

Abstract
Generating human single-nucleotide polymorphisms (SNPs) is no longer a rate-limiting step for genetic studies of disease. The number of SNPs in public databases already exceeds 200,000, and the total is expected to exceed 1,000,000 within a year. Rather, progress is limited by the inability to genotype large numbers of SNPs. Current genotyping methods are suitable for studying individual loci or at most a handful at a time. Here, we describe a method for parallel genotyping of SNPs, called single base extension–tag array on glass slides, SBE-TAGS. The principle is as follows. SNPs are genotyped by single base extension (SBE), using bifunctional primers carrying a unique sequence tag in addition to a locus-specific sequence. Because each locus has a distinct tag, the genotyping reactions can be performed in a highly multiplexed fashion, and the resulting product can then be “demultiplexed” by hybridization to the reverse complements of the sequence tags arrayed on a glass slide. SBE-TAGS is simple and inexpensive because of the high degree of multiplexing and the use of an easily generated, generic tag array. The method is also highly accurate: we genotyped over 100 SNPs, obtaining over 5,000 genotypes, with approximately 99% accuracy.
The recent dramatic advances in genetics and genomics have opened unprecedented opportunities for understanding the consequences of genetic variation. It is now possible to generate extensive collections of single-nucleotide polymorphisms (SNPs), for both humans and experimental organisms (1). Genotyping large numbers of these SNPs in appropriate samples should lead to insights into complex genetic traits, including many common human diseases (2–4). However, these studies will require, at a minimum, the genotyping of hundreds or thousands of SNPs in thousands of samples (2). Thus, fulfilling this promise requires technologies to genotype SNPs efficiently and inexpensively.
A wide variety of technologies have been used to genotype SNPs, including single base extension [SBE, also called minisequencing or template-directed incorporation (5–8)], 5′ exonuclease assays such as TaqMan (9), oligonucleotide ligation (10), molecular beacons (11), differential hybridization (12, 13), and cleavage by a flap endonuclease [Invader (14)]. These methods have been successfully used to genotype small numbers of SNPs singly or at most a few at a time; however, most of these methods are difficult to adapt to studies involving hundreds or thousands of SNPs. To perform these large-scale genetic studies efficiently, highly parallel methods of genotyping SNPs are required.
We set out to design an efficient method for genotyping hundreds of SNPs in parallel. We chose SBE as a platform because it is compatible with multiplex genotyping and because primer extension with DNA polymerase can distinguish single nucleotide differences with high accuracy (6). In this paper, we describe a simple and inexpensive method, single base extension–tag array on glass slides (SBE-TAGS), by which many SNPs can be genotyped simultaneously with SBE; the multiplex SBE reaction is deconvoluted by hybridization to a generic, easily prepared microarray. SBE-TAGS has advantages over two recently described microarray-based methods that use SBE (15, 16): a single generic SBE-TAGS microarray can be used to genotype many different sets of SNPs, and the array can be inexpensively generated in a research laboratory setting. We show that the SBE-TAGS method provides highly accurate genotypes for over 100 SNPs and is thus suitable for a wide range of genetic studies that require large-scale SNP genotyping.
Materials and Methods
Samples.
DNA from inbred mouse strains (A/J, C57BL/6J, DBA/2J, and CAST/Ei) and F1 offspring (DBA/2J × C57BL/6J, A/J × C57BL/6J, CAST/Ei × DBA/2J, and CAST/Ei × C57BL/6J) was obtained as described (17). Human DNA was prepared from an ethnically diverse panel of cell lines from the Coriell cell repository as described previously (18).
Generic Tag Array.
We identified unique sequence tags from the bacteriophage λ genome, using PRIMER3.0 (http://www.genome.wi.mit.edu). The parameters were as follows: length, 25–27 bases; Tm, approximately 55°C at salt concentration of 50 mM; low self-similarity (fewer than three bases at primer ends, fewer than eight internal bases); G+C content of 40–60%; and no more than two consecutive identical bases. From this list of tags, the primers were chosen to have low similarity to each other. These tags were then compared, using blast (19), to The Institute for Genomic Research (http://www.tigr.org/tdb/hgi) and internal Whitehead Institute databases to generate 166 sequences not homologous to known human genes; the final list of tags can be found at www.genome.wi.mit.edu/publications/SBE-TAGS.
We reserved one tag for use as a positive control. The full-length reverse complements of the remaining tags were spotted on the slide to create the generic tag array. Spotted oligonucleotides were synthesized with a stretch of 15 dT residues at the 5′ end to facilitate attachment to the slide and were therefore 40–42 bases long. Unmodified oligonucleotides (100 μM in 3× SSC) mixed with the positive control oligonucleotide (10 μM) were spotted in quadruplicate onto poly-l-lysine-coated microscope slides (Labscientific, Livingston, NJ) or silane-treated slides (Telechem, Sunnyvale, CA), using a GMS417 microarrayer (Affymetrix, Woburn, MA) at a rate of three hits per spot, in a humidified chamber (40% relative humidity). After spotting, slides were prepared as described previously (20), with the denaturation step omitted, and stored under low humidity (Sanpia Dry Keeper). To determine cross-hybridization, the tags were divided into 18 groups of eight “rows” and 10 “columns.” Each group was labeled by using the SBE protocol below, with 2 μg of λ DNA as a template, except that all four 2′,3′-dideoxynucleoside triphosphates (ddNTPs) were labeled with carboxytetramethylrhodamine (TAMRA). For 2 of the 165 spots tested, fluorescent signal intensity that was >10% of the average positive signal was observed at incorrect spot locations (representing cross-hybridization), and the corresponding two sequence tags were not used.
SNPs and Primer Design.
The 72 mouse SNPs have been described previously (17). The set of 76 human SNPs was chosen from among those identified in a screen of 106 genes (18). Locations of polymorphisms, PCR primers, and SBE primers are available from the website (www.genome.wi.mit.edu/SNP/mouse). For the human loci, PCR primers were chosen using PRIMER3.0 to have a Tm near 60°C, a G+C content of 20–80%, and a length of 16–25 bases with a product size of 30–120 bases. PCR primers for the mouse loci have been described previously (17). SBE primers were designed to be 16–25 bases long with a Tm of approximately 55°C, terminating at the base 5′ to the SNP. All human primers can be found on our website. By convention, the SBE primer was designed on the coding strand where possible.
Pairing SBE Primers with Tags and Grouping SBE Primers.
Each SBE primer (length 16–25 bases) was initially paired randomly with one of the 163 tags (length 25–27 bases) to generate a hybrid tag-SBE primer sequence containing the tag at the 5′ end (length 41–52 bases). An algorithm to predict foldback of these long primers was developed empirically based on examination of results from a test set of 96 tag-SBE primers; the optimal parameters were based on linear regression. The algorithm calculates a score for each potential site of internal priming by comparing the sequence at the 3′ end of the primer with potential complementary sequences in the body of the primer:
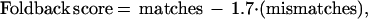 |
with an additional 0.8 subtracted for a mismatch in the penultimate base. To qualify as a complementary sequence, the final base in the primer must match and a minimum of one base is required in the hairpin turn between the two complementary sequences. Primers with foldback scores greater than 3 were redesigned by pairing the SBE primer with a different tag. If the foldback occurred within the SBE primer itself, the SBE primer was designed on the other strand, if possible.
Primers giving signal in SBE reactions lacking a template were considered to have foldback and were redesigned by pairing the primer with a different sequence tag. To prevent cross-priming in multiplex SBE, SBE primers were grouped so that no pair of primers gave a foldback score of >8, using the above algorithm (the cut-off score is higher for cross-priming than for foldback, because the degree of complementarity required to observe a monomolecular foldback event is less than that required for a bimolecular cross-priming event). To test for cross-priming within a group, multiplex SBE was performed with no template. Primers giving signal were redesigned using an SBE primer on the other strand; in all cases, this redesign successfully prevented cross-priming.
Multiplex PCR and SBE.
For the human SNPs, individual PCR products were amplified in 15 μl [1× PCR buffer II: 1.5 mM MgCl2/0.1 mM dNTPs/0.75 unit of AmpliTaq Gold (Perkin–Elmer)/84 nM primers/12.5 ng of DNA]. After incubation for 10 min at 96°C, 35 cycles of 96°C for 30 s, 50°C for 1 min, and 72°C for 1 min were performed. Approximately 30 PCR products were pooled, and 10 μl of the pooled product was used as a template for multiplex SBE. To remove excess dNTPs and primers, a mix of 10 μl was added, containing 2 units of shrimp alkaline phosphatase (Roche Molecular Biochemicals) and 4 units of exonuclease I (Amersham) in 75 mM Tris⋅HCl, pH 8.5/7.5 mM MgCl2 with incubation at 37°C for 45 min followed by 96°C for 15 min. Ten microliters of the treated PCR products was added to 5 μl of SBE mix containing 100 mM Tris⋅HCl (pH 9.5), 4 mM MgCl2, 1 pmol of each tag-SBE primer, 2 units of Thermosequenase (Amersham Pharmacia), and 5 pmol each of TAMRA-ddATP, TAMRA-ddCTP, cyanine 5 (Cy5)-ddGTP, and carboxy-X-rhodamine (ROX)-ddUTP (NEN Life Sciences). For multiplex PCR, amplification and streptavidin-biotin purification of PCR products were performed as described previously (17), and purified multiplex PCR products were resuspended in 15 μl of the SBE mix containing 50 mM Tris (pH 9.5), 2 mM MgCl2, plus primers, enzyme, and dye-ddNTPs as above. The SBE reaction consisted of 30 cycles of primer extension (96°C for 30 s, 50°C for 30 s, and 60°C for 1 min).
SBE products were added to the microarray in a total volume of 20 μl containing 1.33× SSC, 0.067% SDS, 0.033 mg/ml salmon sperm DNA, and 56 nM 5-carboxyfluorescein (FAM)-labeled positive control oligonucleotide. After the addition of a coverslip (Lab Scientific), the slide was placed in a small, sealed hybridization chamber (Eastern Tool, Cambridge, MA) with 55 μl of water to limit evaporation and hybridized at 50°C for 4 h. The arrays were washed in 2× SSC/0.1% SDS for 5 min at room temperature, followed by three brief rinses in 2× SSC and a final 1-min rinse in 0.2× SSC. Slides were centrifuged at 500 rpm for 15 min to remove residual wash solution.
Analysis of Tag Array Data.
Arrays were scanned at 20-μm resolution, using the ScanArray 5000 (GSI Lumonics, Billevica, MA) with an external argon laser at the following settings: FITC, laser 100%, photomultiplier tube (PMT) 90%; TAMRA, laser 80%, PMT 80%; ROX, laser 90%, PMT 90%; Cy5, laser 80%, PMT 80%. The FITC laser was used to measure the FAM fluorescent signal. After the local background in each of the channels was subtracted, the signal was normalized to the intensity of the FAM-labeled positive control oligonucleotide at each spot. Spots where the positive control signal was quite faint (<10% of the average for the slide) were treated as missing.
Because the multiple fluors have overlapping excitation and emission spectra, a matrix was applied to correct for the small amount of “cross-talk” (signal from one dye detected with more than one filter set). This matrix is dependent on the equipment and settings used and must be determined empirically for each scanning protocol. To determine this 3 × 3 correction matrix, a slide with three “pure dye” samples, each containing one of the three dyes (TAMRA, ROX, and Cy5), was scanned using each of the three filter sets. For each sample, the data from each filter set were expressed as the fraction of the signal intensity obtained with the filter set corresponding to the dye present in the sample. These data were then arranged as a 3 × 3 matrix with the diagonal elements equal to 1. The correction matrix is simply the inverse of this 3 × 3 matrix. The normalized data from the TAMRA, ROX, and Cy5 channels were multiplied by the correction matrix to generate corrected “pure dye” values. To correct for the different brightness and efficiency of incorporation of the dye terminators, we multiplied the Cy5 matrix-corrected signal by 0.5 and the TAMRA matrix-corrected signal by 1.5 for SNPs containing a C. These values were also determined empirically, using known heterozygous samples (data not shown).
After these adjustments, the relative fractions of the total signal contributed by each of the two expected dyes were determined, and the following genotype score was calculated:
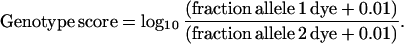 |
The genotype score ranges from approximately −2 to 2, with heterozygotes having values between −1 and +1 and homozygotes generally above +1 or below −1.
To generate criteria for determining genotypes, boundary conditions were determined as follows. For each SNP, we analyzed data from all of the genotyped samples; for the human SNPs, we also included three composite “reference” samples, each consisting of examples of one of the three possible genotypes for as many SNPs as possible. Only slides and data points with signal above threshold were used. An automated clustering algorithm was used to group the scores for each SNP into clusters (representing genotypes) and define boundaries between these clusters; if the algorithm failed to find adequately separated genotype clusters, the SNP was not analyzed further. Upper and lower limits for the boundaries were set at ±1. These limits were chosen because we found that heterozygote samples rarely gave scores outside this range (a score of ±1 corresponds to a ratio of the two dyes of 10:1).
Slides and data points with signal above threshold were subjected to automated genotype calling, using these boundaries. Each SNP was represented by four replicate spots on the array; for a genotype to be called, the genotypes at all four replicate spots had to agree. Slides with low overall hybridization signals were excluded (approximately 5–10% of slides), and all spots on these slides were considered failures. For each slide, the average signal over the four replicate spots was required to be >25% of the average signal for those spots taken over all slides. Although this threshold eliminates only 1–2% of data points, application of this threshold was important because spots with unexpectedly low signal sometimes represented failed reactions accompanied by high background, resulting in a score unrelated to the true genotype. Spots above this signal threshold with all four genotype scores above the first boundary were called allele 1 homozygotes, spots with four genotype scores below the second boundary were called allele 2 homozygotes, and spots with four genotype scores between the two boundaries were called heterozygotes. Although we used four replicate spots in these experiments, examination of our data suggests that duplicate spots would give similar results, as long as they were well separated spatially on the array.
Results
Design of the SBE-TAGS Method.
SBE (5, 21) involves extension of a primer located adjacent to the position of a SNP, using DNA polymerase in the presence of fluorescently labeled ddNTPs (Fig. 1 Upper). We modified the method by uniquely marking each primer with distinct sequence tags to the 5′ end, allowing separation of the multiplex SBE reaction by hybridization to a microarray (Fig. 1 Lower). Because the sequence tags are unrelated to the particular set of SNPs to be genotyped, we were able to design a generic microarray that could be used for any collection of SNPs.
Figure 1.

Schematic depiction of SBE-TAGS genotyping. (Upper) In SBE, a hybrid primer containing a generic sequence tag followed by a locus-specific sequence is hybridized adjacent to the SNP and extended with fluorescent dideoxynucleotides. (Lower) Multiple SBE reactions are performed in solution; each SBE primer is marked by a different unique sequence tag. The multiplex reaction is analyzed after hybridization to a generic tag array. The array is generated by spotting the reverse complements of the sequence tags onto a glass microscope slide.
To select tags, we first identified sequences that were predicted to have similar melting temperatures but not to cross-hybridize with one another (see Materials and Methods). The tags were then tested experimentally for cross-hybridization, by labeling small subsets of the tags and hybridizing them to a microarray containing the reverse complements of the entire set. A set of 163 suitable tags was selected in this fashion. We then considered how to assign tags to specific SBE primers to maximize the success of multiplex genotyping. Two potential problems must be avoided. The primers should not have self-complementarity between their 3′ end and an internal sequence, because this could promote unimolecular self-priming events or “foldback” (17); this is an important issue, given that the hybrid primers are relatively long (≈45 bases). Similarly, the set of primers should not have cross-complementarity, to avoid “cross-priming.” Both problems can be recognized by testing for the incorporation of fluorescently labeled nucleotides in the absence of genomic template. With random matching of SBE primers and tags, 25% of the oligonucleotides showed foldback or cross-priming. We designed an algorithm to match SBE primers with tags and to assemble them into multiplex sets (see Materials and Methods); this algorithm reduced the occurrence to less than 5%.
We chose a set of dye-labeled ddNTPs to allow all possible SNPs to be genotyped in the same reaction. It suffices to use three dyes, with the same dye attached to both ddATP and ddCTP. For A/C polymorphisms, the SBE primer can be designed on the opposite strand, converting the SNP to a G/T polymorphism. We tested a variety of dye-ddNTP combinations and found that Cy5-ddGTP, ROX-ddUTP, TAMRA-ddATP, and TAMRA-ddCTP gave adequate spectral separation and minimal misincorporation (data not shown).
Accuracy of the SBE-TAGS Method.
To test the SBE-TAGS genotyping method, we designed hybrid tag-SBE primers for a set of 76 human SNPs that could be amplified using a single set of PCR conditions. We individually amplified these 76 SNPs from 82 samples and combined the amplicons from each sample into three pools, each containing approximately 25 SNPs. For each sample, multiplex SBE reactions were then performed on the three pools, and the resulting products were hybridized to glass slides containing the generic tag array. We used a clustering algorithm (see Materials and Methods) to call the genotypes; representative SNP loci are shown in Fig. 2. For 60 of 76 SNPs, the data met stringent quality control criteria for signal intensity and clustering and were subjected to automatic genotype calling. Sixteen SNPs were excluded: 2 gave no signal and 14 showed poor separation between genotype clusters. Notably, this poor separation does not reflect a problem related to the array, because these 14 assays also performed poorly in a different SBE-based genotyping method [fluorescence polarization (8)]; our previous experience with SBE suggests that designing the SBE primer on the opposite strand (where possible) would rescue a substantial fraction of these assays (S.B., J.N.H., and P.S., unpublished results). For these 60 SNPs, 96% of genotypes were successfully called (4,709/4,920 possible genotypes).
Figure 2.

Clustering of genotyping scores for 10 representative SNPs. SNPs were randomly selected from among those demonstrating all three genotypes. Each symbol represents the average genotype score and signal intensity of the quadruplicate spots for a particular locus. ♦, Homozygotes for allele 1; ■, heterozygotes; ▴, homozygotes for allele 2; x, no genotype called. Shown on the y axis is the normalized signal, and on the x axis, the genotyping score. Dotted lines represent the boundaries between genotypic classes generated by the clustering algorithm; these boundaries were subsequently used for automated genotyping.
The genotypes obtained with SBE-TAGS were extremely accurate, as determined by comparison with genotypes previously obtained by fluorescence polarization (8) (shown to be 99% accurate by comparison with direct sequencing; S.B., J.N.H., and P.S., unpublished results). Where the two methods gave different genotypes, the discrepancy was resolved by direct sequencing. Of 1,244 genotypes for which data were available from both methods, the methods agreed in 1,226 cases. In the 18 discrepant cases, SBE-TAGS was correct 13 times, for an accuracy of 99.6% (1,239/1,244).
We next attempted to further increase the efficiency and throughput of SBE-TAGS by employing multiplex reactions for the PCR amplification, in addition to the SBE. For this purpose, we used a set of 72 mouse SNPs known to differ between the A/J and C57BL/6J strains and for which multiplex PCR primers had been designed previously (17). The SNPs were divided into three groups for multiplex amplification followed by multiplex SBE. The resulting products were then pooled and hybridized to the generic tag array. We genotyped five strains of mice: three homozygotes (A/J, C57BL/6J, DBA/2J) and two F1 hybrids (A/J × C57BL/6J and DBA/2J × C57BL/6J). Each mouse was genotyped in quintuplicate (Fig. 3). A total of 53 SNPs met our stringent quality control criteria and were subjected to automatic genotype calling (6 gave poor signal and 13 showed insufficient separation between genotype clusters). The call rate was lower than that for the set of human SNPs (Table 1), likely because of variable and/or less efficient amplification at the multiplex PCR step. The genotypes were compared with the known genotypes for these strains (17), and the accuracy rate was 98% (Table 1).
Figure 3.

Examples of data from SBE-TAGS genotyping. Data are shown for two spots on the array for each of three mice. B6, B6 × A/J, A/J: each set of three squares represents data on DNA from the indicated mouse. Cyanine 5, ROX, TAMRA: For each dye, the color intensity shown represents the “pure dye” signal strength after matrix correction to eliminate “cross-talk” between dyes (see Materials and Methods). B6, C57BL/6J.
Table 1.
Results of genotyping using SBE-TAGS
| SNPs | No. of genotypes
|
Accuracy, % | |
|---|---|---|---|
| Attempted | Called | ||
| Human (60) | 4,920 | 4,709 | 99.6 |
| Mouse (53) | 1,325 | 1,062 | 98 |
To calculate accuracy, a substantial fraction of the genotypes determined by automated, blinded SBE-TAGS genotyping were also determined by using an independent method; the small number of discrepancies were resolved by direct sequencing.
SBE-TAGS is thus highly accurate. As with all such genotyping methods, inaccurate genotypes can result if the binding sites for the PCR or SBE primers are themselves polymorphic. To explore this issue, we genotyped F1 mice from crosses between Mus musculus domesticus and Mus musculus castaneus, the genomes of which diverge by 0.5% (17). Five strains of mice were genotyped in quadruplicate: three homozygotes (C57BL/6J, DBA/2J, CAST/Ei) and two F1 hybrids (C57BL/6J × CAST/Ei and DBA/2J × CAST/Ei). Most of the loci could still be genotyped successfully, but we observed a slightly higher error rate. Direct sequencing revealed that these additional errors indeed occurred at five loci where the PCR or SBE primers were located over sites of sequence difference between the two subspecies (data not shown). These results underscore the importance of gathering and using sequence information about nearby polymorphisms.
Discussion
Many genetic studies require the genotyping of hundreds of SNPs in thousands of individuals, but such studies are currently difficult and expensive, even for well-equipped laboratories. The SBE-TAGS method should make such experiments possible. The ability to genotype SNPs with multiplex reactions and easily generated generic arrays makes the approach rapid and inexpensive. The arrays can be readily generated by depositing unmodified oligonucleotides on a glass slide with routine spotting equipment. The materials cost of the array is approximately $2, with half the cost for the treated glass slides and half for the oligonucleotides. Other reagent costs are relatively low, because of the high degree of multiplexing. In this regard, the method compares favorably with methods for genotyping individual SNPs, such as Invader (14) and TaqMan (9).
In parallel with our development of SBE-TAGS, colleagues at Affymetrix and Case Western Reserve have been pursuing a related approach and have recently described their results (15). This approach is similar to SBE-TAGS but relies on microarrays synthesized by photolithography (“DNA chips”; refs. 22 and 23) and a two-color detection scheme that requires distinct single base extension reactions for each pair of bases. In contrast, SBE-TAGS uses inexpensive spotted microarrays that require no special equipment for processing, and the three-color detection scheme allows all types of SNPs to be genotyped in a single tube.
Other SNP genotyping methods using microarray technology have been described. One method (16) uses arrays containing locus-specific SBE primers (rather than using a generic array) and requires that the SBE reaction occur on a solid support (which is somewhat more difficult than performing the reactions in solution, as in SBE-TAGS). An array-based method using allele-specific ligation has also been described (24), but the throughput of this method remains uncertain, and the requirement for locus-specific fluorescent primers adds to the cost. Finally, high-density microarrays have been used to genotype SNPs in parallel by differential hybridization to oligonucleotide probes (12). Unlike SBE-TAGS, however, this method requires the synthesis of a different array for each set of SNPs. Thus, SBE-TAGS compares favorably with other array-based genotyping methods.
We believe SBE-TAGS can make large-scale SNP genotyping accessible to a wide range of researchers. Because of its accuracy, ease, and potential for high throughput, SBE-TAGS should facilitate genetic studies that previously required a very substantial commitment of resources, including linkage disequilibrium mapping to pursue promising findings from linkage analysis, extensive candidate gene association studies, loss of heterozygosity studies, and genotyping of large crosses in experimental organisms.
Acknowledgments
We thank G. Tsan for technical assistance, J. Ireland for initial SBE primer design, and C. Cowles and J. Singer for providing the F1 DNAs. The research was supported by a research contract from Bristol-Myers Squibb, Millennium Pharmaceuticals, and Affymetrix (E.S.L.). J.N.H. is the recipient of a Howard Hughes Medical Institute Postdoctoral Fellowship for Physicians. P.S. is the recipient of a Young Investigator Award from the National Alliance for Research on Schizophrenia and Depression.
Abbreviations
- SNPs
single-nucleotide polymorphisms
- SBE
single base extension
- SBE-TAGS
single base extension–tag array on glass slides
- ddNTP
2′,3′-dideoxynucleoside triphosphate
- TAMRA
carboxytetramethylrhodamine
- ROX
carboxy-X-rhodamine
- FAM
5-carboxyfluorescein
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.210394597.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.210394597
References
- 1.Altshuler, D., Pollara, V. J., Cowles, C., Van Etten, W. J., Baldwin, J., Linton, L. & Lander, E. S. Nature (London), in press. [DOI] [PubMed]
- 2.Risch N, Merikangas K. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 3.Lander E S. Science. 1996;274:536–539. doi: 10.1126/science.274.5287.536. [DOI] [PubMed] [Google Scholar]
- 4.Collins F S, Guyer M S, Charkravarti A. Science. 1997;278:1580–1581. doi: 10.1126/science.278.5343.1580. [DOI] [PubMed] [Google Scholar]
- 5.Syvanen A C, Aalto-Setala K, Harju L, Kontula K, Soderlund H. Genomics. 1990;8:684–692. doi: 10.1016/0888-7543(90)90255-s. [DOI] [PubMed] [Google Scholar]
- 6.Pastinen T, Kurg A, Metspalu A, Peltonen L, Syvanen A C. Genome Res. 1997;7:606–614. doi: 10.1101/gr.7.6.606. [DOI] [PubMed] [Google Scholar]
- 7.Chen X, Zehnbauer B, Gnirke A, Kwok P Y. Proc Natl Acad Sci USA. 1997;94:10756–10761. doi: 10.1073/pnas.94.20.10756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen X, Levine L, Kwok P Y. Genome Res. 1999;9:492–498. [PMC free article] [PubMed] [Google Scholar]
- 9.Livak K J, Marmaro J, Todd J A. Nat Genet. 1995;9:341–342. doi: 10.1038/ng0495-341. [DOI] [PubMed] [Google Scholar]
- 10.Tobe V O, Taylor S L, Nickerson D A. Nucleic Acids Res. 1996;24:3728–3732. doi: 10.1093/nar/24.19.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tyagi S, Bratu D P, Kramer F R. Nat Biotechnol. 1998;16:49–53. doi: 10.1038/nbt0198-49. [DOI] [PubMed] [Google Scholar]
- 12.Wang D G, Fan J B, Siao C J, Berno A, Young P, Sapolsky R, Ghandour G, Perkins N, Winchester E, Spencer J, et al. Science. 1998;280:1077–1082. doi: 10.1126/science.280.5366.1077. [DOI] [PubMed] [Google Scholar]
- 13.Howell W M, Jobs M, Gyllensten U, Brookes A J. Nat Biotechnol. 1999;17:87–88. doi: 10.1038/5270. [DOI] [PubMed] [Google Scholar]
- 14.Mein C A, Barratt B J, Dunn M G, Siegmund T, Smith A N, Esposito L, Nutland S, Stevens H E, Wilson A J, Phillips M S, et al. Genome Res. 2000;10:330–343. doi: 10.1101/gr.10.3.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan J B, Chen X, Halushka M K, Berno A, Huang X, Ryder T, Lipshutz R J, Lockhart D J, Chakravarti A. Genome Res. 2000;10:853–860. doi: 10.1101/gr.10.6.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pastinen T, Raitio M, Lindroos K, Tainola P, Peltonen L, Syvanen A C. Genome Res. 2000;10:1031–1042. doi: 10.1101/gr.10.7.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindblad-Toh K, Winchester E, Daly M J, Wang D G, Hirschhorn J N, Laviolette J P, Ardlie K, Reich D E, Robinson E, Sklar P, et al. Nat Genet. 2000;24:381–386. doi: 10.1038/74215. [DOI] [PubMed] [Google Scholar]
- 18.Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Shaw N, Lane C R, Lim E P, Kalyanaraman N, et al. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- 19.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 20.DeRisi J L, Iyer V R, Brown P O. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 21.Landegren U, Nilsson M, Kwok P Y. Genome Res. 1998;8:769–776. doi: 10.1101/gr.8.8.769. [DOI] [PubMed] [Google Scholar]
- 22.Lipshutz R J, Fodor S P, Gingeras T R, Lockhart D J. Nat Genet. 1999;21:20–24. doi: 10.1038/4447. [DOI] [PubMed] [Google Scholar]
- 23.Chee M, Yang R, Hubbell E, Berno A, Huang X C, Stern D, Winkler J, Lockhart D J, Morris M S, Fodor S P. Science. 1996;274:610–614. doi: 10.1126/science.274.5287.610. [DOI] [PubMed] [Google Scholar]
- 24.Gerry N P, Witowski N E, Day J, Hammer R P, Barany G, Barany F. J Mol Biol. 1999;292:251–262. doi: 10.1006/jmbi.1999.3063. [DOI] [PubMed] [Google Scholar]