

Abstract
The neural crest gives rise to numerous cell types, including Schwann cells, neurons, and melanocytes. The extent to which adult neural crest-derived cells retain plasticity has not been tested previously. We report that cutting adult mouse sciatic nerve induces pigmentation around nerve fascicles, among muscle bundles, and in the hypodermis. Pigmented cells are derived from adult nerve, because pigmentation occurs even when nerve fragments are grafted into tyrosinase null albino mice. Pigmentation defects are pervasive in patients with neurofibromatosis type 1 (NF1). Mice hemizygous for Nf1 mutations show enhanced pigmentation after nerve lesion and occasionally form pigmented and unpigmented tumors. TheNf1 nerve and the Nf1 host environment both contribute to enhanced pigmentation. Grafted purifiedNf1 mutant glial cells [S100+–p75NGFR+–GFAP+–EGFR+or S100+–p75NGFR+–GFAP+–EGFR−] mimic nerve-derived pigmentation. The NF1 protein, neurofibromin, is a Ras-GAP that acts downstream of a few defined receptor tyrosine kinases, including [β-common (βc)] the shared common receptor for granulocyte and monocyte colony-stimulating factor, interleukin-3 (IL3), and IL5. Cytokines in the environment have the potential to suppress pigmentation as shown by nerve injury experiments in null mice; when is βc absent orNf1 is mutant, melanogenesis is increased. Thus, the adult nerve glial cell phenotype is maintained after nerve injury by response to cytokines, through neurofibromin.
Keywords: Schwann cell, melanocyte, NF1, transdifferentiation, GMCSF, injury, stem cell
Neural crest-derived cells differentiate into numerous derivatives, including neurons, melanocytes (pigment cells of the skin and iris), and peripheral nerve glia, called satellite cells and Schwann cells (Le Dourain and Kalcheim, 1999). The fates of neural crest cells, as with other embryonic cell types, become progressively restricted as development proceeds. Neural crest cells that form the dorsal root ganglia (DRGs) normally develop into sensory neurons, satellite cells, and Schwann cells but not pigment cells. It is not known to what extent developmental restrictions can be reversed or whether unrestricted cells persist in adult nerves.
Melanocytes and Schwann cells can arise from a bipotential Schwann cell–melanocyte precursor (Dupin et al., 2000). Avian DRG and spinal nerve glia can be stimulated to make pigment at early developmental stages (Nichols and Weston, 1977; Nichols et al., 1977; Ciment et al., 1986; Stocker et al., 1991; Nataf and Le Douarin, 2000). Differentiated melanocytes from embryonic day 7 (E7) avian skin can lose melanocytic properties and exhibit glial markers (Dupin et al., 2000). This is believed to result from transdifferentiation, that is, loss of differentiation properties followed by acquisition of markers specific to a different cell phenotype (Eguchi and Kodama, 1993). Persistence of stem cells in peripheral nerve may also account for phenotype plasticity. Neural crest stem cells persist in nerve and can form neurons, glia, and smooth muscle–mesenchymal cells through embryo day 14 (Morrison et al., 1999). Neither stem cells nor transdifferentiation of Schwann cells has been observed in adult peripheral nerve.
Injury to adult peripheral nerve causes loss of differentiated Schwann cell phenotypes (Fawcett and Keynes, 1990; Scherer, 1997) and partially recapitulates the developmental cytokine environment (Grotendorst, 1992). Here we used sciatic nerve injury in genetically altered mice to test whether peripheral nerve cells can be induced to differentiate along alternative pathways.
Neurofibromatosis type 1 (NF1) patients develop neurofibromas, peripheral nerve tumors containing mainly Schwann cells, with infrequent pigmented cells, and show skin hyperpigmentation (Riccardi, 1992; Fetsch et al., 2000). Neurofibromin, the product of theNf1 gene, is expressed in neural crest cells and Schwann cells (Daston and Ratner, 1992; Daston et al., 1992; Stocker et al., 1995), suggesting a role in the Schwann cell–melanocyte lineages. Neurofibromin is an essential Ras-GAP in certain cell types, acting downstream of particular tyrosine kinase receptors (Cichowski and Jacks, 2001). Ras-GTP levels, in response to particular cytokines and growth factors, are abnormally high in Nf1 mutant cells (Kim et al., 1995; Vogel et al., 1995; Lakkis et al., 1999; Wehrle-Haller et al., 2001). Nf1 mutant hemaotpoetic cells show enhanced response to granulocyte and monocyte colony-stimulating factor (GMCSF) and interleukin-3 (IL3), which signal through a common β receptor [β-common (βc)] (Bollag et al., 1996; Largaespada et al., 1996; Zhang et al., 1998; Birnbaum et al., 2000; Ingram et al., 2000). Neurofibromin-dependent changes in the GMCSF–IL3 signaling pathway could be relevant to peripheral nerve cells because both cytokines are upregulated after nerve lesion (Saada et al., 1996).
We demonstrate that wounding causes pigmentation of nerve-derived glial cells and that the βc receptor, viaNf1, normally suppresses melanogenesis after injury.
MATERIALS AND METHODS
Mouse strains. Male wild-type andNf1 heterozygous (+/−) mice were backcrossed onto the C57BL/6 background; they were derived and genotyped as described previously (Brannan et al., 1994). Mice null for Nf1 diein utero. Mice null for βcwere on the C57BL/6 background and were genotyped as described previously (Nishinakamura et al., 1995). In some experiments, albino C57BL/6 mice with a spontaneous mutation in tyrosinase C57BL/6J-TYRC-2J were used (The Jackson Laboratory, Bar Harbor, ME).
Morphology of the embryonic and adult sciatic nerve.Pregnant mice at day 18–20 of gestation were anesthetized by inhalation of metafane, and the embryos were removed and transcardially perfused for 5 min with EM fixative. Mice at postnatal day 0 (P0), P1, P3, P7, and P30 were anesthetized with sodium pentobarbital and perfused, as were 10-month-old mice. Sciatic nerves were dissected, postfixed, and processed for semithin sections, and EMs (n = 3 each age group per genotype) were evaluated.
Nerve wounding. Wild-type and Nf1+/− mice (3–4 months old) were anesthetized with avertin (0.5–0.8 ml, i.p.). Skin overlying the sciatic nerve was cut, the muscles were parted, and the sciatic nerve was exposed. After surgeries, muscles were realigned, and the skin was closed with surgical staples. Animals were allowed to survive 7 d to 1 year after nerve surgery and were then anesthetized with sodium pentobarbital and perfused with 4% paraformaldehyde. Sciatic nerve, skin, or muscle were dissected, postfixed, and processed for paraffin embedding and light microscopy, postfixed in glutaraldehyde and processed for electron microscopy, or cryopreserved and sectioned. For nerve crush, sciatic nerve 2 mm medial from the sciatic notch was crushed by mechanical pressure three times with a sharp pair of #5 Dumont tweezers. Animals were evaluated after 6 (n = 9 per genotype) or 10 (n = 9 per genotype) weeks after crush. For nerve transection, the sciatic nerve was cut 2 mm medial to the sciatic notch, and cut ends of the nerve were sutured together using prolene sutures (7–0). Animals were allowed to survive for 1 (n = 48 per genotype), 3 (n = 22 per genotype), or 6 (n = 6 per genotype) months or 1 year (n = 4 per genotype) after surgery. For nerve transection and deflection, the sciatic nerve was cut 2 mm medial to the sciatic notch, and the cut ends were deflected to opposite sides and then secured under muscle masses. Animals were allowed to survive for 1 (n = 15 per genotype) or 3 (n = 15 per genotype) months.
Quantification of pigmentation. For counting pigmented patches, after perfusion, the animal's skin was removed, and the gross thigh area was viewed under a dissecting microscope (Wild) at 40× magnification. Pigmented patches appeared as collections of streaks on the fascia overlying muscle and nerve that likely represent clones of differentiated melanocytes. Each streak was counted in each animal. This analysis represents an underestimate of total melanocytes.
To measure melanosomes, electron micrographs were generated of hypodermal pigmented cells and skin melanosomes, >235 melanosomes were traced, and areas were measured on a Zidas imaging pad.
Histology and immunocytochemistry. Paraffin sections (5- to 6-μm-thick) were cut and processed for hematoxylin and eosin (H&E) or Gomori's trichrome. For immunostaining, these sections were stained with polyclonal rabbit anti-S100 (1:5000; Dako, Carpinteria, CA) to mark Schwann cells, or mouse monoclonal anti-neurofilament (15GI; 1:1) or polyclonal anti-neurofilament (NF 178.3; 1:500; a gift from L. Parysek, University of Cincinnati, College of Medicine, Cincinnati, OH) to mark axons. Bromodeoxyuridine (BrdU) labeling was as described by Weiler and Farbman (1997). Paraffin sections were processed using an anti-BrdU kit (Zymed, San Francisco, CA). The number of BrdU-positive nuclei were counted in triplicate sections from each animal. To stain macrophages, unfixed nerves were sectioned on a cryostat and stained with F40 as described previously (Perry et al., 1995).
Nerve grafts. Adult mice were anesthetized, and sciatic fragments (1 cm) were removed. Nerves were rinsed in DMEM, incubated in DMEM with 10% FBS and 10 μg/ml Hoechst 33342 dye (Sigma, St. Louis, MO) for 1 hr at 37°C, rinsed in DMEM, and chilled on ice for 30 min. A second mouse (recipient) was anesthetized, the sciatic nerve was exposed, the nerve was transected, and Hoechst dye-labeled nerve fragment was grafted between the cut ends of the recipient sciatic nerve by suturing one end of recipient nerve to donor-labeled nerve on each side. This method is based on that of Aguayo et al. (1979). The grafted nerve was then transected, and each end of the dye-labeled nerve fragment was deflected. Animals (n = 9 per genotype) were killed 30 d after surgery and fixed by perfusion, the pigmented area was excised and cryoprotected, and sections were cut and then analyzed for pigmentation and dye-labeled nuclei.
Cell grafting. Wild-type, Nf1+/− andNf1−/− Schwann cells were prepared from E12.5 DRGs (Kim et al., 1995) and used at passage 2–3. Morphologically transformed hyperproliferative cells derived from −/− Schwann cells (−/−TXF) were purified as described previously (Kim et al., 1997) and used at passage 2–4. Wild-type and Nf1−/− fibroblasts were prepared from E12.5 torsos as described previously (Atit et al., 1999) and used at passage 2. Twenty thousand cells were plated onto matrigel-coated transwell filters (Kim et al., 1997) for 48 hr and then labeled with Hoechst dye (see above). Control filters processed in parallel were analyzed to ensure that cells were labeled before grafting. Filters were grafted around the transected area of the nerve (n = 4 per cell type into wild-type mice;n = 4 into heterozygous mice). After 30 d, animals were fixed by perfusion, and cryostat sections from pigmented areas were evaluated for pigmentation and for dye-positive nuclei.
Characterization of Nf1−/− TXF cells. Cells plated onto LabTek (Andover, MA) slides were immunostained after fixation in 4% paraformaldehyde with rat anti-mouse p75NGF receptor (NGFR) (1:10; a kind gift from D. Anderson, California Institute of Technology, Pasadena, CA), rabbit anti-S100 (1:200; Dako), rabbit anti-GFAP (1:1000; Dako), and mouse anti-smooth muscle actin (SMA) (Sigma). Cells were rinsed, incubated in FITC-labeled secondary antibody (1:200; Jackson ImmunoResearch, West Grove, PA), rinsed, and cover glassed. Immunostaining was visualized by fluorescence microscopy.
RESULTS
To test the hypothesis that trauma can alter the differentiation of neural crest-derived cells in peripheral nerves, we examined the effects of three injury paradigms (nerve crush, nerve cut and suture, and nerve cut with deflection of cut ends) on pigmentation.
Severe nerve injury induces pigmentation around peripheral nerves
One month after adult mouse sciatic nerves were cut and cut ends were resutured (n = 48 per genotype), pigmented cells were observed on gross dissection. Clusters of highly pigmented cells extended up to 1 cm proximal and 1 cm distal to the transection site (Fig. 1). Wild-type mice consistently showed barely detectable groups of pigmented cells (Fig.1A). Nf1 mutant mouse littermates showed a slightly enhanced pigmentation response (Fig. 1B). Pigmented patches were counted under a dissecting microscope in six wild-type and 10 Nf1+/− animals, as described in Materials and Methods. The number of patches in wild type was 46.3 ± 7.6, whereas in mutants it was 112 ± 30.1 (p = 0.0001). An increase in pigmentation and a difference between wild-type and mutant animals was easily detectable after nerve cut and deflection (n = 15 per genotype) (Fig.1C,D). Quantification of pigmented patches in three animals of each genotype showed 133.3 ± 15.5 (wild type) and 360.3 ± 51.4 (Nf1+/−; p =0.006). Of 15 mutant animals subjected to nerve cut with deflection, eight showed massive pigmentation that was too great to quantify, as documented in Figure 1D; 0 of 15 wild-type mice showed this response.
Fig. 1.
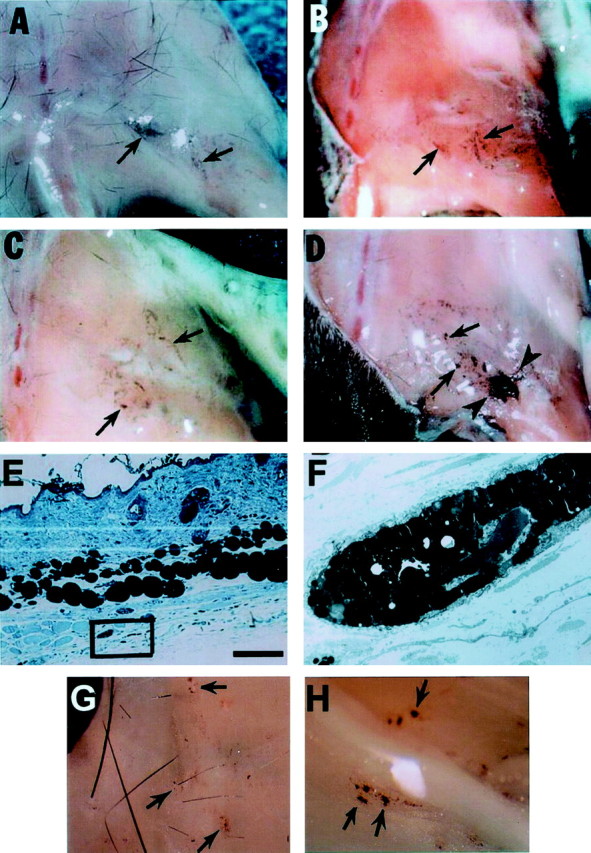
Hyperpigmentation in C57BL/6 adult mice after nerve injury. A–D, Gross photographs of mice hindlimb areas 1 month after nerve transection and/or deflection. Skin has been removed. A, Wild-type mouse with nerve cut shows little pigmentation (arrows). B,Nf1+/− mouse with nerve cut shows slightly more pigmentation compared with A. C, Wild-type mouse with nerve transection and deflection of the nerve shows some pigmentation. D, Nf1+/− mouse with nerve transection and deflection shows dramatic clusters of highly pigmented cells (arrowheads). E, Toluidine blue-stained plastic section of skin from the lesion area of an Nf1+/− mouse 3 months after nerve cut. Clusters of pigmented cells present in the facia underneath the hypodermis are within the black box. Scale bar, 100 μm. This area was evaluated at the EM level and is shown in F, showing cytoplasmic pigment granules characteristic of a melanocyte (magnification, 2700×). G, Gross photograph of a ventral view of mutant mouse skin over lesioned area 1 month after nerve transection showing pigmentation (arrows).H, In a mutant mouse 1 month after nerve transection, spots of pigmentation extended ventral to the sciatic nerve (arrows).
Pigmented cell clusters were also detectable on the ventral portion of the dermis (Fig. 1G) and on the ventral surface of theNf1+/− nerve (Fig. 1H). Wild-type animals after nerve cut and deflection rarely showed pigment in these locations (data not shown). Semithin plastic sections showed that pigmented cells in skin were subjacent to the hypodermis (Fig.1E). Electron microscopy confirmed that the pigmented cells were melanocytes (Fig. 1F), with characteristic developing and mature melanonosomes. The size of the melanosomes in these cells was not different from that of melanosomes from skin hair follicles in wild-type animals, indicating that the cells were melanocytes and not melanophages (data not shown).
Mice were studied at various times after nerve cut and resuture. Increased pigmentation was detectable by 7 d after nerve cut and reached maximal levels at 21–30 d. Variable amounts of pigmentation were detected 3 months after injury. In some animals, dramatic melanogenesis was observed, whereas in others little pigmentation was present. By 6–12 months after injury, pigmentation was no longer present (data not shown).
Nerve crush does not initiate pigmentation
Crush injury causes a transient breech of the blood–nerve barrier and severs axons; Schwann cells proliferate, myelin is digested by resident and recruited macrophages, and proximal stumps of neurons regrow axons that regenerate and reinnervate distant targets. Sciatic nerves analyzed after crush injury did not show nerve abnormality 6 or 10 weeks after injury as assessed by gross analysis or H&E staining of paraffin sections. No differences between wild-type andNf1+/− mutants were evident in Schwann cell proliferation after crush injury, as assessed by staining with anti-BrdU (data not shown), and myelin degradation proceeded on schedule. No pigmented cells were noted on gross or microscopic inspection, indicating that a crush injury is not sufficient to cause the pigmentation response. Thus, the Nf1+/− response is an amplified response to wounding, and the response is modulated by the severity of the injury.
To determine whether increased macrophage numbers might explain why the cut-and-deflected nerves showed increased melanogenesis compared with nerve crush, the F40 antibody was used to stain macrophages in distal nerve segments 7 d after injury when macrophage numbers are at their peak (Perry et al., 1995). Similar staining intensity was observed in the distal stumps of crushed and cut nerves (data not shown), indicating that altered macrophage numbers is unlikely to explain the differences observed.
To determine whether increased pigmentation in Nf1 mutants results from peripheral nerve abnormality, sciatic nerve structure was analyzed at embryonic day 18 through postnatal day 30 in plastic sections and by electron microscopy. No abnormalities were detected in nerves of mutant animals (data not shown). Significantly, no pigmented cells were noted within or surrounding the nerve. These data extend and are consistent with previous studies (Brannan et al., 1994; Jacks et al., 1994; Cichowski et al., 1999).
Tumor formation after nerve lesion
Unpigmented and pigmented tumors were occasionally associated with lesioned Nf1 nerves. Unpigmented tumors were detected in 3 of 22 mice studied at 3 months after nerve cut. Peripheral nerve tumors never developed in wild-type mice, with or without wounding. Unpigmented tumors were at least 0.5 cm distant from the lesion sites and associated with nerves (Fig.2A). In sections, these tumors were found to be unencapsulated and diffusely invasive (Fig.2D), with significant amounts of Gomori's trichrome-positive matrix (Fig. 2G,I), S100-positive cells [presumed Schwann cells] (60%) (Fig.2H), S100-negative cells (40%) (methyl green-H), and a few neurofilament-positive axons (Fig. 2J). The high ratio of S100-positive cells to neurofilament-positive axons suggests that many S100-positive cells were free of axons, as reported for human neurofibromas. In 3 of 22 mice 3 months after nerve cut and in 25 mice after cut and deflection, heavily pigmented tumors were detected proximal or distal to the lesion sites (Fig.2B). No pigmented tumors were evident in wild-type littermates after nerve injury. In semithin plastic sections, capsules were apparent on pigmented tumors (Fig.2C,E,F). These tumors have not been studied further.
Fig. 2.

Pigmented and unpigmented tumors inNf1+/− mutant mice 3 months after nerve transection.A, Hindlimb portion of a fixed mutant mouse, 3 months after nerve cut (skin has been removed). An unpigmented tumor, delineated by arrows, is on the left side of the midline of the body. Pigmentation is evident in muscle lateral to the tumor.B, Hindlimb portion of an Nf1+/− mouse with a pigmented tumor, delineated by arrowheads.Arrows point to pigmented spots adjacent to the tumor.C, Toluidine blue-stained semithin plastic section showing the pigmented tumor in B, with capsule. Scale bar, 100 μm. The tumor is shown at higher magnification inE (scale bar, 50 μm) and F (scale bar, 20 μm).D, A nonpigmented tumor in paraffin section after H&E staining, showing tumor infiltration between muscles and lack of a capsule. G, Gomori's trichrome staining of an adjacent section reveals that this tumor is full of collagen-rich matrix and highlights collagen-rich bundles infiltrating nearby muscle (arrows). Scale bar, 100 μm. H, Anti-S100 staining (brown; DAB reaction product) of an adjacent section of the same tumor as D andE, at higher magnification. The section was counterstained with methyl green to show S100-negative cells.J, Anti-neurofilament staining of an adjacent section reveals the presence of axons within the tumor. Scale bars:H, J, 10 μm; I, 50 μm.
Pigmented cells that arise after nerve injury are derived from the adult nerve
The data presented above suggest the hypothesis that pigmented cells result from an abnormal response of endogenous melanocytes to wound cytokines. An alternative possibility is that cells within the adult nerve migrate away from the nerve and become pigmented. To distinguish between these possibilities, nerve fragments from wild-type and mutant animals were labeled with Hoechst dye, a fluorescent nuclear marker, and then grafted into wild-type or mutant host animals. Grafted nerve fragments were then lesioned, and, 30 d later, pigmented cells were evaluated for the presence of the marker dye (Fig.3A) (see Fig. 5A). Surprisingly, we observed numerous examples of cells containing dye-labeled nuclei with adjacent cytoplasm containing pigment granules. Two representative examples are shown in Figure 3, B–D andE–G. Not all dye-labeled cells became pigmented, indicating that only some grafted cells developed into mature melanocytes containing pigment. These data strongly suggest that some cells derived from the adult nerve form pigment in the wound environment.
Fig. 3.
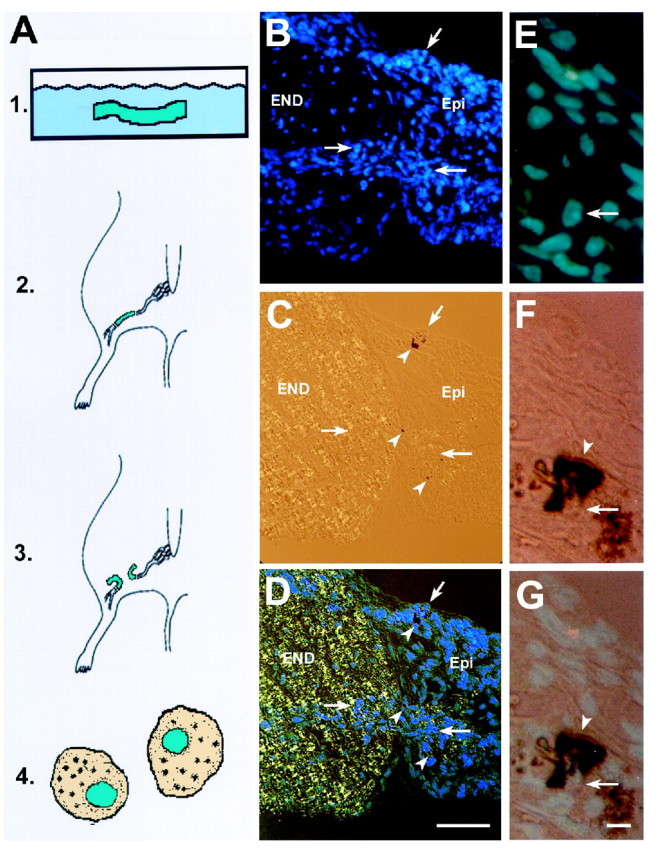
Cells from adult nerve can become pigmented.A, Schematic diagram of nerve grafting technique.1, Sciatic nerve fragments (1 cm) from adult mice were labeled with Hoechst dye in vitro. 2, Hoechst-labeled nerve fragments were transplanted into recipient adult mouse nerve by suturing the dye-labeled nerve fragment between the cut ends of the host nerve. 3, The dye-labeled nerve fragment was cut, and the cut ends were deflected. 4, Thirty days later, animals were killed, areas of pigmentation were cut on a cryostat, and dye-positive cells were evaluated for pigmentation.B–D, Cryosection from a mouse in which anNf1+/− nerve segment was grafted into anNf1+/− host. Dye-positive blue cells are detectable within the endoneurium (END) and adjacent epineurium (Epi). Pigmented cells (arrowheads) are in the epineurium. B, Hoechst-labeled cells (blue nuclei; arrows) in endoneurium and epineurium were visualized with a 4′,6′-diamidino-2-phenylindole (DAPI) filter. C, Note the black (pigmented) cells visualized by bright-field microscopy. D, Bright field and DAPI. E–G, Cryosections of labeled cells at higher magnification; sections were taken from muscle distant from the transection site, 1 month after dye-labeled nerve graft and nerve transection. These are from a different animal thanB–D. Both donor and recipient wereNf1+/−. Scale bar (in D):B–D, 100 μm. E, Hoechst-labeled cells (blue nuclei) in the muscle using a DAPI filter.Arrow represents the area of pigmented cells seen inF and G. F, Black, pigmented cells shown in bright field. G, Bright field and DAPI showing blue nucleus closely surrounded by pigment granules. Scale bar (in G); E–G, 20 μm.
Fig. 5.

Hyperpigmentation after nerve and cell grafting: In A, 1 cm lengths of sciatic nerve from animals of designated genotype (under x-axis) were Hoechst labeled and then grafted into recipient hosts of designated genotype (underarrows). Two thousand to 4000 dye-labeled cells were counted in sections from each animal. Each section was analyzed using bright-field optics and then DAPI filter and fluorescence optics for pigmented cells surrounding labeled nuclei and total pigmented cells. Data are shown as an average of results from two animals for each condition, except Nf1+/− into Nf1+/−, for which four animals were used. The percentage of pigmented dye-labeled nuclei for each animal was similar (Nf1+/+into Nf1+/+: 0, 0; Nf1+/+ intoNf1+/−: 2, 5; Nf1+/−into Nf1+/+: 5, 9; Nf1+/− intoNf1+/−: 44.6, 46.4, 57, 53.9). InB, wild-type fibroblasts (Fb) or wild-type Schwann cells (SC), Nf1heterozygous (SC+/−) or null (SC−/−) or Nf1 −/−TXF (−/−TXF) were dye labeled and grafted around lesion areas of Nf1heterozygous host animals (+/−). Results shown are averages of results from sections taken from three recipient animals for +/+ cells, two animals for Nf1+/− andNf1−/− Schwann cells, and from five animals forNf1 −/−TXF. Analysis was as in A. The percentage of pigmented dye-labeled nuclei for each animal was similar (Nf1+/+ Schwann cells and fibroblasts intoNf1+/−: 0, 0, 0; Nf1+/− Schwann cells into Nf1+/−: 4.9, 4.0; Nf1−/− Schwann cells into Nf1+/−: 6.8, 7.8; Nf1−/−TXF into Nf1+/−: 8.3, 17.1, 9.3, 13.5).
To rule out the possibility that dye-labeled cells were simply in close proximity to pigmented cells, wild-type or Nf1 adult nerve fragments were grafted from black C57BL/6 mice into albino mice that carried a null allele for the tyrosinase (tyr) gene (Fig.4A). The albino mice shown in Figure 4A are white because their melanocytes cannot make pigment. Therefore, if any black pigment arises in the mutant mice after a nerve graft, pigmented cells must derive from the graft. The mutation causing the lack of pigment in these mice arose on the C57BL/6 background, making them syngenic with theNf1 mice. Thirty days after nerve injury, mice were evaluated for pigmentation. Black patches were visible in all recipient albino mice in muscle overlying nerve (Fig.4C,D), and in adjacent skin (Fig.4B), supporting the view that pigmented cells derive from nerve. Wild-type nerve fragments grafted into the albino mice also resulted in appearance of pigmented cells, but, as anticipated, no black patches were present when nerve was cut and deflected in albino mice without nerve graft (data not shown; n = 3). Thus, cells with the potential to form melanocytes exist in adult wild-type and Nf1 mutant nerve.
Fig. 4.

Pigmentation in an albino mouse (tyr−/−) after a nerve graft from C57BL/6Nf1+/− mouse. A, Dorsal view of the white skin of an albino host, showing absence of black hairs, 1 month after grafting of a 2.0 mm segment of nerve from a C57BL/6Nf1+/− mouse, followed by cut and deflection of the grafted nerve. The skin remains white, indicating absence of pigmented melanocytes in this mutant. B, Ventral side of the skin overlying the graft region showing pigmented spots in the skin.C, D, Two examples of pigmented cells (brown–black pigment granules) in cryosections within muscle overlying the grafted nerve segment after staining of sections with H&E to highlight nuclei and cytoplasm. In C, note the clusters of pigmented cells (arrows). InD, note the individual cells with visible nuclei and pigment granules in the cytoplasm (arrows). Scale bar (in D): C, D, 10 μm.
In experiments in which C57BL/6 nerve was grafted into albino mice, the pigmentation phenotype was not as dramatic as observed when the mutant nerve was transected and deflected in animals with a normal tyrosinase gene, as shown in Figure. 1. This observation suggested the possibility that cells within the nerve in conjunction with a mutant environment amplify the pigmentation response. To test this, a series of grafting experiments were performed in which Hoescht-labeled nerve fragments, 1 cm in length, were grafted into wild-type or mutant animals. The percentage of dye-labeled cells containing pigment (in fascia around nerve and around muscles) was expressed as a percentage of total dye-labeled cells (note that single cells were counted, not streaks as for Fig. 1). As shown in Figure5A, the percentage of dye-positive cells containing pigment in wild type to wild-type grafts was extremely low; no pigmented cells were detected in 10,000 dye-labeled cells analyzed. In contrast, when graft alone or host alone was Nf1 heterozygous, 6.3 and 5% of the dye-labeled cells contained pigment granules, respectively. Strikingly, when both graft and host were Nf1 heterozygous, a 10-fold increase in dye and pigment-laden cells was observed; 55.2% of labeled cells were pigmented. The data confirm a role for both nerve-derived cells and for the host environment in the pigmentation phenotype.
Glial peripheral nerve cells retain the ability to become pigmented
To identify nerve-derived cell populations that might contribute to the pigmentation response, Schwann cells or fibroblasts were cultured on filters and then grafted into recipient Nf1heterozygous animals, around cut and resutured nerves. The filter provided a solid support and allowed implantation of a similar number of cells per genotype. Thirty days after implantation, blocks of tissue containing pigmented cells were removed for sectioning and analysis. Neither wild-type Schwann cells nor fibroblasts generated detectable numbers of dye-positive pigmented cells (Fig. 5B).Nf1 mutant (−/−) fibroblasts also did not produce dye-positive pigmented cells (data not shown). These results suggested that another cell population in the nerve contributes to the pigmented cell population. In an attempt to identify such a cell, we used Schwann cells from Nf1 mutant animals that became pigmented with increased frequency. All Nf1 Schwann cell preparations tested yielded dye-labeled pigmented cells. Nf1 heterozygous and null Schwann cells purified from DRG neurons showed 4.5 and 7.4% dye-positive pigmented cells, respectively. A population of rapidly proliferating cells was identified previously in dorsal root ganglion cultures from Nf1−/− and, to a lesser extent, from cultures from Nf1+/− embryos (Kim et al., 1997). When these cells, designated −/−TXF, were grown on filters and grafted around cut nerves of four host animals, an average of 12.1% of the dye-labeled cells were pigmented. The number of pigmented cells derived from dye-labeled −/−TXF cells differed from theNf1+/− cells in a Student's t test (two-tail, two-sample unequal variance; p = 0.03). TheNf1−/− cells showed a trend toward difference from the −/−TXF cells (p = 0.10) but did not reach statistical significance. This series of experiments demonstrates that glial cells, not fibroblasts, have the potential to become pigmented in the wound environment. In addition, loss-of-function mutations atNf1 in glial cells increase the percentage of cells that form pigment in the wound environment.
Immunohistochemical characterization of −/−TXF peripheral nerve cells that become pigmented
Wild-type Schwann cells and Nf1 −/−TXF cells were compared in immunohistochemistry (Fig.6). S100, p75, and GFAP marked wild-type mouse Schwann cells as expected. The −/−TXF cells expressed all three Schwann cell markers. SMA is a marker for embryonic fibroblasts and for smooth muscle cells. Embryonic fibroblasts as expected expressed SMA and low levels of S100 (expressed by some fibroblast and muscle cells) but not p75 or GFAP. Neither wild-type Schwann cells nor Nf1−/−TXF cells expressed SMA. This marker analysis demonstrates that −/−TXF cells are within the glial cell lineage. In three independent preparations, every cell expressed all three markers (data not shown). The high percentage of cells expressing all three antigens rules out the possibility that a small but significant portion of the cells have characteristics differences from −/−TXF cells.
Fig. 6.

Characterization of −/−TXF glial cells. Photomicrographs of immunostained cells. +/+ Schwann cells (SC) and fibroblasts (Fib) were used as positive and negative controls. A, D,G, and J represent −/−TXF Schwann cells, B, E, H, andK are +/+ Schwann cells, and C,F, I, and L are −/− fibroblasts [with the same pattern of expression as +/+ fibroblasts (data not shown)]. A–C show anti-p75NGFR,D–F show anti-S100, G–I show anti-GFAP, and J–L show anti-SMA immunostaining. The −/−TXF Schwann cells are p75NGFR, S100, and GFAP positive. +/+ Schwann cells are also positive for p75NGFR and S100 but show much weaker staining for GFAP (visible in only the boxed inset inH; a longer exposure than H, surroundinginset). Exposure times for G–I were matched. Fibroblasts are strongly positive for SMA. Scale bar, 50 μm.
Suppression of pigmentation occurs through the βcsubunit of the GMCSF–IL5 receptor
The GMCSF–IL3 receptor βc pathway is activated after nerve injury (Saada et al., 1996) and has been linked to Nf1 signaling (Birnbaum et al., 2000). We hypothesized that the GMCSF–IL3 receptor βc effect might underlie heightened response of Nf1 mutant cells to nerve lesion. We tested the effects of βc loss by crossingNf1 mutant mice with those null for loss of βc (Nishinakamura et al., 1995). Data shown in Figure 7A represent quantitative analysis of pigmented patches visible on muscles in wild-type and mutant for Nf1, βc, or both. Loss of βc increased pigmentation in mice to levels comparable with Nf1 mutation. Conversely, in the absence of βc, the effects ofNf1 mutation were completely reversed. These data show that βc is necessary for the enhanced pigmentation in Nf1 mutants.
Fig. 7.
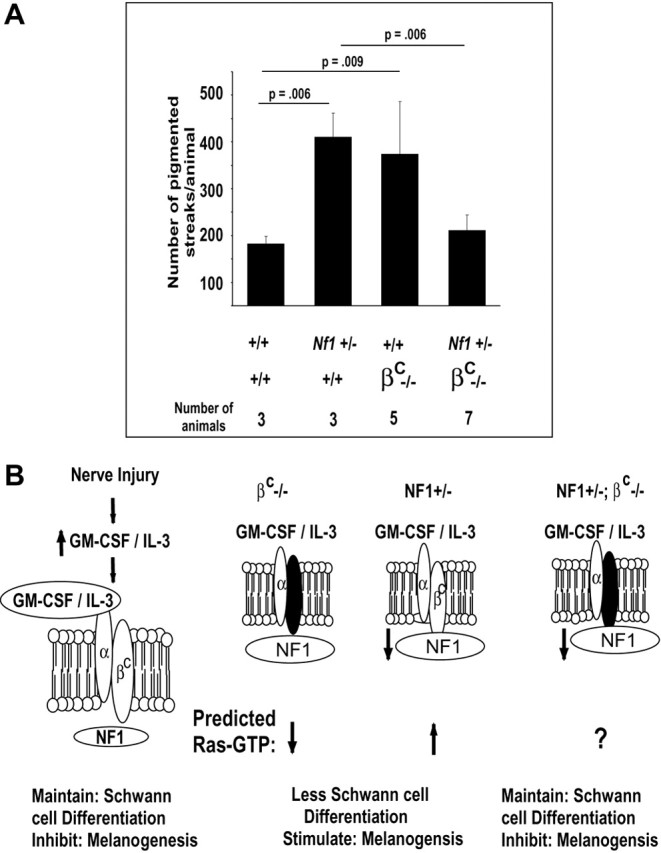
The Nf1 mutant phenotype is rescued by loss of the βc receptor. A, Nerves were cut and deflected in groups of animals of the genotypes designated under the x-axis. Three weeks after surgery, the number of visible pigmented patches–streaks was counted in muscle. Statistical analysis was using Student's t test and, for each pairwise comparison, is designated by linesconnecting each two groups. B, A model depicts the pathway that inhibits pigmentation after nerve injury. On theleft, after injury, levels of cytokines, including GMCSF and IL3, increase (Saada et al., 1996). Cytokines binds α subunits, which interact with βc, resulting in activation of downstream signaling, including Ras activation (Bagley et al., 1997), maintenance of Schwann cell differentiation, and inhibition of most melanogenesis. The Nf1 Ras-GAP mediates the βc signaling, likely by modulating the duration or extent of Ras signaling, but perhaps in addition through other signaling cascades. In the middle panels, when βc is absent or Nf1 is mutant, melanogenesis is increased. On the right, when βc is absent andNf1 is mutant in affected cells, we postulate (? symbol) that Ras activation is close to wild-type levels, so that wild-type levels of pigmentation are observed.
DISCUSSION
Previous studies suggested that a bipotential glial–melanocyte progenitor exists transiently in developing peripheral nerves (Nichols and Weston, 1977; Nichols et al., 1977; Ciment et al., 1986; Stocker et al., 1991; Dupin et al., 2000; Nataf and Le Douarin, 2000). Our data show that melanocytes can also be generated from adult peripheral nerve cells during nerve injury and that a pathway involving the IL3–GMCSF receptor βc and neurofibromin suppresses this pigmentation (Fig. 7B). Based on our findings, we propose that injury causes accumulation of cytokines that direct lineage switching by glia.
Our data support a model in which glial cells migrate from the endoneurium into epineurial fascia and the hypodermis and then become pigmented. Pigmentation does not occur after nerve crush injury, when glial cells remain in basal lamina tubes, but does result when peripheral nerves are cut and cells escape from nerve ends (Fawcett and Keynes, 1990; Sunderland, 1991). This finding indicates that a pathway out of the endoneurium is required for cells to switch to a pigmenting phenotype, because normal cells cannot penetrate a basal lamina (Erickson, 1987). We cannot completely exclude the possibility that cells in the epineurium pigment in the wound environment. However, pigmentation is greatly enhanced when the basal lamina constraining endoneurial cells is lost (after nerve cut and deflection), and cells normally encased in the endoneurium can escape. In addition, purified glial cells grafted into the wound environment become pigmented. These data argue strongly that epineurial cells are not the sole source of pigment-forming cells.
Pigmented cells were never observed within the endoneurium. Factors inside the endoneurium could enhance the Schwann cell phenotype and inhibit melanocyte formation, and/or the endoneurium could lack melanogenic factors. GMCSF and IL3 are released by endoneurial nerve fibroblasts after nerve injury (Saada et al., 1996). Our genetic data (see below) suggest that these factors suppress melanogenesis in the endoneurium. However, it remains possible that Nf1 and/or the βc signaling cascade affect other Schwann cell functions, such as proliferation, migration, and/or differentiation, all likely required before melanogenesis. However, because we reliably detected Hoechst-stained nuclei even 30 d after implantation, it is likely that implanted cells divided very little; dye is diluted with each round of cell division. Some evidence suggests migration by Schwann cells. For example, large groups of Hoechst-labeled cells were often observed in the muscle and hypodermis. This implies migration of streams of cells from the nerve.
Grafting wild-type nerve fragments into Nf1 mutants increased pigmentation by wild-type cells. Dye-labeled (grafted wild type) cells were scored for pigment, excluding Nf1 mutant cells as a source of pigmented cells. It is possible that migration promoting and/or melanogenic factors produced by Nf1 mutant cells in denervated skin or muscle act on grafted wild-type cells.Nf1 mutant Schwann cells increase expression of several growth factors (Mashour et al., 2001). Stem cell factor (c-kit ligand) enhances melanocyte migration in an Nf1-dependent manner (Wehrle-Haller et al., 2001), and Nf1 loss partially rescues pigmentation defects caused by c-kit mutation (Ingram et al., 2000). This factor may therefore contribute to the noncell autonomousNf1 effect on melanogenesis.
Although wild-type nerve cells formed pigment, <1 in 10,000 wild-type cultured Schwann cells became pigmented after grafting into wounded nerve. Cells that pigment in wild-type nerves may be progenitor cells that do not survive under culture conditions favoring embryonic Schwann cell development. It seems more likely that pigmented cells de-differentiate from Schwann cells and re-differentiate (transdifferentiate) at low incidence in the wound environment because uniformly S100-, p75-, and GFAP-positive Nf1 mutant Schwann cells become pigmented at detectable frequency. Melanocytes can transdifferentiate into glia (Dupin et al., 2000). We have so far failed in forcing melanogenesis by mutant mouse Schwann cells in vitro; melanogenesis by mouse Schwann cells has never been reported, even very early in development. Enhanced pigment formation byNf1 mutant Schwann cells may result from enhanced lineage plasticity. Nf1 −/−TXF cells characterized here, which form pigment cells, have characteristics of Schwann cell progenitors and of Schwann cells. Nf1 −/−TXF cells are like Schwann cell progenitors in their cobblestone-like growth pattern (Dong et al., 1995; Kim et al., 1997, 1999) and expression of p75 (shown here). However, they are maintained in neuregulin, a sufficient signal to promote Schwann cell differentiation from Schwann cell precursors (Shah et al., 1994; Dong et al., 1995). They express GFAP, which is normally highly expressed only in nonmyelinating Schwann cells, but also express the epidermal growth factor receptor that is normally expressed by neural crest cells but not Schwann cells (DeClue et al., 2000). Analysis of the relationship of Nf1 −/−TXF cells to Schwann cell progenitors and the influence of Nf1 mutation on Schwann cell lineage plasticity is of interest in light of our observations. Nf1 mutation alters hematopoetic progenitors (Zhang et al., 1998; Birnbaum et al., 2000; Ingram et al., 2000) and is associated with defects in developing neurons (Brannan et al., 1994;Vogel et al., 1995; Lakkis et al., 1999).
Increased plasticity of Nf1 mutant Schwann cells, or their precursors, could be relevant to peripheral nerve tumorigenesis (Cichowski et al., 1999; Vogel et al., 1999; Zhu et al., 2002). We occasionally observed nerve tumor formation after nerve injury toNf1+/− mice, although these mice, unlike human NF1 patients, do not form neurofibromas (Brannan et al., 1994; Jacks et al., 1994; Zhu et al., 2002). The mechanism underlying tumor formation in our study is unknown. However, our data support the hypothesis that, when the cytokine environment is permissive, as it is after wounding, both tumors and aberrant pigmentation can occur. In humans with NF1, pigmented cells occur within neurofibromas, and hyperpigmented skin frequently overlies plexiform neurofibromas of childhood (Fetsch et al., 2000) (B. Korf, personal communication). Both could result through transdifferentiation or from differentiation of a progenitor cell population to glial and melanocyte derivatives.
Our findings suggest a novel, nonhematopoietic function of βc in nerve response to injury, consistent with a role for the βcligands GMCSF and IL3 in maintaining skin homeostasis after wounding (Szabowski et al., 2000). GMCSF, IL5, and IL3 all use the βc receptor. Each associates with a unique α subunit that confers binding (for review, see Bagley et al., 1997). One or more of these ligands may suppress melanogenesis through activation of βc. Additional experiments will be required to identify the relevant ligand(s). In the hematopoietic system, GMCSF plays a key role upstream of Nf1signaling (Birnbaum et al., 2000).
Like βc mutants, Nf1 mutants showed increased injury-induced pigmentation. This finding suggests a role for Nf1 in suppression of the melanogenic phenotype.Nf1 appears to act downstream of βc, because loss of βc together with hemizygosity forNf1 inhibited the melanogenesis enhanced by either alone. Neurofibromin is a Ras-GTPase activating protein (Ras-GAP) (for review, see Cichowski and Jacks, 2001). The effects of Nf1 mutation on pigmentation could be Ras mediated. In hematopoetic cells, GMCSF stimulates Ras activation, and Ras activation in response to GMCSF or IL3 is augmented in cells lacking Nf1 (Bollag et al., 1996;Largaespada et al., 1996). Schwann cells lacking Nf1 have elevated levels of Ras-GTP (Kim et al., 1995). Loss of βc is predicted to cause decreased Ras signaling in cells, because the receptor that normally couples to Ras activation is missing. Decreased neurofibromin, in contrast, results in increased Ras-GTP or longer-duration Ras-GTP signals after receptor activation. However, βc andNf1 mutants both show increased pigmentation after nerve injury. One interpretation of these data are that precise levels of Ras-GTP or duration of Ras-GTP in peripheral nerve cells are required after nerve injury to maintain Schwann cell phenotype. In PC12 cells, Ras-GTP duration is believed to determine whether neurites grow or not (Qui and Green, 1992). In a 3T3 cell assay, transforming activity of mutant Ras proteins correlates with their intrinsic GTPase activity (Donovan et al., 2002). In addition, deletion of the wild-type allele in a Ras mutant tumor may be essential for tumorigenesis (Osaka et al., 1997; Sugio et al., 1997). Loss of βcand mutation at Nf1 together may normalize Ras-GTP levels by summing a positive and negative effect on Ras signaling (Fig.7B, ? symbol), thus reducing pigmentation. Testing the role of Ras in injury-induced pigmentation in adult nerves will be of interest in light of our findings.
Footnotes
This work was supported by National Institutes of Health Grants R01-NS28840 (N.R.) and R01-AR45429 (R.E.B.) and the Department of Defense Program on Neurofibromatosis (N.R.). We thank G. DeCourten-Meyers for tumor assessment, W. Thompson, M. Rao, K. Shannon, and W. Pavan for helpful discussions, and Roger West for gross photography. We gratefully acknowledge R. Murray (DNAX, Palo Alto, CA) for βc-deficient mice, N. Copeland and C. Brannan for NF1 mice, and C. Stiles and L. Sherman for thoughtful manuscript critique.
Correspondence should be addressed to Nancy Ratner, Department of Cell Biology, Neurobiology, and Anatomy, University of Cincinnati, College of Medicine, 3125 Eden Avenue, Cincinnati, OH 45267-0521. E-mail:nancy.ratner@uc.edu.
REFERENCES
- 1.Aguayo AJ, Bray GM, Perkins SC. Axon-Schwann cell relationships in neuropathies of mutant mice. Ann NY Acad Sci. 1979;317:512–531. [PubMed] [Google Scholar]
- 2.Atit RP, Crowe MJ, Greenhalgh DG, Wenstrup RJ, Ratner N. The Nf1 tumor suppressor regulates mouse skin wound healing, fibroblast proliferation, and collagen deposited by fibroblasts. J Invest Dermatol. 1999;112:835–842. doi: 10.1046/j.1523-1747.1999.00609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bagley CJ, Woodcock JM, Stomski FC, Lopez AF. The structural and functional basis of cytokine receptor activation: lessons from the common beta subunit of the granulocyte-macrophage colony-stimulating factor interleukin-3 (IL-3) and IL-5 receptors. Blood. 1997;89:1471–1482. [PubMed] [Google Scholar]
- 4.Birnbaum RA, O'Marcaigh A, Wardak Z, Zhang YY, Dranoff G, Jacks T, Clapp DW, Shannon KM. Nf1 and Gmcsf interact in myeloid leukemogenesis. Mol Cell. 2000;5:189–195. doi: 10.1016/s1097-2765(00)80415-3. [DOI] [PubMed] [Google Scholar]
- 5.Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P, Lange BJ, Freedman MH, McCormick F, Jacks T, Shannon K. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet. 1996;12:144–148. doi: 10.1038/ng0296-144. [DOI] [PubMed] [Google Scholar]
- 6.Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW, Buchberg AM, Jenkins NA, Parada LF, Copeland NG. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994;8:1019–1029. doi: 10.1101/gad.8.9.1019. [DOI] [PubMed] [Google Scholar]
- 7.Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104:593–604. doi: 10.1016/s0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- 8.Cichowski K, Shih TS, Schmitt E, Santiago S, Reilly K, McLaughlin ME, Bronson RT, Jacks T. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999;286:2172–2176. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- 9.Ciment G, Glimelius B, Nelson DM, Weston JA. Reversal of a developmental restriction in neural crest-derived cells of avian embryos by a phorbol ester drug. Dev Biol. 1986;118:392–398. doi: 10.1016/0012-1606(86)90009-6. [DOI] [PubMed] [Google Scholar]
- 10.Daston MM, Ratner N. Neurofibromin a predominantly neuronal GTPase activating protein in the adult is ubiquitously expressed during development. Dev Dynam. 1992;5:216–226. doi: 10.1002/aja.1001950307. [DOI] [PubMed] [Google Scholar]
- 11.Daston MM, Scrable H, Nordlund M, Sturbaum AK, Nissen LM, Ratner N. The protein product of the neurofibromatosis type 1 gene is expressed at highest abundance in neurons Schwann cells and oligodendrocytes. Neuron. 1992;8:415–422. doi: 10.1016/0896-6273(92)90270-n. [DOI] [PubMed] [Google Scholar]
- 12.DeClue JE, Heffelfinger S, Benvenuto G, Ling B, Li S, Rui W, Vass WC, Viskochil D, Ratner N. Epidermal growth factor receptor expression in neurofibromatosis type 1-related tumors and NF1 animal models. J Clin Invest. 2000;105:1233–1241. doi: 10.1172/JCI7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong Z, Brennan A, Liu N, Yarden Y, Lefkowitz G, Mirsky R, Jessen KR. Neu differentiation factor is a neuron-glia signal and regulates survival proliferation and maturation of rat Schwann cell precursors. Neuron. 1995;15:585–596. doi: 10.1016/0896-6273(95)90147-7. [DOI] [PubMed] [Google Scholar]
- 14.Dong Z, Sinanan A, Parkinson D, Parmantier E, Mirsky R, Jessen KR. Schwann cell development in embryonic mouse nerves. J Neurosci Res. 1999;56:334–348. doi: 10.1002/(SICI)1097-4547(19990515)56:4<334::AID-JNR2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 15.Donovan S, Shannon KM, Bollag G. GTPase activating proteins: critical regulators of intracellular signaling. Biochim Biophys Acta. 2002;1602:23–45. doi: 10.1016/s0304-419x(01)00041-5. [DOI] [PubMed] [Google Scholar]
- 16.Dupin E, Glavieux C, Vaigot P, Le Douarin NM. Endothelin 3 induces the reversion of melanocytes to glia through a neural crest-derived glial-melanocytic progenitor. Proc Natl Acad Sci USA. 2000;97:7882–7887. doi: 10.1073/pnas.97.14.7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eguchi G, Kodama R. Transdifferentiation. Curr Opin Cell Biol. 1993;5:1023–1028. doi: 10.1016/0955-0674(93)90087-7. [DOI] [PubMed] [Google Scholar]
- 18.Erickson CA. Behav of neural crest cells on embryonic basal laminae. Dev Biol. 1987;120:38–49. doi: 10.1016/0012-1606(87)90101-1. [DOI] [PubMed] [Google Scholar]
- 19.Fawcett JW, Keynes RJ. Peripheral nerve regeneration. Annu Rev Neurosci. 1990;13:43–60. doi: 10.1146/annurev.ne.13.030190.000355. [DOI] [PubMed] [Google Scholar]
- 20.Fetsch JF, Michal M, Miettinen M. Pigmented (melanotic neurofibroma: a clinicopathologic and immunohistochemical analysis of (19) lesions from 17 patients. Am J Surg Pathol. 2000;24:331–343. doi: 10.1097/00000478-200003000-00001. [DOI] [PubMed] [Google Scholar]
- 21.Grotendorst GR. Chemoattractants and growth factors in wound healing: biochemical and clinical aspects (Cohen IK, Lindbald WJ, eds), pp 237–246. Harcourt Brace Jovanovich; Philadelphia: 1992. [Google Scholar]
- 22.Ingram DA, Yang FC, Travers JB, Wenning MJ, Hiatt K, New S, Hood A, Shannon K, Williams DA, Clapp DW. Genetic and biochemical evidence that haploinsufficiency of the Nf1 tumor suppressor gene modulates melanocyte and mast cell fates in vivo. J Exp Med. 2000;191:181–188. doi: 10.1084/jem.191.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet. 1994;7:353–361. doi: 10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
- 24.Kim H, Rosenbaum T, Marchionni M, Ratner N, DeClue JE. Schwann cells from neurofibromin deficient mice exhibit activation of p21ras inhibition of cell proliferation and morphological changes. Oncogene. 1995;11:325–335. [PubMed] [Google Scholar]
- 25.Kim HA, Ling B, Ratner N. Nf1-deficient mouse Schwann cells are angiogenic and invasive and can be induced to hyperproliferate: reversion of some phenotypes by an inhibitor of farnesyl protein transferase. Mol Cell Biol. 1997;17:862–872. doi: 10.1128/mcb.17.2.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lakkis MM, Golden JA, O'Shea KS, Epstein JA. Neurofibromin deficiency in mice causes exencephaly and is a modifier for Splotch neural tube defects. Dev Biol. 1999;212:80–92. doi: 10.1006/dbio.1999.9327. [DOI] [PubMed] [Google Scholar]
- 27.Largaespada DA, Brannan CI, Jenkins NA, Copeland NG. Nf1 deficiency causes Ras-mediated granulocyte/macrophage colony stimulating factor hypersensitivity and chronic myeloid leukaemia. Nat Genet. 1996;12:137–143. doi: 10.1038/ng0296-137. [DOI] [PubMed] [Google Scholar]
- 28.Le Douarin N, Kalcheim C. The neural crest, Ed 2. Cambridge UP; New York: 1999. [Google Scholar]
- 29.Mashour GA, Ratner N, Khan GA, Wang H-L, Martuza RL, Kurtz A. The angiogenic factor midkine is aberrantly expressed in NF1-deficient Schwann cells and is a mitogen for neurofibroma-derived cells. Oncogene. 2001;20:97–105. doi: 10.1038/sj.onc.1204026. [DOI] [PubMed] [Google Scholar]
- 30.Morrison SJ, White PM, Zock C, Anderson DJ. Prospective identification isolation by flow cytometry and in vivo self-renewal of multipotent mammalian neural crest stem cells. Cell. 1999;96:737–749. doi: 10.1016/s0092-8674(00)80583-8. [DOI] [PubMed] [Google Scholar]
- 31.Nataf V, Le Douarin NM. Induction of melanogenesis by tetradecanoylphorbol-13 acetate and endothelin 3 in embryonic avian peripheral nerve cultures. Pigment Cell Res. 2000;13:172–178. doi: 10.1034/j.1600-0749.2000.130309.x. [DOI] [PubMed] [Google Scholar]
- 32.Nichols DH, Weston JA. Melanogenesis in cultures of peripheral nervous tissue. I. The origin and prospective fate of cells giving rise to melanocytes. Dev Biol. 1977;60:217–225. doi: 10.1016/0012-1606(77)90120-8. [DOI] [PubMed] [Google Scholar]
- 33.Nichols DH, Kaplan RA, Weston JA. Melanogenesis in cultures of peripheral nervous tissue. II. Environmental factors determining the fate of pigment-forming cells. Dev Biol. 1977;60:226–237. doi: 10.1016/0012-1606(77)90121-x. [DOI] [PubMed] [Google Scholar]
- 34.Nishinakamura R, Nakayama N, Hirabayashi Y, Inoue T, Aud D, McNeil T, Azuma S, Yoshida S, Toyoda Y, Arai K. Mice deficient for the IL-3/GM-CSF/IL-5 beta c receptor exhibit lung pathology and impaired immune response while beta IL3receptor-deficient mice are normal. Immunity. 1995;2:211–222. doi: 10.1016/1074-7613(95)90046-2. [DOI] [PubMed] [Google Scholar]
- 35.Osaka M, Matsuo S, Koh T, Sugiyama T. Loss of heterozygosity at the N-ras locus in 7, 12-dimethylbenz[a] anthracene-induced rat leukemia. Mol Carcinog. 1997;18:206–212. [PubMed] [Google Scholar]
- 36.Perry VH, Tsao JW, Fearn S, Brown MC. Radiation-induced reductions in macrophage recruitment have only slight effects on myelin degeneration in sectioned peripheral nerves of mice. Eur J Neurosci. 1995;7:271–280. doi: 10.1111/j.1460-9568.1995.tb01063.x. [DOI] [PubMed] [Google Scholar]
- 37.Qui MS, Green SH. PC12 cell neuronal differentiation is associated with prolonged p21ras activity and consequent prolonged ERK activity. Neuron. 1992;9:705–717. doi: 10.1016/0896-6273(92)90033-a. [DOI] [PubMed] [Google Scholar]
- 38.Riccardi VM. Neurofibromatosis: phenotype natural history and pathogenesis, Ed 2. Johns Hopkins UP; Baltimore, MD: 1992. [Google Scholar]
- 39.Saada A, Reichert F, Rotshenker S. Granulocyte macrophage colony stimulating factor produced in lesioned peripheral nerves induces the up-regulation of cell surface expression of MAC-2 by macrophages and Schwann cells. J Cell Biol. 1996;133:159–167. doi: 10.1083/jcb.133.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scherer SS. The biology and pathobiology of Schwann cells. Curr Opin Neurol. 1997;10:386–397. doi: 10.1097/00019052-199710000-00006. [DOI] [PubMed] [Google Scholar]
- 41.Shah NM, Marchionni MA, Isaacs I, Stroobant P, Anderson DJ. Glial growth factor restricts mammalian neural crest stem cells to a glial fate. Cell. 1994;77:349–360. doi: 10.1016/0092-8674(94)90150-3. [DOI] [PubMed] [Google Scholar]
- 42.Stocker KM, Sherman L, Rees S, Ciment G. Basic FGF and TGF-beta 1 influence commitment to melanogenesis in neural crest-derived cells of avian embryos. Development. 1991;111:635–645. doi: 10.1242/dev.111.2.635. [DOI] [PubMed] [Google Scholar]
- 43.Stocker KM, Baizer L, Coston T, Sherman L, Ciment G. Regulated expression of neurofibromin in migrating neural crest cells of avian embryos. J Neurobiol. 1995;27:535–552. doi: 10.1002/neu.480270408. [DOI] [PubMed] [Google Scholar]
- 44.Sugio K, Molberg K, Albores-Saavedra J, Virmani AK, Kishimoto Y, Gazdar AF. K-ras mutations and allelic loss at 5q and 18q in the development of human pancreatic cancers. Int J Pancreatol. 1997;21:205–217. doi: 10.1007/BF02821606. [DOI] [PubMed] [Google Scholar]
- 45.Sunderland S. Nerve injuries and their repair, p 538. Churchill Livingstone; Edinburg: 1991. [Google Scholar]
- 46.Szabowski A, Maas-Szabowski N, Andrecht S, Kolbus A, Schorpp-Kistner M, Fusenig NE, Angel P. c-Jun and JunB antagonistically control cytokine-regulated mesenchymal-epidermal interaction in skin. Cell. 2000;103:745–755. doi: 10.1016/s0092-8674(00)00178-1. [DOI] [PubMed] [Google Scholar]
- 47.Vogel KS, Brannan CI, Jenkins NA, Copeland NG, Parada L. Loss of neurofibromin results in neurotrophin-independent survival of embryonic sensory and sympathetic neurons. Cell. 1995;82:733–742. doi: 10.1016/0092-8674(95)90470-0. [DOI] [PubMed] [Google Scholar]
- 48.Vogel KS, Klesse LJ, Velasco-Miguel S, Meyers K, Rushing EJ, Parada LF. Mouse tumor model for neurofibromatosis type 1. Science. 1999;286:2176–2179. doi: 10.1126/science.286.5447.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wehrle-Haller B, Meller M, Weston JA. Analysis of melanocyte precursors in Nf1 mutants reveals that MGF/KIT signaling promotes directed cell migration independent of its function in cell survival. Dev Biol. 2001;232:471–483. doi: 10.1006/dbio.2001.0167. [DOI] [PubMed] [Google Scholar]
- 50.Weiler E, Farbman AI. Proliferation in the rat olfactory epithelium: age-dependent changes. J Neurosci. 1997;17:3610–3622. doi: 10.1523/JNEUROSCI.17-10-03610.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang YY, Vik TA, Ryder JW, Srour EF, Jacks T, Shannon K, Clapp DW. Nf1 regulates hematopoietic progenitor cell growth and ras signaling in response to multiple cytokines. J Exp Med. 1998;187:1893–1902. doi: 10.1084/jem.187.11.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–922. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]