Cys-Aaa- sequence) are devoted to metal ion coordination and about 50% to further intra- and interchain interactions. After several trials, an undecapeptide that contains the α-turn motif at the N-terminal part and that extends by six more amino acids at the C-terminal part was selected.
The design began by initially fixing the backbone conformation. Two template undecapeptide structures were possible alternatives: [Cys6-Ala16]- and [Cys39-Phe49]-Rd. They differ in composition and partly in folding. Fig. 3 A and B describe the symmetric dimers with Λ configuration obtained by C2 symmetry on [Cys6-Ala16]-Rd and [Cys39-Phe49]-Rd, respectively. The extended conformation at the Glu12-Ala16 segment projects the peptide chain away from its symmetrical copy, whereas the 310 helical turn at the Pro45-Phe49 segment folds the peptide chain back toward its symmetrical copy. [Cys39-Phe49]-Rd peptide segment was chosen as a template structure for the subsequent design.
Figure 3.

Stereo view of the symmetric dimers with Λ configuration obtained by the application of a C2 symmetry to [Cys6-Ala16]-Rd (A) and [Cys39-Phe49]-Rd (B).
The [Cys39-Phe49]-Rd segment cannot dimerize, as depicted in Fig. 3A, because this arrangement contains severe atom overlaps: Pro40 bumps with the backbone atoms of Glu48′ (prime numbering indicates symmetrical duplicate) and Phe49 sidechain overlaps the sidechain of Phe49′. Substitution of Pro40 with Thr abolishes the Pro40—Glu48′ bumps and provides favorable inter- and intrachain interactions: Thr Oγ atom is H-bonded to the Glu48′ C′O group in the model. A Thr to Pro substitution can be considered a conservative substitution, because this residue is present at the corresponding position of the other β-hairpin ([Cys6-Ala16]-Rd). In the Rd structure, Thr7 NH is H-bonded to Glu48 C′O. Substitution of Phe49 with an Ile residue eliminates the Phe49–Phe49′ steric clash and provides favorable interchain hydrophobic interactions. The Ile sidechain is smaller and sufficiently flexible to allow suitable filling of space. To reinforce dimer formation (positive design), Val41 was replaced with a Lys residue, because its Nζ may form an ion pair with Glu48′. Lys Nζ is also H-bonded to the Gly43 C′O group. This interaction seemed particularly important to us for the overall stability of the symmetric dimer and to stabilize the Λ isomer (see below for additional elements of negative design). Great care was given to reinforce the β-turn type III conformation, present in the Rd Pro45-Phe49 segment. Asn substitutes Pro45 and was preferred to a Pro residue, because the side chain is exposed to the solvent and because the backbone NH can H-bond the Glu48 Oɛ atom, and the Nδ atom can H-bond Glu48 Oɛ and Ser47 Oγ in the model. Aib replaces Lys46, because Aib residue is well known to strongly stabilize type III β-turns at position i+1 or i+2 of the turn (46). Cys39, Cys42-Gly43-Ala44, and Ser47-Glu48 appeared fully compatible with the amino acid substitutions so far described. The N- and C-terminal residues were acylated and amidated, respectively, to avoid perturbing effects of the end charges. The final sequence of METP peptide compared with Rd (5–16) and Rd (38–49) is reported below:
METP Ac-Cys-Thr-Lys-Cys-Gly-Ala-Asn-Aib-Ser-Glu-Ile-NH2
Rd (5–16)-Val-Cys-Thr-Val-Cys-Gly-Tyr-Glu-Tyr-Asp-Pro-Ala-
Rd (38–49)-Val-Cys-Pro-Val-Cys-Gly-Ala-Pro-Lys-Ser-Glu-Phe-.
The model structure was subjected to extensive energy minimization. The minimized structure (Fig. 4) differs slightly from the starting model. The Ac-Cys-Thr-Lys-Cys-Gly- segment adopts a type I-αRS α-turn, -Ala-Asn- is in a β-extended conformation, and -Aib-Ser-Glu-Ile-NH2 is in a right-handed 310 helical turn.
Figure 4.

Energy minimized structure of Fe(II)-METP complex.
The METP peptide sequence is incompatible with the fixed backbone conformation for the undesired Δ isomer (negative design). The peptide, in the particular conformation chosen in the starting model, interacts poorly with the symmetric counterpart: no van der Waals contacts and very few intra- and interchain H-bonds are present. Particularly important are the charged amino acids. Both Lys residues are on one side of the molecule, and both Glu residues are on the opposite side. The hydrophobic amino acids are no longer clustered, and their sidechains are exposed to the solvent. This undesired Δ-isomer, however, was subjected to energy minimization. The structure converged to a minimum that differs significantly from the initial structure (data not shown).
Metal Binding.
The METP peptide binds cobalt, zinc, and iron ions. Co(II) was selected for initial binding studies, because cobalt complexes show distinctive UV-vis spectra, which are sensitive to the metal coordination geometry and to the nature of the ligands (47). In addition, the spectroscopic properties of Co(II)-Rds were previously examined (30, 31). Fig. 5 shows the absorption spectrum obtained when a 10% excess amount of CoCl2 was added to the reduced peptide in water solution and when the pH was raised to the final value of 7.5. The spectrum exhibits absorption maxima and molar extinction coefficients in good agreement with those reported in the literature for the Co(II) derivative of Rd (30, 31) (Table 1). The intensities of the d-d transitions in the visible region are characteristic of Co(II) coordination in a tetrahedral geometry (47) and exclude higher coordination numbers. The intense ligand-to-metal charge transfer (LMCT) band at 350 nm indicates thiolate ligation, and its intensity is in good agreement with those observed in proteins and model systems (30, 31, 38, 39). The position of the absorption maxima of both d-d and LMCT transitions, which are sensitive to the identity of the ligands and move toward lower energies as the number of thiolate ligands increases, is consistent with a (Sγ-Cys)4 coordination environment (38–41).
Figure 5.

Optical absorption spectrum of Co(II)-METP complex.
Table 1.
UV-vis spectral data of METP complexes and Rd
| Compound | Wavelength, nm [ɛ × 10−3 (M−1⋅cm−1)] | |||
|---|---|---|---|---|
| Fe(II)-METP* | 310 (4.72) | 332 (3.31) | ||
| Rdred*† | 311 (10.8) | 333 (6.30) | ||
| Fe(III)-METP‡ | 358 (7.66) | 486 (4.42) | 565 (2.50) | |
| Rdox*† | 380 (10.8) | 490 (8.85) | 565 (≈4.0) | |
| Co(II)-METP§ | 353 (3.57) | 624 (0.420) | 685 (0.617) | 735 (0.582) |
| Co(II)-Rd*¶ | 350 (4.70) | 620 (0.564) | 685 (0.616) | 748 (0.517) |
A pH titration from 4 to 8.5 causes an increase of the absorption intensities of both the ligand-to-metal charge transfer and d-d bands, which indicates Cys deprotonation and concomitant complexation. The Cys residues are deprotonated with a midpoint pH value of 6.6 at 1.0 mM METP peptide concentration and 0.55 mM CoCl2 concentration.
The stoichiometry of the Co(II)-METP complex formation and the binding constant was determined by direct spectrophotometric titration, as shown in Fig. 6. The best fit of the data to Eq. 1, with [P]t = 0.754 mM, gave a METP peptide/Co(II) ratio of 2:1 and a dissociation constant of 53.5 (± 2.8) μM at pH 7.5 (Supplementary Material; see www.pnas.org).
Figure 6.

(Left) Spectrophotometric titration of METP peptide with Co(II). The saturation fraction 2[CoP2]/[P]t is plotted against the concentration ratio [Co]t/[P]t. Solid line shows the best fit of data points to Eq. 1. (Right) Spectrophotometric competition experiment of Co(II)-METP complex with Zn(II). The saturation fraction 2[CoP2]/[P]t is plotted against the concentration ratio [Zn]t/[P]t. Solid line shows the best fit of data points to Eq. 2.
The affinity of the peptide for Zn(II) was determined by competition experiments. Zn(II) readily displaces Co(II) from the complex: the absorption intensities of the UV-vis bands of the cobalt complex decrease sharply on Zn(II) addition. METP peptide binds Zn(II) more tightly than Co(II); in the presence of three equivalents of Co(II), at a peptide concentration of 0.571 mM (Fig. 6), a dissociation constant of 2.7 (±0.1) μM was obtained (see Supplementary Material). The observed lower affinity of METP peptide for Co(II), with respect to Zn(II), is fully consistent with a tetrahedral coordination site (40, 41).
The iron complex was obtained by addition of (NH4)2Fe(SO4)2 (1.1 equivalent) to the reduced METP peptide and was ascertained by UV-vis and CD spectroscopies. The electronic absorption spectrum of the Fe(II)-METP complex closely resembles that of Rds (23) (Fig. 7 and Table 1). A red color of the Fe(III) complex immediately appeared on exposure of the Fe(II) complex to air. However, the color faded very rapidly (in less than 1 min), and it was impossible to record the spectrum of the Fe(III) complex. This behavior was observed in other peptide-based Rd mimetics (35) and in the Rd-like site engineered into the B1 domain of protein G (38). An autoredox reaction may explain the observed instability of Fe(III) tetrathiolate complexes (34):
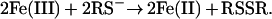 |
A few stable [Fe(III)(SR)4](1−) compounds containing organic ligands of a different nature were obtained in organic solvents (34, 37). Addition of N,N-diisopropylethylamine (to deprotonate the Cys residues) to a CH3OH solution of the reduced METP peptide in the presence of FeCl3 led to a deep red complex formation. The absorption spectrum of the oxidized complex shows the typical ligand-to-metal charge transfer transitions in the visible region (Fig. 7; Table 1). The similarity between the optical spectrum of Fe(III)-METP complex and those reported for oxidized Rds strongly supports our model. The Fe(III) center is tetrahedrically coordinated by four cysteine thiolates, as in the core structure of oxidized Rds (23, 29). It should be noted that bleaching of the solution was quite slow in CH3OH and results in a reddish-brown precipitate. Interestingly, addition of Fe(II) [as (NH4)2Fe(SO4)2] and N,N-diisopropylethylamine to the CH3OH solution of METP peptide resulted in the instantaneous formation of the Fe(III) complex. This result can be explained by considering the effect of the solvent polarity on the Fe(III)/Fe(II) redox potential in the Fe-(Sγ-Cys)4 site (24). The effective net charge of the oxidized Fe(III)-(Sγ-Cys)4 site is −1, whereas that of the reduced Fe(II)-(Sγ-Cys)4 site is −2. Thus, it is expected that solvents of high polarity stabilize the reduced state.
Figure 7.

Optical absorption spectrum of Fe(II)- (dotted line) and Fe(III)- (continuous line) METP complexes.
All these data show that METP peptide coordinates both Fe(II) and Fe(III) with a tetrahedral (Sγ-Cys)4 geometry, thus resembling one of the distinctive features of Rds. However, the interconversion between the two iron oxidation state was irreversible.
CD Spectroscopy.
The effect of metal binding on the structure of METP peptide was examined by CD spectroscopy in both the far UV and the visible region (Fig. 8). The CD spectrum of the fully reduced METP peptide in aqueous solution resembles that of unordered peptides (48), exhibiting a strong negative band at 198 nm and a shoulder with a small negative value at 220 nm (Fig. 8A). The coordination of Zn(II) induces significant changes on the structure of the peptide. The CD spectrum shows a double minima at 225 and 203 nm and a maximum at about 190 nm (Fig. 8A). This spectral pattern reflects the presence of α-helix turns, 310 helical turns, or type I (III) β-turns (48). A similar spectrum was observed for the Co(II)-METP complex (data not shown). These features suggest that the peptide chain folds into a turned conformation on metal binding, as in the design. This behavior was also observed on Fe(III) coordination in CH3OH solution (Fig. 8B). The slight differences between the CD spectra of the zinc and iron complexes can be ascribed to the different solvents. The same solvent-inducing effect can be observed for the fully reduced unbound peptide.
Figure 8.

CD spectra of METP peptide and its metal complexes. (A) Far UV CD spectrum in aqueous solution of reduced METP peptide (dotted line) and Zn(II)-METP complex (continuous line). (B) Far UV CD spectrum in CH3OH solution of reduced METP peptide (dotted line) and Fe(III)-METP complex (continuous line). (C) Near UV-vis spectrum in aqueous solution of Co(II)-METP complex. (D) Near UV-vis spectra of Fe(II)- (dotted line; in aqueous solution) and Fe(III)- (continuous line; in CH3OH solution) METP complexes.
Cotton effects in the near UV and visible regions are found for Co(II)- and Fe(II/III)-METP complexes. The CD spectrum of Co(II)-METP complex (Fig. 8C) exhibits a spectral pattern very similar to that reported for the Co(II) reconstituted Rd (30). The spectral pattern for both Fe(II)- and Fe(III)-METP complexes (Fig. 8D) strictly resembles that found for the natural and synthetic Rds (32). The overall similarity between the CD spectra of the Co(II)-, Fe(II)-, and Fe(III)-METP complexes and those of natural and cobalt reconstituted Rds fully confirms the same structural organization around the metal center in both the model system and its natural counterpart.
NMR Characterization.
Fig. 9 shows the amide proton region of Zn(II)-METP complex. The presence of only one set of signals supports the C2 symmetry of the complex, as expected from the design.
Figure 9.

Amide proton region of the one-dimensional spectrum of Zn(II)-METP complex in CD3OH solution at 298 K. NH Assignment is reported by sequence number.
All the NMR data [nuclear Overhauser effects, coupling constants, amide, and αCH proton chemical shifts; see Supplementary Material (www.pnas.org)] indicate that the complex adopts a well-defined conformation with structural elements very similar to those observed in Zn(II) reconstituted Rds (25, 26) and as expected from the design. Strong NHi-NHi+1 connectivities observed for residues Lys3-Gly5 and Glu10-Ile11 indicate that the segments Cys1-Gly5 and Ser9-Ile11 comprise tight turns. The value of 3J(NH-αCH) coupling constants are in full agreement with the φ angles predicted in the model METP complex (see Supplementary Material). Finally, the pattern of the amide and αCH proton chemical shifts is similar to that observed for Zn(II)-Rd (25, 26). We are presently refining a starting model by restrained molecular dynamic simulations.
Conclusions
This work describes in a rational mode the concept of diastereomerism that may occur when polypeptides coordinate to metal ions. We also extend our earlier work (15), which utilizes local pseudo-C2 symmetry around the metal center to construct symmetric molecules with minimized size.
We achieved the design of a miniaturized Rd, named METP, starting from the analysis of the x-ray structure of Rd from D. vulgaris (19). In the miniaturization process, we selected the type and number of constituents to be assembled, to include all the elements of the primary and secondary coordination shell to the metal. An undecapeptide, which self assembles in the presence of tetrahedrally coordinating ions as a unique diastereomeric form (Λ), was designed.
The peptide was obtained easily and in good yields. It avidly binds Co(II), Zn(II), Fe(II), and Fe(III), in the expected 2:1 stoichiometry. The geometry around the metal ion was found to be as designed, resembling the Rd-like tetrahedral environment. The UV-vis spectra, as well as the Cotton effects in the near UV and visible regions of Co(II)-, Fe(II)- and Fe(III)-METP complexes, are very similar to those of natural and Co(II) reconstituted Rds (23, 29, 30, 32). Further, the analysis of the far UV CD spectra of free and metal-bound METP peptide and the NMR characterization of Zn(II)-METP complex indicate that METP folds into the designed turned conformation on metal coordination.
METP can be considered a good starting model to reproduce several spectroscopic properties of Rds for its ability to bind Fe(II) and Fe(III) with the same tetrahedral geometry. The different stability of Fe(II)- and Fe(III)-METP complexes in different solvent systems can be ascribed to different redox Fe(III)/Fe(II) potentials in different solvents. It has been widely reported (24) that both the solvent effect and the backbone and polar side-chain contributions account for the differences in the redox potentials between homologous natural Rds. An increased solvent accessibility at the Fe-(Sγ-Cys)4 site is an important factor that affects the redox potential of Rds. Thus, the presence of a solvent exposable Fe-(Sγ-Cys)4 site in the METP model could account for the lower stability of the Fe(III) complex in aqueous solution. This has been found already by Farinas and Regan (38), who reported the de novo design of a Rd-like iron site engineered into the B1 domain of protein G. The region in which the metal-binding site was inserted provides minimal protection from the solvent, thus causing instability of the Fe(III) center. By contrast, the lone example, to the best of our knowledge, of a designed Fe-(Sγ-Cys)4 site capable of undergoing successive cycles of oxidation and reduction was reported by Benson et al. (39). They introduced a tetrahedral tetratiolate iron center into wild-type thioredoxin by converting the active-site disulfide (Cys32-Cys35) into a metal-based redox center.
Taken together, these results demonstrate the success of the approach in the construction of miniaturized Rds. METP represents a suitable prototype for the development of Rd models showing a higher degree of functional similarity with respect to natural counterparts. Furthermore, by changing the nature of the coordinating residues, it will be possible to develop models of many tetrahedral metal-binding sites such as, for example, the Cys2His2 or His3Cys zinc-binding motifs and the copper-binding domain of blue copper proteins (6). The synthesis and characterization of novel METP analogs, including additional design constraints, are already in progress.
Supplementary Material
Acknowledgments
We thank Prof. Liberato Ciavatta for many useful suggestions and Prof. William F. DeGrado for critically reading the manuscript. This work was supported by the Italian Ministry of University and Scientific Research (PRIN 9803184222).
Abbreviations
- METP
miniaturized electron transfer protein
- UV-vis
UV-visible
- CD
circular dichroism
- Rds
rubredoxins
References
- 1.Cunningham B C, Wells J A. Curr Opin Struct Biol. 1997;7:457–462. doi: 10.1016/s0959-440x(97)80107-8. [DOI] [PubMed] [Google Scholar]
- 2.DeGrado W F, Summa C M, Pavone V, Nastri F, Lombardi A. Annu Rev Biochem. 1999;68:779–819. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 3.Regan L. Trends Biochem Sci. 1995;20:280–285. doi: 10.1016/s0968-0004(00)89044-1. [DOI] [PubMed] [Google Scholar]
- 4.Hellinga H W. Folding Des. 1998;3:R1–R8. doi: 10.1016/S1359-0278(98)00001-7. [DOI] [PubMed] [Google Scholar]
- 5.Lu Y, Valentine J S. Curr Opin Struct Biol. 1997;7:495–500. doi: 10.1016/s0959-440x(97)80112-1. [DOI] [PubMed] [Google Scholar]
- 6.Lippard S J, Berg J M. Principles of Bioinorganic Chemistry. Mill Valley, CA: University Science Books; 1994. pp. 349–378. [Google Scholar]
- 7.Choma C T, Lear J D, Nelson M J, Dutton P L, Robertson D E, DeGrado W F. J Am Chem Soc. 1994;116:856–865. [Google Scholar]
- 8.Gibney B R, Mulholland S E, Rabanal F, Dutton P L. Proc Natl Acad Sci USA. 1996;93:15041–15046. doi: 10.1073/pnas.93.26.15041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnold P A, Shelton W R, Benson D R. J Am Chem Soc. 1997;119:3181–3182. [Google Scholar]
- 10.Sakamoto S, Ueno A, Mihara H. J Chem Soc Perkin Trans. 1998;2:2395–2404. [Google Scholar]
- 11.Nastri F, Lombardi A, Morelli G, Maglio O, D'Auria G, Pedone C, Pavone V. Chem Eur J. 1997;3:340–349. [Google Scholar]
- 12.D'Auria G, Maglio O, Nastri F, Lombardi A, Mazzeo M, Morelli G, Paolillo L, Pedone C, Pavone V. Chem Eur J. 1997;3:350–362. [Google Scholar]
- 13.Lombardi A, Nastri F, Sanseverino M, Maglio O, Pedone C, Pavone V. Inorg Chim Acta. 1998;276:301–313. [Google Scholar]
- 14.Nastri F, Lombardi A, D'Andrea L D, Sanseverino M, Maglio O, Pavone V. Biopolymers. 1998;47:5–22. doi: 10.1002/(SICI)1097-0282(1998)47:1<5::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 15.Lombardi A, Summa C, Geremia S, Randaccio L, Pavone V, DeGrado W F. Proc Natl Acad Sci USA. 2000;97:6298–6305. doi: 10.1073/pnas.97.12.6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Summa C M, Lombardi A, Lewis M, DeGrado W F. Curr Opin Struct Biol. 1999;9:500–508. doi: 10.1016/S0959-440X(99)80071-2. [DOI] [PubMed] [Google Scholar]
- 17.Sieker L C, Stenkamp R E, LeGall J. Methods Enzymol. 1994;243:203–216. doi: 10.1016/0076-6879(94)43016-0. [DOI] [PubMed] [Google Scholar]
- 18.Bau R, Rees D C, Kurtz D M, Scott J A R. J Biol Inorg Chem. 1998;3:484–493. [Google Scholar]
- 19.Dauter Z, Sieker L C, Wilson K S. Acta Crystallogr B. 1992;48:42–59. doi: 10.1107/s0108768191010613. [DOI] [PubMed] [Google Scholar]
- 20.Misaki S, Morimoto Y, Ogata M, Yagi T, Higuchi Y, Yasuoka N. Acta Crystallogr D. 1999;55:408–413. doi: 10.1107/s0907444998011810. [DOI] [PubMed] [Google Scholar]
- 21.Watenpaugh K D, Sieker L C, Jensen L H. J Mol Biol. 1980;138:615–633. doi: 10.1016/s0022-2836(80)80020-9. [DOI] [PubMed] [Google Scholar]
- 22.Bertini I, Kurtz D M, Eidsness M K, Jr, Liu G, Luchinat C, Rosato A, Scott R A. J Biol Inorg Chem. 1998;3:401–410. [Google Scholar]
- 23.Im S-C, Sykes A G. J. Chem. Soc. Dalton Trans. 1996. 2219–2222. [Google Scholar]
- 24.Zeng Q, Smith E T, Kurtz D M, Jr, Scott R A. Inorg Chim Acta. 1996;242:245–251. [Google Scholar]
- 25.Richie K A, Teng Q, Elkin C J, Kurtz D M., Jr Protein Sci. 1996;5:883–894. doi: 10.1002/pro.5560050510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blake P R, Park J-B, Zhou Z H, Hare D R, Adams M W, Summers M F. Protein Sci. 1992;1:1508–1521. doi: 10.1002/pro.5560011112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maher M J, Xiao Z, Wilce M C J, Guss M, Wedd A G. Acta Crystallogr D. 1999;55:962–968. doi: 10.1107/s0907444999001900. [DOI] [PubMed] [Google Scholar]
- 28.Kummerle R, Zhuang-Jackson H, Gaillard J, Moulis J M. Biochemistry. 1997;36:15983–15991. doi: 10.1021/bi971636e. [DOI] [PubMed] [Google Scholar]
- 29.Eidsness M K, Burden A e, Richie K A, Kurtz D M, Scott R A, Smith E T, Ichiye T, Beard B, Min T, Kang C. Biochemistry. 1999;38:14803–14809. doi: 10.1021/bi991661f. [DOI] [PubMed] [Google Scholar]
- 30.May S W, Kuo J Y. Biochemistry. 1978;17:3333–3338. doi: 10.1021/bi00609a025. [DOI] [PubMed] [Google Scholar]
- 31.Moura I, Teixeira M, LeGall J, Moura J J G. J Inorg Biochem. 1991;44:127–139. doi: 10.1016/0162-0134(91)84025-5. [DOI] [PubMed] [Google Scholar]
- 32.Christensen M, Hammerstad-Pedersen J M, Holm A, Iversen G, Jensen M H, Ulstrup J. Eur J Biochem. 1994;224:97–101. doi: 10.1111/j.1432-1033.1994.tb19999.x. [DOI] [PubMed] [Google Scholar]
- 33.Christensen H E M, Hammerstad-Pedersen J M, Holm A, Roepstorff P, Ulstrup J, Vorm O, Ostergard S. FEBS Lett. 1992;312:219–222. doi: 10.1016/0014-5793(92)80939-e. [DOI] [PubMed] [Google Scholar]
- 34.Millar M, Lee J F, O'Sullivan T, Koch S A, Fikar R. Inorg Chim Acta. 1996;243:333–343. [Google Scholar]
- 35.Ueyama N, Nakata M, Nakamura A. Bull Chem Soc Jpn. 1981;54:1727–1730. [Google Scholar]
- 36.Anglin J R, Davison A. Inorg Chem. 1975;14:234–237. [Google Scholar]
- 37.Lane R W, Ibers J A, Frankel R B, Papaefthymiou G C, Holm R H. J Am Chem Soc. 1977;99:84–98. doi: 10.1021/ja00443a017. [DOI] [PubMed] [Google Scholar]
- 38.Farinas E, Regan L. Protein Sci. 1998;7:1939–1946. doi: 10.1002/pro.5560070909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benson D E, Wisz M S, Liu W, Hellinga H W. Biochemistry. 1998;37:7070–7076. doi: 10.1021/bi980583d. [DOI] [PubMed] [Google Scholar]
- 40.Berg J M, Merkle D L. J Am Chem Soc. 1989;111:3759–3761. [Google Scholar]
- 41.Krisek B A, Merkle D L, Berg J M. Inorg Chem. 1993;32:937–940. [Google Scholar]
- 42.Wuthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. [Google Scholar]
- 43.Pavone V. Int J Biol Macromol. 1988;10:238–240. [Google Scholar]
- 44.Pavone V, Gaeta G, Lombardi A, Nastri F, Maglio O, Isernia C, Saviano M. Biopolymers. 1996;38:705–721. doi: 10.1002/(SICI)1097-0282(199606)38:6%3C705::AID-BIP3%3E3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 45.Gunasekaran K, Ramakrishnan C, Balaram P. Protein Eng. 1997;10:1131–1141. doi: 10.1093/protein/10.10.1131. [DOI] [PubMed] [Google Scholar]
- 46.Pavone V, Di Blasio B, Santini A, Benedetti E, Pedone C, Toniolo C, Crisma M. J Mol Biol. 1990;214:633–635. doi: 10.1016/0022-2836(90)90279-U. [DOI] [PubMed] [Google Scholar]
- 47.Bertini I, Luchinat C. Adv Inorg Biochem. 1984;6:71–111. [PubMed] [Google Scholar]
- 48.Fasman G D, editor. Circular Dichroism and Conformational Analysis of Biomolecules. New York: Plenum; 1996. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.