

Abstract
Allyl isothiocyanate, the pungent principle of wasabi and other mustard oils, produces pain by activating TRPA1, an excitatory ion channel on sensory nerve endings. Isothiocyanates are membrane-permeable electrophiles that form adducts with thiols and primary amines, suggesting that covalent modification, rather than classical lock-and-key binding, accounts for their agonist properties. Indeed, we show that thiol reactive compounds of diverse structure activate TRPA1 in a manner that relies on covalent modification of cysteine residues within the cytoplasmic N terminus of the channel. These findings suggest an unusual paradigm whereby natural products activate a receptor through direct, reversible, and covalent protein modification.
Keywords: chemical modification, irritants, natural products, pain
Plants have evolved ingenious defensive strategies to ward off herbivorous predators. Such protective mechanisms often involve the production of chemical irritants that activate sensory nerve fibers of offending creatures to produce discomfort or pain (1, 2). For example, plants of the genus Brassica (mustard), Allium (onion), and Cinnamomum (cinnamon) produce pungent isothiocyanate, thiosulfinate, and α,β-unsaturated aldehyde compounds, respectively, which elicit acute pain and neurogenic inflammation by activating TRPA1, a nonselective cation channel expressed by sensory neurons of the pain pathway (3–7). Each of these compounds is capable of forming covalent adducts with thiols, primary amines, and to a lesser extent, hydroxyl groups (8), raising questions as to how electrophiles with such indiscriminate reactivity function as reversible and specific ion-channel agonists.
Here we provide evidence to support a model whereby a variety of structurally distinct environmental irritants activate TRPA1 through a mechanism primarily involving covalent modification of specific cysteine side chains located within the putative cytoplasmic N-terminal domain of the channel. Pharmacological analysis of various sulfhydryl reactive agents provides a biochemical rationale for the reversible properties of mustard oil and other naturally occurring TRPA1 agonists. Importantly, TRPA1 mutants that are insensitive to these electrophilic agonists retain their ability to function as “receptor operated” channels, indicating that distinct biochemical pathways can contribute to TRPA1 activation by environmental or endogenous stimuli. These studies provide a mechanistic framework to further elucidate biochemical and structural parameters underlying activation of this member of the TRP ion channel family.
Results
The diverse chemical nature of TRPA1-activating irritants (Fig. 1a) suggests that reactivity, rather than structure per se, constitutes the critical determinant of TRPA1 agonist activity. To clarify this issue, we first asked whether benzyl thiocyanate (BTC) is able to activate TRPA1. BTC is isosteric with the TRPA1 agonist benzyl isothiocyanate (BITC), and, thus, whereas both compounds possess thiocyanate functional groups of similar size, the single-bond character of the S–C linkage in BTC reduces its electrophilicity compared with BITC (Fig. 1b). As previously shown, perfusion of TRPA1-expressing Xenopus oocytes with BITC (20 μM) produced robust membrane currents (3). In contrast, BTC had little, if any, effect, consistent with the idea that agonist actions of isothiocyanates reflect their reactive properties, rather than their affinity for a specific ligand-binding pocket.
Fig. 1.

Chemical reactivity, rather than structure, confers TRPA1 agonist properties. (a) Chemical structures of electrophilic TRPA1 agonists AITC (mustard oil), diallyl disulfide (DADS), and cinnamaldehyde. (b) Structural similarities between benzyl isothiocyanate (BITC, Left Upper) and benzyl thiocyanate (BTC, Left Lower) do not reflect agonist properties. Representative trace from an oocyte expressing human TRPA1 (n = 5; Right). Benzyl- and allyl isothiocyantes evoke large, reversible inward currents at –80 mV, whereas BTC does not (20 μM each). (Scale bars: 200 nA and 1 min.) (c) Precomplexed conjugates of AITC (compounds A and B, 200 μM each) do not activate TRPA1 expressed in HEK293 cells transfected with TRPA1, as measured by Fura-2/AM ratiometric calcium imaging. Traces represent normalized average response of 250 cells over two trials. Thiourea A and dithiocarbamate B are synthetic compounds designed to mimic adducts likely to form when AITC is introduced into the cellular environment. Response to AITC (200 μM) verifies expression of function channels. Mean peak response ± SEM: A, 1.04 ± 0.11; B, 1.10 ± 0.03; AITC, 4.63 ± 0.12.
Perhaps isothiocyanate electrophiles function as prodrugs that must be converted to an active species in situ. In this case, an endogenous nucleophile, such as glutathione or spermine, would form a covalent complex with isothiocyanate to produce an active dithiocarbamate or thiourea. To test this possibility, we used calcium imaging to determine whether two model compounds, allyl isothiocyanate complexed to N-acetyl cysteine or N-Boc-diamino hexane (Fig. 1c), could activate TRPA1 in transfected HEK293 cells (9, 10). Neither compound possessed agonist activity, and because a prospective isothiocyanate conjugate must contain similar structural features, a prodrug scenario seems unlikely.
We next pursued the hypothesis that TRPA1 activation by isothiocyanates (or other electrophilic agonists) occurs via direct protein modification. To test this possibility, we asked whether TRPA1 could be activated with a cysteine-reactive electrophile that is structurally unrelated to any natural product agonist. We chose the maleimide functional group because it is widely used to form covalent linkages with sulfhydryl side chains and, in contrast to isothiocyanates (11), forms adducts that are irreversible under physiological conditions (Fig. 2a). Indeed, TRPA1-expressing oocytes or HEK293 cells were rapidly and robustly activated by N-methyl maleimide (NMM) within a concentration range (10–200 μM) similar to that required for activation by allyl isothiocyanate (AITC) (Fig. 2 b–d). NMM-induced responses were attenuated by the pore blocker ruthenium red and showed outward rectification and single-channel conductance properties that also are characteristic of AITC-evoked currents (Fig. 2 b and c and see below). However, in striking contrast to responses elicited by AITC, currents evoked by NMM persisted, even after extensive washout. These observations are consistent with a channel activation mechanism involving irreversible covalent modification of a cysteine side chain by the maleimide functional group.
Fig. 2.

Synthetic cysteine-modification reagent NMM irreversibly activates TRPA1. (a) A chemical model of reversible and irreversible agonist action by AITC and NMM, respectively. (b) Representative trace from an oocyte expressing human TRPA1, showing reversible activation by AITC and irreversible activation by NMM (50 μM each, –80 mV) (n = 9). Pore blocker ruthenium red is used to demonstrate channel specificity (RR, 10 μM). Control injected oocytes showed no response (data not shown). (Scale bars: 50 nA and 1 min.) (c) Current–voltage relationships of TRPA1 responses to AITC (red trace) and NMM (blue trace) shown in b, as assessed by 1-s voltage ramps. (d) Calcium imaging of HEK293 cells transfected with TRPA1 shows robust responses to NMM (20 μM). (e) Average traces from Fura-2 ratiometric imaging of 20 capsaicin-responsive trigeminal neurons derived from matched littermates of wild-type (+/+) and TRPA1-null (−/−) mice. Responses to NMM and AITC (20 μM each) overlap in wild-type neurons (blue trace), whereas NMM fails to activate neurons derived from TRPA1-null mice (red trace). Mean peak responses to NMM: 2.32 ± 0.2 (+/+), 1.05 ± 0.1 (−/−).
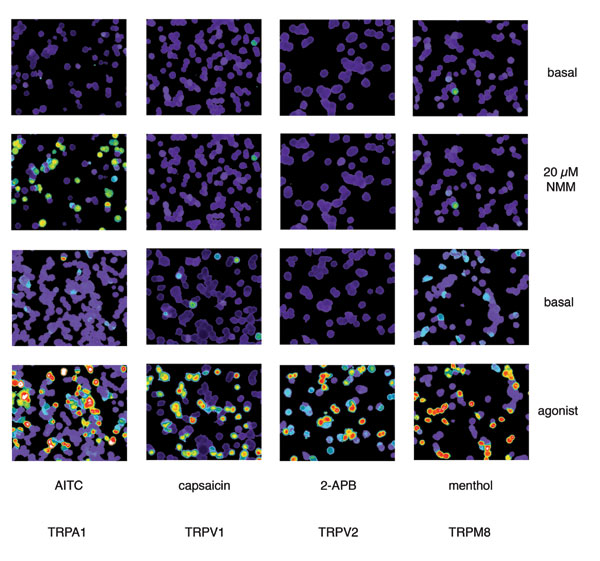
Although NMM is highly reactive and indiscriminate with respect to protein targets, its effects were specific in that no responses were observed with vector-transfected controls, or in cells expressing other TRP family members, including the heat-activated channels TRPV1 and TRPV2, and the cold- and menthol-activated channel, TRPM8 [supporting information (SI) Fig. 5]. These results indicate that acute exposure to AITC or NMM produces specific, TRPA1-mediated effects on membrane conductance, despite indiscriminate reactivity of these agents with available sulfhydryl groups. This is also the case for native TRPA1 receptors, where calcium imaging showed robust and overlapping responses to AITC and NMM in trigeminal neurons cultured from wild-type, but not TRPA1-deficient mice (Fig. 2e).
AITC and NMM may activate TRPA1 through a mechanism involving direct covalent modification of specific cysteine residues within the channel protein. To address this possibility, we generated a series of cysteine-to-serine or -alanine substitutions at 13 of 21 positions that are invariant among human, rat, and mouse TRPA1 sequences (Fig. 3a). These mutants were expressed in HEK293 cells, and the response to Cys-modification reagents was assessed by using calcium imaging. Replacement of 10 cysteine residues had little or no effect on channel function; however, substitutions at three closely spaced positions (C619, C639, and C663) located within the cytoplasmic N terminus resulted in partial, but significant and cumulative, decreases in electrophile-evoked responses. The resulting triple mutant, TRPA1-3C, exhibited pharmacological properties consistent with the specific ablation of cysteine modification-mediated channel gating. Thus, TRPA1 was activated by the membrane-permeable, sulfhydryl reactive agent MTSEA (but not by MTSET, a membrane impermeant variant; Fig. 3b), whereas the triple mutant was MTSEA-insensitive, and responded to AITC slowly only and at concentrations in excess of the wild-type EC50, as will be further described below. The observation that wild-type TRPA1 is activated by MTSEA and not MTSET correlates agonist properties with membrane permeability and cysteine reactivity, consistent with the notion that TRPA1 gating by electrophilic agonists is mediated by the identified cytosolic cysteines. Mutant TRPA1-3C channels also were unresponsive to NMM as determined by a variety of methods, including calcium imaging in transfected HEK293 cells (SI Fig. 6), whole-cell voltage-clamp recordings in oocytes (Fig. 3c), or single-channel analysis of cell-attached membrane patches (Fig. 3d). Despite this insensitivity to sulfhydryl reactive agonists, the TRPA1-3C mutant retained sensitivity to structurally unrelated agonists, such as δ-9-tetrahydrocannabinol (THC) or 2-aminophenyl borane (2-APB) (see below), demonstrating a specific requirement for these three cysteine residues (C619, C639, and C663) in mediating sensitivity to electrophilic agonists.
Fig. 3.

Three cysteine residues in the intracellular N terminus of TRPA1 confer sensitivity to NMM. (a) Cysteine mutations conferring sensitivity to NMM cluster within a 50-aa stretch between the cluster of 18 ankyrin repeats and the first putative transmembrane region. Red dots denote residues required for full response to sulfhydryl modification reagents. Blue dots denote sites where mutations had no effect. (b) Activation of wild-type hTRPA1 (black trace) and triple mutant hTRPA1-3C (green trace) by sulfhydryl-selective reagents show expected properties for an intracellular reactive site. Membrane-impermeable reagent MTSET (200 μM) fails to activate either channel, whereas membrane-permeant MTSEA (200 μM) activates wild-type hTRPA1 only. AITC (200 μM) is used as a positive control. Traces represent normalized average calcium response of transfected HEK293 cells (n = 350) as assessed by Fura-2/AM ratiometric calcium imaging. Mean peak responses ± SEM: MTSET, 1.09 ± 0.01 (WT), 1.02 ± 0.0003 (3C); MTSEA, 2.34 ± 0.07 (WT), 1.03 ± 0.01 (3C); AITC, 4.73 ± 0.05 (WT), 4.87 ± 0.50 (3C). (c) Representative traces (–80 mV) from an oocyte expressing wild-type hTRPA1 (black) compared with NMM-insensitive triple mutant hTRPA1-3C (green). THC (400 μM, with 200 μM β-cyclodextran carrier) elicits robust, reversible inward currents in both cases, whereas NMM (50 μM) does not appreciably activate the triple mutant (n = 5 for each trace). (Scale bars: 200 nA and 1 min.) (d) NMM (50 μM, trace 1) activates human TRPA1 in a similar manner to AITC (20 μM, trace 2). For traces 1 and 2, HEK293 cells stably expressing hTRPA1 were recorded under cell-attached configuration at a holding potential of –60 mV, with sampling frequency of 5 kHz, filtering at 1 kHZ. Single-channel conductances (±SD) were as follows: 102.7 ± 12.9 for NMM (n = 4) and 101.8 ± 10.9 pS (n = 4) for AITC. Agonists were delivered by backfilling pipet with drug. For traces 3 and 4, oocytes injected with TRPA1-3C were recorded under similar conditions. Activation of TRPA1 by NMM (50 μM) is specifically abrogated in triple mutant hTRPA1-3C (trace 3), whereas AITC (500 μM) still can activate this current (trace 4). Single-channel conductance (±SD) was 107.7 ± 10.7 pS (n = 6) with 500 μM AITC backfilled in the pipet. (Scale bars: 10 pA and 200 msec.) (e) Triple mutant TRPA1-3C retains receptor-operated channel function. Average traces of calcium response from transfected HEK 293 cells (n = 50, two trials), as assessed by Fura-2/AM imaging. Upon application of bradykinin (BK) (100 nM), cells cotransfected with BK2R and wild-type hTRPA1 (black trace) or triple mutant hTRPA1-3C (green trace) show sustained elevated calcium levels in excess of cells transfected with BK2R alone (red trace). Mean peak responses: 2.52 ± 0.18 (WT + BK2R), 2.80 ± 0.23 (3C + BK2R), and 1.29 ± 0.20 (BK2R alone). (f) Triple mutant hTRPA1-3C is not responsive to diallyl disulfide (DADS) (600 μM). Representative trace from oocytes expressing triple mutant hTRPA1-3C (n = 5). Response to AITC (200 μM) is used as a positive control. (Scale bars: 100 nA and 1 min.)
Previous studies have shown that TRPA1 functions as a canonical “receptor-operated” TRP channel that can be activated downstream of phospholipase C-coupled receptors (3, 4). We therefore asked whether this signaling pathway requires the three cysteine residues necessary for activation by NMM. Using calcium imaging, we compared populations of HEK293 cells transfected with the bradykinin receptor (BK2R) alone and in combination with TRPA1 or TRPA1-3C. Cells expressing BK2R alone showed a characteristic calcium store release transient, whereas in those expressing both BK2R and TRPA1, an enhanced calcium response was observed reflecting calcium influx through activated TRPA1 channels. Similar results were obtained with the TRPA1-3C mutant, indicating that residues required for normal sensitivity to sulfhydryl-reactive agonists are not involved in PLC-mediated channel activation. We also observed PLC-mediated TRPA1 activation directly in whole-cell patch-clamp recordings from HEK293 cells transfected with the m1 muscarinic acetylcholine receptor (m1AChR) and TRPA1-3C (3) (SI Fig. 7). Taken together, these results show that TRPA1 can be activated by independent mechanisms having distinct structural requirements.
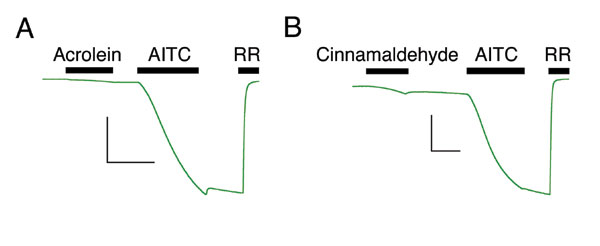
In addition to AITC, TRPA1 is activated by a variety of other natural product or environmental irritants, such as diallyl disulfide (DADS) from garlic, cinnamaldehyde from cinnamon, and acrolein, a common air pollutant. Like AITC or NMM, these agents show preferential reactivity with sulfhydryl groups, and, therefore, we asked whether they share the same requirement for the cysteine cluster identified above. As predicted, the TRPA1-3C mutant showed little, if any, activation by these compounds (Fig. 3f and SI Fig. 8), consistent with a gating mechanism involving covalent modification at these sites.
As shown above, the TRPA1-3C mutant is largely insensitive to electrophiles with a preference for cysteine modification. Interestingly, however, we noted that this mutant retained a weakened, but finite, sensitivity to isothiocyanates that was apparent on prolonged exposure to relatively high concentrations (>100 μM) of AITC (Fig. 3 b and d). At the single-channel level, these responses resembled those of wild-type TRPA1 channels, having a similar unitary conductance (Fig. 3d). However, electrophysiological analysis in oocytes showed an ≈10-fold difference in potency when comparing AITC-evoked activation of wild-type TRPA1 and TRPA1-3C (EC50 = 64.5 ± 3.12 versus 602 ± 44.7 μM, respectively) (Fig. 4a). Another important distinction between the wild-type and mutant response was the fact that the latter was irreversible on washout (Fig. 4b). A similar shift in the magnitude and kinetics of the TRPA1-3C response could be observed with calcium imaging of transfected HEK293 cells (Fig. 4c). These results suggest that AITC primarily targets the three cysteines that are essential for activation by sulfhydryl-selective electrophiles, but that a component of isothiocyanate sensitivity may involve modification of other residues. Because isothiocyantes also can modify primary amines, we reasoned that this residual response could reflect irreversible covalent protein modification of lysine side chains (Fig. 4d). Numerous lysines lie within close proximity of the C619, C639, and C663 cluster (Fig. 3a), and, therefore, we asked whether any of these residues might account for the AITC-evoked response observed with the TRPA1-3C mutant. Indeed, calcium imaging in transfected HEK293 cells showed that replacement of lysine-708 with arginine (K708R) rendered the TRPA1-3C mutant completely insensitive to AITC, without affecting responses to the control agonist, 2-APB (Fig. 4c). Similar results were obtained when voltage-clamp analysis in oocytes was used to assess a lysine-to-glutamine substitution at this same position (K708Q) (Fig. 4b). As a control, we found that TRPA1 channels bearing the single lysine mutation (K708R) retained sensitivity to AITC or NMM, as well as to ruthenium red blockade (SI Fig. 9).
Fig. 4.

Residual AITC response of triple mutant TPPA1-3C is mediated by irreversible modification of lysine-708. (a) AITC dose–response curves for TRPA1 and TRPA1-3C expressed in oocytes (n = 5 for each data point). Points were obtained by measuring response to 30-sec pulse of test dose at +80 mV and normalized to maximum response elicited by 5 mM AITC. EC50 values were obtained by using curves fit with the least-squares approximation method. (b) Representative traces (–80 mV) from oocytes expressing wild-type hTRPA1 (black trace), triple mutant hTRPA1-3C (green trace), and quadruple mutant TRPA1-3C–K708Q (red trace) (n = 5 each). Response to AITC (200 μM) is weaker and irreversible in triple mutant and completely ablated in quadruple mutant. Response to THC (400 μM) and blockade by ruthenium red (10 μM) are used as positive controls. (Scale bars: 200 nA and 1 min.) (c) Average traces of calcium response from transfected HEK293 cells (n = 250, two trials), as assessed by ratiometric Fura-2/AM imaging. Cells transfected with quadruple mutant hTRPA1-3C–K708R (red trace) are not sensitive to AITC (150 μM) but retain capacity to respond to 2-APB (200 μM), whereas cells transfected with triple mutant TRPA1-3C (green trace) or wild-type TRPA1 (black trace) respond to both drugs. Mean peak responses: AITC, 4.58 ± 0.04 (WT), 3.54 ± 0.01 (3C), 1.17 ± 0.04 (K708R); 2-APB, 4.43 ± 0.04 (WT), 5.19 ± 0.05 (3C), 5.30 ± 0.05 (K708R). (d) A chemical model for irreversible activation of TRPA1-3C involving the formation of a stable thiourea linkage between AITC and a lysine side chain.
Discussion
Our findings elucidate the biochemical mechanism whereby natural product and environmental irritants interact with TRPA1. Although we have not demonstrated chemical labeling of specific cysteine residues per se, our site-directed mutagenesis and pharmacological experiments provide compelling evidence that C619, C639, and C663 (and to a lesser extent K708) are directly modified by electrophilic agonists, which gate TRPA1 through reactivity-based, reversible, covalent modification of the cytoplasmic N-terminal tail of the channel protein. This proposed mechanism is shared by other TRPA1 agonists derived from a variety of plant (e.g., garlic and cinnamon) and synthetic (air pollutant and chemotherapeutic agents) origin. This is in contrast to other natural-product TRP-channel activators, such as capsaicin, menthol, and camphor, which lack a reactive functional group and likely interact with their targets (TRPV1, TRPM8, and TRPV3, respectively) through a more classical noncovalent mechanism.
Recent work has established that TRPA1 acts as both a ligand-gated and receptor-operated excitatory channel. Indeed, this ability to function as a polymodal signal detector is shared with a number of other TRP family members and represents a mechanism whereby these channels can integrate information from numerous physiological inputs (12–14). Our analysis of TRPA1 mutants clearly shows that channels lacking response to cysteine-reactive agonists, such as NMM and diallyl disulfide, retain sensitivity to receptor-mediated PLC activation, a process that may result from the consequent elevation in cytoplasmic calcium levels (3, 7). Regardless of the exact mechanism of PLC-evoked channel activation, we demonstrate that gating by electrophilic agonists is structurally separable from that governing the receptor-mediated pathway.
Although numerous drugs and natural products covalently cross-link to their biological target, the TRPA1 agonists described above are anomalous because of their indiscriminate reactivity and apparent lack of structural specificity. Thus, we surmise that small hydrophobic augmentations of the channel protein at the identified nucleophilic sites (primarily C619, C639, and C663) are sufficient to trigger robust channel opening. These modifications may induce channel gating through altered subunit–subunit or subunit–lipid interactions or by modulating the association of TRPA1 with other cellular proteins or factors. In any case, our analysis suggests that the N-terminal cytoplasmic domain of TRPA1 plays an important role in regulating channel activity through one or more of these mechanisms. As such, this region of the channel serves as a biosensor for the detection of a large class of noxious environmental agents. In addition to the identified cysteine cluster, the N terminus of TRPA1 bears several noteworthy structural features consistent with regulatory function. The sequence begins with 18 ankyrin repeat domains, which are known to mediate protein–protein interaction (15). Moreover, TRPA1 has been postulated to serve as a mechanosensor based, in large part, on biophysical and modeling studies, suggesting that this ankyrin repeat domain may act as a gating spring coupling channel activation to changes in tension at the cell surface (16–19). Finally, consistent with a proposed role of calcium in regulating TRPA1 function, the N-terminal ankyrin repeat stretch also contains a canonical calcium-binding EF-hand domain beginning at residue 470. It remains to be determined whether chemical modification of the cysteine cluster leads to channel opening through functional interaction with the ankyrin repeat or EF-hand domains. In addition, more extensive structure-activity analyses will be required to determine how hydrophobicity or other chemical attributes of modifying electrophiles influence agonist activity. Such studies also will shed light on the range of amino acid side-chain modifications that favor channel gating.
The mechanism of AITC action is, in many ways, similar to a variety of endogenous cellular signaling processes, such as phosphorylation or disulfide bonding, in which small, reversible covalent modifications produce conformational changes in protein structure that have a range of functional consequences. In fact, previous studies have shown that mechanisms involving reversible cysteine modification may regulate the activity of other ion channels, such as cyclic nucleotide-gated channels (20), NMDA-type glutamate-gated channel (21) or skeletal muscle ryanodine receptors (22). In these cases, oxidative modification of cytoplasmic cysteines by nitric oxide or hydrogen peroxide has been proposed to serve as an endogenous pathway of channel activation or modulation (23). Although we have not observed appreciable activation of TRPA1 on exposure to NO or H2O2 (not shown), it remains possible that the sites of AITC modification play a role in the physiological regulation of TRPA1 by other endogenous factors having appropriate electrophilic properties. In any event, the results described above provide chemical and genetic strategies for probing previously undescribed mechanisms of TRP channel activation and physiological pathways regulating TRPA1 function in vivo.
Materials and Methods
Neuronal Cell Culture and Calcium Imaging.
Trigeminal ganglia were dissected as described in ref. 3. Calcium imaging by using fura-2/AM (Molecular Probes, Eugene, OR) was performed as described in ref. 3, and analysis was conducted with automated routines written in Igor Pro (Wavemetrics, Lake Oswego, OR). Ratios expressed in graphs are normalized to baseline calcium levels. Extracellular Ringer's solution contained 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM d-glucose, and 10 mM Hepes buffered to pH 7.4 with NaOH.
Patch-Clamp Recordings.
HEK293 cells and oocytes were recorded in cell-attached patch clamp configurations. Pipet and bath solutions contained 10 mM Hepes, 150 mM CsCl, 1 mM MgCl2, and 1 mM EGTA buffered to pH 7.4 with CsOH. Agonists were backfilled and allowed to diffuse to the pipet tip.
Expression in HEK293 Cells and Oocytes.
HEK293 cells were plated on poly-(d)-lysine-coated chamber slides (Nalge-Nunc, Rochester, NY). Cells were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) by using 5–25 ng/cm2 construct plasmid; pcDNA3 vector was added to bring the total amount of plasmid DNA to 100 ng/cm2. After transfection (16 h), cells were loaded with fura-2/AM (10 μM) for 60 min at 23°C and imaged in Ringer's solution. For oocyte expression, constructs were linearized with DraIII or AvrII and transcribed with T7 polymerase (Ambion, Austin, TX). Oocytes were injected with 50–300 ng of RNA and recorded 24–72 h after injection. Bath solution contained 10 mM Hepes, 120 mM NaCl, 2 mM KCl, 0.2 mM EGTA, and 2 mM MgCl2 buffered to a final pH of 7.45 with NaOH. Optimal expression levels were achieved by using the triple mutant C619A C639A C663A in ooctyes and C619S C639S C663S in HEK293 cells, and the corresponding quadruple mutants were derived accordingly.
Molecular Biology.
Mutations in TRPA1 were made by using the QuikChange site-directed mutagenesis protocol (Stratagene, La Jolla, CA).
Chemicals.
Compounds A and B were synthesized in a single-coupling step from AITC and known compound N-Boc-(1, 6)-diamino hexane or commercial N-acetyl cysteine. Compounds were purified by silica gel chromatography with gradient elution with ethyl acetate/hexane and characterized by 1H NMR, consistent with published spectra. All other chemicals were purchased from Sigma (St. Louis, MO).
Supplementary Material
Acknowledgments
We thank Drs. R. Nicoll and J. Wells for helpful discussions and criticism, Dr. J. Taunton for providing access to chemical synthesis facilities, and Dr. I. Meng for assistance and advice with cannabinoid pharmacology. This work was supported by a Ruth Kirschstein NRSA Postdoctoral Fellowship (to A.H.), a Beginning Grant-in-Aid from the American Heart Association (to H.-h.C.), a Burroughs Welcome Fund Career Award in Biomedical Sciences (to D.M.B.), and grants from the National Institute of Neurological Disorders and Stroke (to D.J.).
Abbreviations
- AITC
allyl isothiocyanate
- BK2R
bradykin receptor
- BTC
benzyl thiocyanate
- NMM
N-methyl maleimide.
Note.
During preparation of this manuscript, another study (24) reported activation of other TRP channels by nitric oxide-mediated reversible covalent modification (nitrosylation) of cysteine residues within the presumptive pore-loop domain.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0609598103/DC1.
References
- 1.Jordt SE, McKemy DD, Julius D. Curr Opin Neurobiol. 2003;13:487–492. doi: 10.1016/s0959-4388(03)00101-6. [DOI] [PubMed] [Google Scholar]
- 2.Ramsey IS, Delling M, Clapham DE. Annu Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- 3.Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D. Nature. 2004;427:260–265. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- 4.Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, Earley TJ, Patapoutian A. Neuron. 2004;41:849–857. doi: 10.1016/s0896-6273(04)00150-3. [DOI] [PubMed] [Google Scholar]
- 5.Bautista DM, Movahed P, Hinman A, Axelsson HE, Sterner O, Hogestatt ED, Julius D, Jordt S-E, Zygmunt PM. Proc Natl Acad Sci USA. 2005;102:12248–12252. doi: 10.1073/pnas.0505356102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Macpherson LJ, Geierstanger BH, Viswanath V, Bandell M, Eid SR, Hwang S, Patapoutian A. Curr Biol. 2005;15:929–934. doi: 10.1016/j.cub.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 7.Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D. Cell. 2006;124:1269–1282. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 8.Smith M, March J, March J. March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. New York: Wiley; 2001. [Google Scholar]
- 9.Vermeulen M, Zwanenburg B, Chittenden GJF, Verhagen H. Eur J Med Chem. 2003;38:729–737. doi: 10.1016/s0223-5234(03)00141-7. [DOI] [PubMed] [Google Scholar]
- 10.Novak L, Hanania M, Kovacs P, Kolonits P, Szantay C. Synthesis (Stuttg) 2001;39:108–118. [Google Scholar]
- 11.Conaway CC, Krzeminski J, Amin S, Chung FL. Chem Res Toxicol. 2001;14:1170–1176. doi: 10.1021/tx010029w. [DOI] [PubMed] [Google Scholar]
- 12.Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 13.Chuang HH, Neuhausser WM, Julius D. Neuron. 2004;43:859–869. doi: 10.1016/j.neuron.2004.08.038. [DOI] [PubMed] [Google Scholar]
- 14.Voets T, Talavera K, Owsianik G, Nilius B. Nat Chem Biol. 2005;1:85–92. doi: 10.1038/nchembio0705-85. [DOI] [PubMed] [Google Scholar]
- 15.Mosavi LK, Cammett TJ, Desrosiers DC, Peng ZY. Protein Sci. 2004;13:1435–1448. doi: 10.1110/ps.03554604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corey DP, Garcia-Anoveros J, Holt JR, Kwan KY, Lin SY, Vollrath MA, Amalfitano A, Cheung EL, Derfler BH, Duggan A, et al. Nature. 2004;432:723–730. doi: 10.1038/nature03066. [DOI] [PubMed] [Google Scholar]
- 17.Lee G, Abdi K, Jiang Y, Michaely P, Bennett V, Marszalek PE. Nature. 2006;440:246–249. doi: 10.1038/nature04437. [DOI] [PubMed] [Google Scholar]
- 18.Howard J, Bechstedt S. Curr Biol. 2004;14:R224–R226. doi: 10.1016/j.cub.2004.02.050. [DOI] [PubMed] [Google Scholar]
- 19.Michaely P, Tomchick DR, Machius M, Anderson RG. EMBO J. 2002;21:6387–6396. doi: 10.1093/emboj/cdf651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Broillet MC, Firestein S. Neuron. 1996;16:377–385. doi: 10.1016/s0896-6273(00)80055-0. [DOI] [PubMed] [Google Scholar]
- 21.Choi YB, Tenneti L, Le DA, Ortiz J, Bai G, Chen HS, Lipton SA. Nat Neurosci. 2000;3:15–21. doi: 10.1038/71090. [DOI] [PubMed] [Google Scholar]
- 22.Xu L, Eu JP, Meissner G, Stamler JS. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 23.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 24.Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y, Mori Y. Nat Chem Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}