

Abstract
BACKGROUND—Congenital chloride diarrhoea (CLD, OMIM 214700) is a serious inherited defect of intestinal electrolyte absorption transmitted in an autosomal recessive fashion. The major clinical manifestation is diarrhoea with high chloride content which can be balanced by substitution. The molecular pathology involves an epithelial Cl /HCO3 exchanger protein, encoded by the solute carrier family 26, member 3 gene (SLC26A3), previously known as CLD or DRA (downregulated in adenomas). To date, almost 30 different mutations in the SLC26A3 gene have been identified throughout the world. No clear genotype-phenotype correlation has been established. PATIENTS/METHODS—Two siblings presenting with CLD were studied for disease history, supplementation, or other treatments, and for mutations in the SLC26A3 gene. RESULTS—Mutation analysis revealed a homozygous I544N mutation in both patients. However, despite the uniform genetic background of CLD in this family, the clinical picture and outcome of the disease were remarkably different between siblings. The older sibling had a late diagnosis and chronic course of the disease whereas the younger one, who was diagnosed soon after birth and immediately received supplementation therapy, grows and develops normally. CONCLUSION—Time of diagnosis, substitution therapy, compliance, and compensatory mechanisms are more important modulators of the clinical picture of CLD than the type of mutation in the SLC26A3 gene. Keywords: chloride diarrhoea; SLC26A3 gene;
Full Text
The Full Text of this article is available as a PDF (128.1 KB).
Figure 1 .
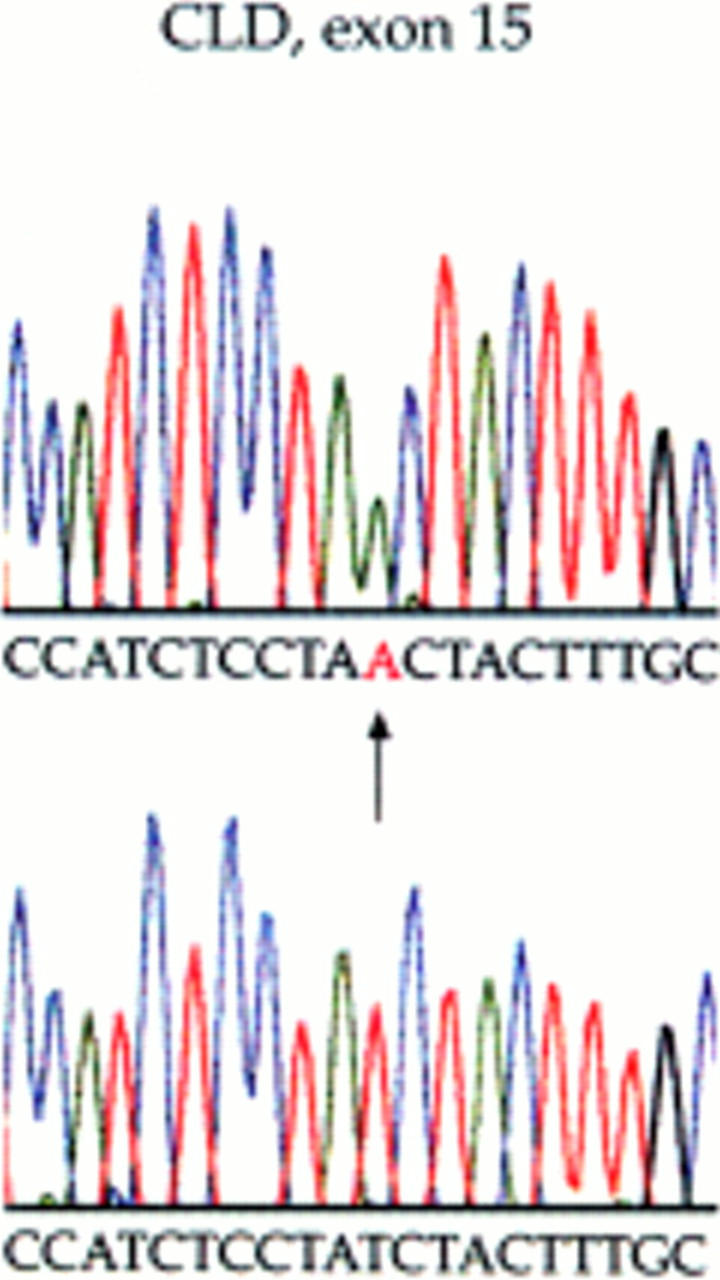
I544N mutation of the SLC26A3 exon 15 identified by sequencing in both siblings. (Top) Homozygous sequence change from patient No 1. (Bottom) Normal sequence from an unaffected control individual. The arrow shows the T to A mutation, changing an isoleucine residue at 544 to asparagine.