

Abstract
Epstein–Barr virus (EBV) infection converts primary human B cells into continuously proliferating lymphoblastoid cell lines (LCLs). To examine the role of EBV nuclear antigen (EBNA) 3C in the proliferation of LCLs, we established LCLs infected with an EBV recombinant that expresses EBNA3C with a C-terminal fusion to a 4-hydroxytamoxifen (4HT)-dependent mutant estrogen receptor, E3C–HT. In the presence of 4HT, LCLs expressed the E3C–HT protein and grew like WT LCLs. When E3C–HT EBV-infected LCLs were transferred to medium without 4HT, E3C–HT protein slowly disappeared, and the LCLs gradually ceased growing. WT EBNA3C expression from an oriP plasmid transfected into E3C–HT LCLs protected the LCLs from growth arrest in medium without 4HT, whereas expression of EBNA3A or EBNA3B did not. The expression of other EBNA proteins and of LMP1, CD21, CD23, and c-myc was unaffected by EBNA3C inactivation. However, EBNA3C inactivation resulted in the accumulation of p16INK4A, a decrease in the hyperphosphorylated form of the retinoblastoma protein, and a decrease in the proportion of cells in S or G2/M phase. These results indicate that EBNA3C has an essential role in cell cycle progression and the growth maintenance of LCLs.
Keywords: oncogenic virus, p16, retinoblastoma protein
Epstein–Barr virus (EBV) causes lymphocyte-proliferative diseases in immune-deficient patients and is associated with Burkitt's lymphoma, Hodgkin's lymphoma, other B and T cell lymphomas, anaplastic nasopharyngeal carcinoma, and some gastric carcinomas (for review, see ref. 1). EBV infection in vitro converts primary human B cells into continuously proliferating lymphoblastoid cell lines (LCLs) (2). In LCLs, EBV expresses six nuclear proteins [EBV nuclear antigens (EBNAs) EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNALP], three integral membrane proteins (LMP1, LMP2A, and LMP2B), two small nonpolyadenylated RNAs (EBER1 and EBER2), and BamA rightward transcripts (for review, see ref. 3). Six of these viral latency proteins, EBNA1, EBNA2, EBNA3A, EBNA3C, EBNALP, and LMP1, are absolutely or critically required for the transformation phenotype (3).
EBNA3A, EBNA3B, and EBNA3C, which are arranged in tandem in the EBV genome, are encoded by genes that are similar in structure (4–8), leading to the proposal that the EBNA3 genes may have arisen from a tandem triplication of an ancestral gene (9–11). The N-terminal amino acids of EBNA3A, EBNA3B, and EBNA3C mediate interaction with a sequence-specific DNA-binding protein, RBP-Jκ (12–16). Reverse genetic experiments with recombinant EBVs indicate that EBNA3A and EBNA3C are essential for the EBV-mediated conversion of primary B cells into LCLs, whereas EBNA3B is dispensable (17–20). The role of EBNA3C in LCL outgrowth and continuous proliferation has been only partially delineated, but, by using a transient transfection reporter assay, EBNA3C has been shown to play a complex regulatory role in the transcription of viral and cellular genes (21–26). EBNA3C has also been shown to interact with histone deacetylase and with the corepressor CtBP (27–29). In addition to its transcriptional functions, it has been reported (30–33) that EBNA3C has cell cycle regulatory functions, presumably mediated by direct protein–protein interactions. EBNA3C expression stimulates cyclin A-dependent kinase activity (30, 31). It also recruits the SCFSKP2 ubiquitin ligase complex and regulates the stability of cell cycle modulatory protein p27 (32). More recently, EBNA3C has been shown to mediate the degradation of the retinoblastoma protein pRb, with the assistance of the SCFSKP2 complex, in transiently or stably transfected cells (33).
The experiments reported here were performed to identify the mechanisms by which EBNA3C contributes to LCL growth maintenance. The EBNA3C ORF was fused in frame to a 4-hydroxytamoxifen (4HT)-dependent mutant estrogen receptor hormone-binding domain, HTER (34), to create an ORF that encodes a conditionally active EBNA3C, E3C–HT, in infected LCLs. We demonstrate that EBNA3C inactivation in LCLs results in growth arrest without affecting the expression of other EBNA proteins or of LMP1. This growth arrest is accompanied by an accumulation of p16INK4A and a decrease in the hyperphosphorylated form of pRb.
Results
Establishment of LCLs by Infection with Recombinant EBV Expressing Conditionally Active EBNA3C.
AK-BAC-GFP, which has a BAC sequence and a NeoR marker for drug selection in mammalian cells (35), was used to construct a recombinant EBV that expresses a conditionally active form of EBNA3C fused to a 4HT-responsive modified estrogen receptor hormone-binding domain, HTER. A DNA fragment containing HTER and the zeocin-resistance gene, a bacterial selection marker, was inserted downstream of the last codon of the EBNA3C ORF of AK-BAC-GFP by GET recombination in Escherichia coli (Fig. 1A). A successfully recombined bacmid that expressed EBNA3C with its C terminus fused in frame to HTER (E3C–HT–zeo BAC) was purified from E. coli, and the zeocin marker was removed in vitro by treatment with Cre recombinase to make E3C–HT BAC (Fig. 1A). Restriction analysis of the E3C–HT bacmid demonstrated the expected digestion patterns with several different enzymes (data not shown). P3HR-1 cells were transfected with the E3C–HT bacmid, followed by selection with G418. A P3HR-1 cell clone containing an intact E3C–HT BAC was induced to produce virus. The resulting virus was used to infect primary human B cells, and LCLs were established in microwells in medium supplemented with 4HT. The LCLs were screened by PCR using primers that distinguish between P3HR-1 DNA (type 2 EBV) and AK-BAC-GFP DNA (type 1 EBV). Thirty percent (9 of 30) of the LCLs were positive for type 1 EBNA3C and negative for type 2 EBNA3C (data not shown).
Fig. 1.
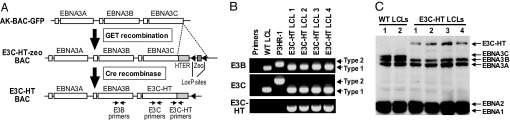
Establishment of LCLs infected with recombinant EBV expressing E3C–HT. (A) Construction of the E3C–HT EBV bacmid. A schematic representation of the EBV genomic EBNA3 region of AK-BAC-GFP is shown. A DNA construct containing the 4HT-responsive mutant estrogen receptor hormone-binding domain (HTER) and a zeocin marker (zeo) flanked by mutant loxP sites was inserted downstream of the EBNA3C ORF in AK-BAC-GFP by GET recombination. The zeocin marker was removed in vitro by treatment with Cre recombinase. The positions of PCR primers are indicated by arrows. (B) PCR analysis of the EBNA3B and EBNA3C genotypes of E3C–HT LCLs. The specific primer pairs distinguish between EBV type 1 and type 2 EBNA3B (E3B) and EBNA3C (E3C), and primers flanking the junction between the EBNA3C and HTER genes (E3C–HT) were used for amplification of fragments from type 1 WT LCLs, type 2 P3HR-1 cells, and four independent E3C–HT LCLs. Control amplifications without template are also shown (Primers). The E3B primers amplify 125- or 149-bp fragments from type 1 or type 2 DNA, respectively. The E3C primers amplify 153- or 246-bp fragments from type 1 or type 2 DNA, respectively. (C) E3C–HT LCLs express a large E3C–HT fusion protein and do not express WT EBNA3C. Total cell lysates of two independent WT LCLs and four independent E3C–HT LCLs were tested for the expression of EBNAs by Western analysis with EBV-immune human serum.
Four independent LCLs infected with type 1 EBNA3C-containing EBV alone were expanded and reanalyzed by PCR for the presence of type 1 E3C–HT and flanking DNA (Fig. 1B). These LCLs contained type 1 EBNA3B and EBNA3C DNA and lacked type 2 EBNA3B and EBNA3C DNA. Thus, the LCLs were infected with type 1 E3C–HT EBV, but not type 2 P3HR-1 EBV. These LCLs contained the E3C–HT fusion construct, as revealed by PCR analysis with a primer set that amplified the junction between the EBNA3C and HTER sequences (Fig. 1B). Western blot analysis using EBV-immune human serum confirmed that the E3C–HT LCLs did not express WT EBNA3C but instead expressed E3C–HT, which is larger than EBNA3C (Fig. 1C).
EBNA3C Is Essential for LCL Growth Maintenance.
Kinetic experiments showed that the expression level of E3C–HT protein was stable in the presence of 4HT. When 4HT was removed from the culture medium, however, E3C–HT protein slowly disappeared and was barely detectable after 2 weeks of cultivation, indicating that it is regulated by 4HT (Fig. 2A). The growth of four independent E3C–HT LCLs was examined in the absence or presence of 4HT. All E3C–HT LCLs grew continuously in medium with 4HT, but the same cells gradually ceased growing in medium without 4HT (Fig. 2B). The growth of control LCLs that expressed WT EBNA3C was unaffected by 4HT (Fig. 2B). To evaluate whether heterologous EBNA3C expression could complement E3C–HT inactivation, E3C–HT LCLs were transfected with an oriP plasmid expressing WT EBNA3C, followed by cultivation in medium with or without 4HT. The LCL transfection efficiency with an EGFP-expressing oriP plasmid was 20–40%, as estimated from the number of EGFP-positive cells. WT EBNA3C-transfected cells grew almost as well in the absence of 4HT as in its presence (Fig. 2D). In contrast, transfection with EBNA3A, EBNA3B, or the control oriP plasmid did not rescue the LCLs from the growth arrest induced by E3C–HT inactivation (Fig. 2D). Importantly, WT EBNA3C expression was stabilized at a higher level in cells growing in the absence of 4HT than in its presence (Fig. 2E). This result demonstrates that WT EBNA3C expression specifically enhances the proliferation of E3C–HT LCLs in the absence of 4HT, indicating that EBNA3C is essential for the maintenance of LCL growth.
Fig. 2.

EBNA3C is essential for LCL growth maintenance. (A) The E3C–HT protein slowly disappears after LCLs are transferred to medium without 4HT. Total cell lysates of E3C–HT LCLs, cultured in the absence (−) or presence (+) of 4HT for the indicated number of days, were immunoblotted with EBV-immune human serum or with an actin antibody. (B) E3C–HT LCLs depend on 4HT for growth. WT LCLs and four independent E3C–HT LCLs were cultured with (+) or without (−) 4HT. Viable cell numbers were determined by trypan blue exclusion, and total viable cell numbers derived from initial cell cultures are plotted. (C) Four independent E3C–HT LCLs were cultured in the absence (−) or presence (+) of 4HT for 17 days. Total cell lysates were prepared and subjected to Western blot analysis. (D) WT EBNA3C expression sustains the growth of E3C–HT LCLs in the absence of 4HT. E3C–HT LCLs were transfected with an oriP plasmid expressing EBNA3A (E3A), EBNA3B (E3B), EBNA3C (E3C), or a control oriP plasmid (Cont.). Five days after transfection (day 0), cells were washed and transferred to medium with (+) or without (−) 4HT, and cell growth was determined as described in B. (E) E3C–HT LCLs express higher levels of transfected WT EBNA3C in the absence of 4HT than in its presence. Total cell lysates were prepared from the cultures shown in D at the indicated time points and tested for EBNA3 expression by Western blot analysis with EBV-immune human serum. Note that transfected WT EBNA3C is derived from the EBV strain B95-8 and therefore migrates between EBNA3A and EBNA3B on an SDS/PAGE gel. HS blot, EBV-immune human serum blot. (F) Total cell lysates were prepared from the cultures shown in D at day 16 and subjected to Western blot analysis.
E3C–HT Inactivation Does Not Affect the Expression of Other EBNA Proteins or LMP1, c-myc, CD21, or CD23.
Because EBNA3C inactivation could affect the growth of LCLs by affecting the expression of other critical EBV latent proteins, we investigated the effect of E3C–HT inactivation on the expression of other EBNA proteins and LMP1. The expression of EBNA1, EBNA2, EBNA3A, EBNA3B, and LMP1 in E3C–HT LCLs did not clearly change over 20 days of E3C–HT inactivation (Fig. 3A). Similar results were repeatedly obtained and were reproduced with four independent E3C–HT LCLs (Fig. 2 C and F). We also evaluated the effect of E3C–HT inactivation on the expression of c-myc, CD21, and CD23, which are regulated by EBNA2 (36–39). The levels of c-myc expression were similar in the absence or presence of 4HT (Fig. 3A). The levels of CD21 and CD23 expression on the surfaces of LCLs were also indistinguishable under both conditions (Fig. 3B). Therefore, E3C–HT inactivation seems to affect the growth of LCLs without changing the expression of other critical viral latent proteins or c-myc.
Fig. 3.

E3C–HT inactivation does not affect the expression of other EBNAs, LMP1, c-myc, CD21, or CD23. (A) E3C–HT LCLs were cultured in the absence (−) or presence (+) of 4HT. Total cell lysates were prepared at the indicated time points and analyzed by Western blot with EBV-immune human serum or LMP1-, c-myc-, or actin-specific antibodies. (B) E3C–HT LCLs were cultured with (+) or without (−) 4HT for 14 days. The cell surface expression of CD21 and CD23 was monitored by flow cytometry (solid lines). Staining with isotype-matched control antibody is indicated by dotted lines. Each experiment was repeated with two independent E3C–HT LCLs with similar results.
EBNA3C Inactivation Results in a Decrease of Cells in S and G2/M Phases Accompanied by a Decrease in Hyperphosphorylated pRb.
The effect of E3C–HT inactivation on the cell cycle distribution of LCLs was assessed (Fig. 4A). In 4HT-containing medium, 54% of E3C–HT LCLs were in G0/G1 phase, 25% were in S phase, 13% were in G2/M phase, and 8% were in sub-G1 phase. The cell cycle profile of E3C–HT LCLs in the presence of 4HT was unchanged with time (data not shown). When LCLs were cultured in medium without 4HT, the proportions of cells in S and G2/M phases progressively fell to 5% and below 3%, respectively, and the proportion in sub-G1 phase progressively increased to 31% (Fig. 4A). Growth and cell cycle distribution of WT LCLs did not change in the absence or presence of 4HT, indicating that 4HT itself has no effect on cell cycle entry and cell survival (Fig. 2B and data not shown). Thus, EBNA3C inactivation inhibits cell cycle progression and results in an increase in the percent of hypodiploid cells. Western blot analysis showed that a fraction of pRb was consistently hyperphosphorylated in the presence of 4HT. By contrast, in the absence of 4HT, hyperphosphorylated pRb progressively diminished (Fig. 4B). The expression levels of hypophosphorylated pRb were similar in the absence and presence of 4HT (Fig. 4B, short exposure). Therefore, EBNA3C inactivation decreases cell cycle progression, which is accompanied by a decrease in hyperphosphorylated forms of pRb.
Fig. 4.

EBNA3C inactivation results in a decrease in the proportion of cells in S and G2/M phases, which is accompanied by a decrease in the level of hyperphosphorylated pRb. (A) E3C–HT LCLs were cultured in medium without 4HT for the indicated number of days. Cells were fixed and stained with propidium iodide, and cell cycle analysis was performed with a FACSCalibur. (B) E3C–HT LCLs were cultured in the absence (−) or presence (+) of 4HT for the indicated number of days. Total cell lysates were prepared, and Western blot analysis was performed with Rb- or actin-specific antibodies. Each experiment was repeated with two independent E3C–HT LCLs with similar results.
EBNA3C Inactivation Results in the Accumulation of p16INK4A.
Because EBNA3C inactivation caused a decrease in pRb phosphorylation, we examined the effect of EBNA3C inactivation on the expression of cyclin-dependent kinase (Cdk) inhibitors. Western blot analysis showed that the p16INK4A protein accumulated in E3C–HT LCLs that were cultured in medium without 4HT (Fig. 5A). The expression of cyclin A gradually decreased after EBNA3C inactivation (Fig. 5A). In contrast, we did not see any clear differences in the expression of p21 and p27 in the absence or presence of 4HT (Fig. 5A). p16INK4A did not accumulate in the E3C–HT LCLs that were transfected with an oriP plasmid expressing WT EBNA3C and cultured in medium without 4HT, indicating that heterologous expression of EBNA3C prevents the accumulation of p16INK4A in LCLs (Fig. 5B). Real-time RT-PCR analysis revealed that the level of p16INK4A transcript was higher in the absence of 4HT than in its presence and that it progressively increased after EBNA3C inactivation. Thus, the accumulation of p16INK4A occurs at the mRNA level (Fig. 5C). Because p16INK4A has previously been shown to disrupt cyclin D/Cdk4 and cyclin D/Cdk6 kinase complexes (40, 41), Cdk4 complexes in E3C–HT LCLs were analyzed in the absence and presence of 4HT. Lysates prepared from LCLs cultured with or without 4HT were immunoprecipitated with an anti-Cdk4 antibody. The immunoprecipitates were then subjected to Western blot analysis with anti-p16 and anti-cyclin D2 antibodies. We examined cyclin D2, but not cyclin D1, because it has been reported that cyclin D1 is not detected in LCLs (42). The expression levels of Cdk4, Cdk6, and cyclin D2 in the LCL lysates were similar in the absence or presence of 4HT (Fig. 5D). Almost identical amounts of Cdk4 were immunoprecipitated from LCLs cultured with or without 4HT. Cdk4 complexes from LCLs cultured without 4HT contained p16INK4A, but Cdk4 complexes from LCLs cultured with 4HT did not (Fig. 5D). In contrast, Cdk4 complexes from LCLs cultured with 4HT contained significantly more cyclin D2 than did LCLs cultured without 4HT (Fig. 5D). These results indicate that the p16INK4A that accumulates in LCLs after EBNA3C inactivation physically associates with Cdk4 and that EBNA3C inactivation results in an increase of the p16INK4A/Cdk4 complex and a decrease of the cyclin D2/Cdk4 complex in LCLs.
Fig. 5.

EBNA3C inactivation results in the accumulation of p16INK4A. (A) EBNA3C inactivation results in an increase in the level of p16 protein in LCLs. E3C–HT LCLs were cultured with (+) or without (−) 4HT for the indicated number of days. Western blot analysis with p16-, p21-, p27-, cyclin A (cycA)-, or actin-specific antibodies was performed. (B) Transfection of WT EBNA3C into E3C–HT LCLs prevents the accumulation of p16 in the absence of 4HT. E3C–HT LCLs were transfected with an oriP plasmid expressing WT EBNA3C (E3C) or a control oriP plasmid (Cont). Cells were cultured in medium with (+) or without (−) 4HT for 21 days and subjected to Western blot analysis with p16- and actin-specific antibodies. (C) EBNA3C inactivation results in an increase in the level of p16 transcript. Total RNA was extracted from E3C–HT LCLs cultured in medium with (+) or without (−) 4HT for 6 or 11 days. Real-time RT-PCR for p16 was performed, and the results were normalized against GAPDH. Reactions were done in triplicate. (D) EBNA3C inactivation results in an increase in the level of the p16/Cdk4 complex and a decrease in the level of the cyclin D2/Cdk4 complex. E3C–HT LCLs cultured with (+) or without (−) 4HT for 20 days were lysed and immunoprecipitated with a Cdk4-specific antibody. Total cell lysates were subjected to Western blot analysis with Cdk4-, Cdk6-, cyclin D2 (cycD2)-, and actin-specific antibodies. Immunoprecipitated samples (Cdk4 IP) were resolved by SDS/PAGE and blotted with Cdk4-, p16-, and cyclin D2-specific antibodies. Each experiment was repeated with two independent E3C–HT LCLs with similar results.
Discussion
We established LCLs expressing a conditionally active form of EBNA3C, E3C–HT, a system that allowed us to clarify the physiological role of EBNA3C in LCL growth maintenance. The level of E3C–HT protein gradually diminished in the absence of 4HT, and it was almost undetectable after 2 weeks of culture without 4HT. Concomitantly with the disappearance of E3C–HT, E3C–HT EBV-infected LCLs ceased growing in the absence of 4HT. Moreover, expression of WT EBNA3C from a transfected oriP plasmid enabled E3C–HT LCLs to grow in the absence of 4HT. These experiments clearly show that EBNA3C has an essential role in maintaining LCL growth.
In LCLs, EBNA3C extensively associates with a cellular transcription factor, RBP-Jκ, and it is presumed to modulate EBNA2–RBP-Jκ-mediated transcription (12–15). In transient transfection assays, EBNA3C expression strongly represses the EBNA2-mediated activation of the EBV Cp EBNA promoter, and it also activates the EBV LMP1 promoter in cooperation with EBNA2 (21–26). However, the effects of EBNA3C on cellular and viral gene expression have never, to our knowledge, been investigated by using a system in which EBNA3C is expressed at physiological levels. The data presented here indicate that EBNA3C inactivation does not clearly affect viral EBNAs or LMP1 expression. c-myc is a well known EBNA2-regulated gene that has a critical role in LCL growth (37–39). Our data show that the expression of c-myc is not affected by EBNA3C inactivation in LCLs. Thus, EBNA3C inactivation seems to inhibit LCL growth without affecting the expression of other EBNAs, LMP1, or c-myc.
It has been reported (30–33) that EBNA3C has cell cycle regulatory functions. It has been shown that transfection of cells with EBNA3C results in the accumulation of cells in G2/M phase (30). Consistently, our data show that cells in G2/M phase decreased after EBNA3C inactivation. EBNA3C has also been shown to stimulate cyclin A-dependent kinase activity without dramatically changing the level of cyclin A (30, 31). In our data, however, EBNA3C inactivation caused a decrease in the level of cyclin A. This may be due to the decrease of cells in S and G2/M phases after EBNA3C inactivation, because cyclin A is expressed mainly in S and G2/M phases (43). Recently, it was reported that EBNA3C recruits the SCFSKP2 ubiquitin ligase complex and mediates the degradation of p27 and pRb (32, 33). Both hyperphosphorylated and hypophosphorylated forms of pRb are reduced in EBNA3C-transfected cells (33). However, in our data, there was no obvious increase in p27 or pRb mediated by EBNA3C inactivation in LCLs, whereas hyperphosphorylated pRb was decreased by EBNA3C inactivation. Differences in the expression levels of EBNA3C and/or differences in cell type are possible explanations for these discrepancies. Alternatively, the increase of hypodiploid cells after EBNA3C inactivation might mask the increase in pRb, because it has been reported that pRb is a substrate of caspases and is degraded in apoptotic cells (44). This may also explain why EBNA3C inactivation in LCLs did not cause the accumulation of hypophosphorylated pRb, even though hyperphosphorylated pRb decreased.
Here, a dominant effect of EBNA3C inactivation in LCLs was the induction of p16INK4A transcription and the accumulation of p16INK4A protein. p16INK4A has been implicated in G1 cell cycle arrest in various cell types, and it specifically associates with Cdk4 and Cdk6 and prevents them from binding D type cyclins (40, 41). The association of p16 with Cdk4 and Cdk6 thus results in the inactivation of Cdk proteins, accumulation of hypophosphorylated pRb, and eventual G1 arrest (40, 41). Immunoprecipitation experiments showed that p16INK4A, which was induced by EBNA3C inactivation, associated with Cdk4 in LCLs. Moreover, the amount of cyclin D2 that coprecipitated with Cdk4 was less in EBNA3C-inactive LCLs than in EBNA3C-active LCLs. These results indicate that EBNA3C inactivation indeed causes an increase in the level of the p16INK4A/Cdk4 complex and a decrease in the level of the cyclin D2/Cdk4 complex in LCLs. Consistently, a decreased level of hyperphosphorylated pRb and a reduced frequency of cells in S and G2/M phases were observed after EBNA3C inactivation. Because the level of p16INK4A mRNA increased after EBNA3C inactivation, EBNA3C may repress p16INK4A expression at the transcriptional level. Nonetheless, it is still unclear whether EBNA3C directly regulates the expression of p16INK4A, and the mechanism by which p16INK4A is induced after EBNA3C inactivation remains to be elucidated. It has been reported that EBNA3C enhances the transformation of rat embryo fibroblasts and relieves p16's suppression of transformation (45). EBNA3C may regulate the p16INK4A-Rb pathway by various mechanisms to maintain the growth of LCLs.
Our data also show that EBNA3C inactivation in LCLs resulted in the increase of hypodiploid cells. This result suggests that EBNA3C contributes to the survival of LCLs. Further investigations are needed to clarify the link between EBNA3C and cell survival.
One important finding of our experiments is that EBNA3C has a role in LCL growth that cannot be filled by EBNA3A or EBNA3B. Transduction of EBNA3C into E3C–HT LCLs prevented growth arrest that was caused by E3C–HT inactivation, whereas the expression of EBNA3A or EBNA3B had no effect. Previous reports of LCLs expressing a conditionally active EBNA3A indicated that EBNA3A, but not EBNA3B or EBNA3C, rescues growth arrest caused by EBNA3A inactivation (16, 19). These results suggest that EBNA3C and EBNA3A have unequivocally distinct functions in supporting LCL growth.
Materials and Methods
Cell Lines.
P3HR-1 (clone 16) is a Burkitt's lymphoma cell line that contains a type 2 EBV genome. IB4 is an LCL transformed with B95-8 strain EBV. LCLs were maintained in RPMI medium 1640 supplemented with 15% FBS, l-glutamine, streptomycin, and penicillin and with or without 400 nM 4HT (Sigma, St. Louis, MO).
Plasmids.
Two loxP sites of pBS246 (Life Technologies, Grand Island, NY) were replaced with mutated loxP sites (2272 loxP) with synthetic oligonucleotides to construct pBS246-mloxp2272. The blunted FokI–BclI fragment of pcDNA4/HisMax (Invitrogen, Carlsbad, CA) containing the zeocin resistance marker gene, zeo, was cloned into the EcoRV site of pBS246-mloxp2272 to make pBS246-mloxp-zeo. A SpeI fragment containing a 4HT-responsive modified estrogen receptor hormone-binding domain, HTER, from pBSKS+ERTM (a gift of T. Littlewood, University of Cambridge, Cambridge, United Kingdom) was cloned into the SpeI site of pBS246-mloxp-zeo to construct pBS246-mloxp-zeo-HTER (34). oriP plasmids expressing Flag, EBNA3A, EBNA3B, or EBNA3C under the control of the SV40 promoter have been described (19). Lytic EBV replication was induced with pSVNaeI Z (19).
Construction of the E3C–HT EBV Bacmid.
AK-BAC-GFP derived from a type 1 EBV Akata strain has been described (35). Minipreparation, maxipreparation, and analysis of bacmid DNA have also been described (35). Construction of the E3C–HT EBV bacmid was performed in E. coli containing AK-BAC-GFP by Redγ, RecE, RecT (GET) recombination as described previously (35, 46). Plasmid pBS246-mloxp-zeo-HTER was PCR-amplified with primers 3CHT-FW, CGTGTTCCCAAAGGACGTGAAGCAGACTGACTACGATGCATCCACTGAAAGTGAGCTGGATCCACGAAATGAAATGG, and 3CHT-RV, CCATGTTTATGTGTCAGTCAAAGATCAAATAGCTAACAGGGGGCACCTTGGATCCCCCCTTTCGTCTTCAAGAATTC, to obtain the linear targeting construct required for GET recombination. The resultant PCR product (1,768 bp long) containing HTER, the zeocin marker surrounded by mutant loxP sites, and 57-bp sequences homologous to the target regions of AK-BAC-GFP at 5′ and 3′ ends was purified, DpnI-digested, and electroporated into recombinase-induced DH10B electrocompetent E. coli (harboring AK-BAC-GFP and pGETrec). The successfully recombined bacmid (E3C–HT–zeo BAC) was purified from zeocin-resistant E. coli and treated with Cre recombinase (Novagen, Madison, WI) in vitro to remove the zeocin marker. The resultant bacmid (E3C–HT BAC) was purified from retransformed E. coli.
Transfection of Bacmids into P3HR-1 Cells.
P3HR-1 c16 cells (5 × 106) were transfected with 5 μg of bacmid DNA via electroporation with a Bio-Rad (Hercules, CA) Gene Pulser II (190 V, 950 μF). Cells were selected in medium containing 1 mg of G418 (Sigma) per milliliter in 96-well tissue culture plates. G418-resistant clones were screened for the presence of intact bacmids. BAC episomes were isolated from P3HR-1 cells by an alkaline lysis procedure and analyzed as described previously (35).
Virus Production and Primary B Lymphocyte Infections.
Lytic EBV infection was induced in P3HR-1cell clones (5 × 106) by transfection with 20 μg of pSVNaeI Z via electroporation. Cell-free virus was prepared by filtration (0.45-μm pore-size filter) of the culture supernatant. Primary human mononuclear cells were infected with diluted virus, and aliquots were distributed into each well of 96-microwell plates in medium with 400 nM 4HT. LCLs were macroscopically visible 3–5 weeks after plating.
PCR Analyses.
Oligonucleotide primers for amplification of distinctive fragments from type 1 versus type 2 EBNA3B or from EBNA3C have been described (19). Oligonucleotides E3C-FW, AAGCTACTGCTGAAGCACAG, and HT-RV, CTGAAGGGTCTAGAAGGATC, were used to amplify the junction between EBNA3C- and HTER-encoding DNA. Cell DNA was prepared for PCR, amplified for 40 cycles with annealing at 58°C, and analyzed by electrophoresis. For real-time RT-PCR, a SYBR Premix Ex Taq kit (Takara Bio, Tokyo, Japan) and a LightCycler (Roche Applied Science, Indianapolis, IN) were used. p16-FW primer, CCCGCTTTCGTAGTTTTCAT, and p16-RV primer, TTATTTGAGCTTTGGTTCTG, were used to quantify the p16 transcript. Reactions were done in triplicate. The results were normalized against GAPDH. The calculation for the real-time RT-PCR data has been described (47).
Western Blotting and Immunoprecipitation.
SDS/PAGE and Western blotting procedures were described previously (19). Anti-c-myc, anti-p16, anti-p21, anti-p27, anti-Cdk4, anti-Cdk6, and anti-cyclin D2 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and mouse anti-human Rb monoclonal antibody G3-245 was purchased from PharMingen (San Diego, CA). For immunoprecipitation, cells were lysed in immunoprecipitation buffer (1% Nonidet P-40/50 mM Tris, pH 7.5/2 mM EDTA/150 mM NaCl, supplemented with protease inhibitors). Lysates were precleared and rotated with anti-Cdk4 antibody, and immune complexes were precipitated with protein A–Sepharose beads. Samples were washed and analyzed by SDS/PAGE and Western blotting.
FACS Analyses for Cell DNA and Surface Expression of CD21 and CD23.
Approximately 106 cells were fixed and stained with propidium iodide (Sigma) or incubated live with CD21 or CD23 antibodies (Dako, Glostrup, Denmark), followed by incubation with biotinylated anti-mouse IgG and streptavidin/phycoerythrin (Dako). Cells were analyzed by using FACSCalibur and Cell Quest software (Becton Dickinson, Franklin Lakes, NJ). The percentages of cells in G0/G1, S, G2/M, and sub-G1 phases were calculated by using FlowJo software (Tree Star, Ashland, OR).
LCL Growth.
LCL cells (1 × 106) were cultured in 25-cm2 culture flasks in 10 ml of complete medium (1 × 105 cells per milliliter) with or without 4HT. Every 3–7 days, the viable cell number was determined by hemocytometer based on trypan blue exclusion, and cultures were diluted to 1 × 105 cells per milliliter with fresh medium to maintain optimum growth. Total viable cell numbers were calculated based on the expansion from the initial 1 × 106 cells.
Complementation Experiments.
E3C–HT LCLs (1 × 107) were transfected with 30 μg of oriP plasmid expressing WT EBNA3C, EBNA3A, EBNA3B, or control oriP plasmid via electroporation (Bio-Rad Gene Pulser II; 210 V, 950 μF). Transfected LCLs were cultured in medium with 4HT for 5 days. Cells were then washed, and 1 × 106 cells were cultured in 10 ml of complete medium with or without 4HT in a 25-cm2 culture flask. Every 4–8 days, viable cell numbers were counted, cultures were split, and total viable cell numbers were calculated relative to the initial culture.
Acknowledgments
We thank E. Kieff for providing oriP plasmids expressing EBNA3A, EBNA3B, and EBNA3C and for helpful suggestions and discussions and H. Higashi and B. Zhao for helpful suggestions. This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (to S.M. and K.T.).
Abbreviations
- LCL
lymphoblastoid cell line
- EBNA
Epstein–Barr virus nuclear antigen
- 4HT
4-hydroxytamoxifen
- Cdk
cyclin-dependent kinase.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
References
- 1.Rickinson AB, Kieff E. In: Fields Virology. Knipe DM, Howley PM, editors. Vol 2. Philadelphia: Lippincott; 2001. pp. 2575–2628. [Google Scholar]
- 2.Henle W, Diehl V, Kohn G, zur Hausen H, Henle G. Science. 1967;157:1064–1065. doi: 10.1126/science.157.3792.1064. [DOI] [PubMed] [Google Scholar]
- 3.Kieff E, Rickinson AB. In: Fields Virology. Knipe DM, Howley PM, editors. Vol 2. Philadelphia: Lippincott; 2001. pp. 2511–2574. [Google Scholar]
- 4.Joab I, Rowe DT, Bodescot M, Nicolas JC, Farrell PJ, Perricaudet M. J Virol. 1987;61:3340–3344. doi: 10.1128/jvi.61.10.3340-3344.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimizu N, Yamaki M, Sakuma S, Ono Y, Takada K. Int J Cancer. 1988;41:744–751. doi: 10.1002/ijc.2910410518. [DOI] [PubMed] [Google Scholar]
- 6.Petti L, Kieff E. J Virol. 1988;62:2173–2178. doi: 10.1128/jvi.62.6.2173-2178.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petti L, Sample J, Wang F, Kieff E. J Virol. 1988;62:1330–1338. doi: 10.1128/jvi.62.4.1330-1338.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ricksten A, Kallin B, Alexander H, Dillner J, Fahraeus R, Klein G, Lerner R, Rymo L. Proc Natl Acad Sci USA. 1988;85:995–999. doi: 10.1073/pnas.85.4.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang H, Cho YG, Wang F. J Virol. 2000;74:5921–5932. doi: 10.1128/jvi.74.13.5921-5932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rivailler P, Jiang H, Cho YG, Quink C, Wang F. J Virol. 2002;76:421–426. doi: 10.1128/JVI.76.1.421-426.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rivailler P, Cho YG, Wang F. J Virol. 2002;76:12055–12068. doi: 10.1128/JVI.76.23.12055-12068.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robertson ES, Grossman S, Johannsen E, Miller C, Lin J, Tomkinson B, Kieff E. J Virol. 1995;69:3108–3116. doi: 10.1128/jvi.69.5.3108-3116.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robertson ES, Lin J, Kieff E. J Virol. 1996;70:3068–3074. doi: 10.1128/jvi.70.5.3068-3074.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waltzer L, Perricaudet M, Sergeant A, Manet E. J Virol. 1996;70:5909–5915. doi: 10.1128/jvi.70.9.5909-5915.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao B, Marshall DR, Sample CE. J Virol. 1996;70:4228–4236. doi: 10.1128/jvi.70.7.4228-4236.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maruo S, Johannsen E, Illanes D, Cooper A, Zhao B, Kieff E. J Virol. 2005;79:10171–10179. doi: 10.1128/JVI.79.16.10171-10179.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tomkinson B, Robertson E, Kieff E. J Virol. 1993;67:2014–2025. doi: 10.1128/jvi.67.4.2014-2025.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomkinson B, Kieff E. J Virol. 1992;66:2893–2903. doi: 10.1128/jvi.66.5.2893-2903.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maruo S, Johannsen E, Illanes D, Cooper A, Kieff E. J Virol. 2003;77:10437–10447. doi: 10.1128/JVI.77.19.10437-10447.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen A, DiVisconte M, Jiang X, Quink C, Wang F. J Virol. 2005;79:4506–4509. doi: 10.1128/JVI.79.7.4506-4509.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johannsen E, Miller CL, Grossman SR, Kieff E. J Virol. 1996;70:4179–4183. doi: 10.1128/jvi.70.6.4179-4183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Radkov SA, Bain M, Farrell PJ, West M, Rowe M, Allday MJ. J Virol. 1997;71:8552–8562. doi: 10.1128/jvi.71.11.8552-8562.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allday MJ, Farrell PJ. J Virol. 1994;68:3491–3498. doi: 10.1128/jvi.68.6.3491-3498.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao B, Sample CE. J Virol. 2000;74:5151–5160. doi: 10.1128/jvi.74.11.5151-5160.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin J, Johannsen E, Robertson E, Kieff E. J Virol. 2002;76:232–242. doi: 10.1128/JVI.76.1.232-242.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosendorff A, Illanes D, David G, Lin J, Kieff E, Johannsen E. J Virol. 2004;78:367–377. doi: 10.1128/JVI.78.1.367-377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radkov SA, Touitou R, Brehm A, Rowe M, West M, Kouzarides T, Allday MJ. J Virol. 1999;73:5688–5697. doi: 10.1128/jvi.73.7.5688-5697.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knight JS, Lan K, Subramanian C, Robertson ES. J Virol. 2003;77:4261–4272. doi: 10.1128/JVI.77.7.4261-4272.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Touitou R, Hickabottom M, Parker G, Crook T, Allday MJ. J Virol. 2001;75:7749–7755. doi: 10.1128/JVI.75.16.7749-7755.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knight JS, Robertson ES. J Virol. 2004;78:1981–1991. doi: 10.1128/JVI.78.4.1981-1991.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knight JS, Sharma N, Kalman DE, Robertson ES. J Virol. 2004;78:12857–12867. doi: 10.1128/JVI.78.23.12857-12867.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knight JS, Sharma N, Robertson ES. Mol Cell Biol. 2005;25:1749–1763. doi: 10.1128/MCB.25.5.1749-1763.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knight JS, Sharma N, Robertson ES. Proc Natl Acad Sci USA. 2005;102:18562–18566. doi: 10.1073/pnas.0503886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanda T, Yajima M, Ahsan N, Tanaka M, Takada K. J Virol. 2004;78:7004–7015. doi: 10.1128/JVI.78.13.7004-7015.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang F, Gregory CD, Rowe M, Rickinson AB, Wang D, Birkenbach M, Kikutani H, Kishimoto T, Kieff E. Proc Natl Acad Sci USA. 1987;84:3452–3456. doi: 10.1073/pnas.84.10.3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaiser C, Laux G, Eick D, Jochner N, Bornkamm GW, Kempkes B. J Virol. 1999;73:4481–4484. doi: 10.1128/jvi.73.5.4481-4484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao B, Maruo S, Cooper A, Chase MR, Johannsen E, Kieff E, Cahir-McFarland E. Proc Natl Acad Sci USA. 2006;103:1900–1905. doi: 10.1073/pnas.0510612103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cooper A, Johannsen E, Maruo S, Cahir-McFarland E, Illanes D, Davidson D, Kieff E. J Virol. 2003;77:999–1010. doi: 10.1128/JVI.77.2.999-1010.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Serrano M, Hannon GJ, Beach D. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 41.Sherr CJ, Roberts JM. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 42.Palmero I, Holder A, Sinclair AJ, Dickson C, Peters G. Oncogene. 1993;8:1049–1054. [PubMed] [Google Scholar]
- 43.Erlandsson F, Linnman C, Ekholm S, Bengtsson E, Zetterberg A. Exp Cell Res. 2000;259:86–95. doi: 10.1006/excr.2000.4889. [DOI] [PubMed] [Google Scholar]
- 44.Tan X, Wang JY. Trends Cell Biol. 1998;8:116–120. doi: 10.1016/s0962-8924(97)01208-7. [DOI] [PubMed] [Google Scholar]
- 45.Parker GA, Crook T, Bain M, Sara EA, Farrell PJ, Allday MJ. Oncogene. 1996;13:2541–2549. [PubMed] [Google Scholar]
- 46.Ahsan N, Kanda T, Nagashima K, Takada K. J Virol. 2005;79:4415–4424. doi: 10.1128/JVI.79.7.4415-4424.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kitagawa N, Goto M, Kurozumi K, Maruo S, Fukayama M, Naoe T, Yasukawa M, Hino K, Suzuki T, Todo S, Takada K. EMBO J. 2000;19:6742–6750. doi: 10.1093/emboj/19.24.6742. [DOI] [PMC free article] [PubMed] [Google Scholar]