

Abstract
Objective: To discuss the acute phase of inflammatory response with a focus on the neutrophilic response and its role in inflammation. We discuss the relative balance between the need for inflammation to stimulate repair and the need to limit inflammation because of the additional damage it causes.
Data Sources: We conducted a MEDLINE search from 1966 to 2005 for literature related to acute inflammation, muscle injury, and repair using combinations of the key words inflammation, neutrophil, macrophage, and cytokines. Additional literature was acquired through cross-referencing of bibliographies of articles obtained through the MEDLINE searches.
Data Synthesis: We reviewed more than 200 relevant articles. Although neutrophils are an important cell population in acute inflammation, few athletic trainers are familiar with the neutrophil's actions or its dichotomous role as both perpetrator of tissue damage and initiator of repair. Neutrophils dominate the early stages of inflammation and set the stage for repair of tissue damage by macrophages. These actions are orchestrated by numerous cytokines and the expression of their receptors, which represent a potential means for inhibiting selective aspects of inflammation.
Conclusions: Neutrophils infiltrate injured tissues but can also be present after noninjurious exercise. These cells have both specific and nonspecific defensive immune system functions that can cause tissue damage in isolation or as sequelae to other tissue injury. It might seem that limiting the action of neutrophils would be clinically beneficial, but these cells are also responsible for initiating the reparative process that is later managed by macrophages. Although achieving a therapeutic balance between limiting inflammation and stimulating repair is important, the duplicitous roles of neutrophils and macrophages in both the inflammation and healing processes create a physiologic paradox for clinicians whose goals are to limit inflammation and to stimulate healing after acute soft tissue injury.
Keywords: acute phase reaction, wound healing, soft tissue
In sports medicine, the term inflammation is often used to describe a series of signs and symptoms after soft tissue or bony injury. Appropriately, this term was originally used to describe the 4 classic signs of the affected tissue's response to trauma: redness, swelling, heat, and pain. 1 Unfortunately, this descriptor was coined without a basis for, or an understanding of, the underlying pathophysiologic processes that created it. As clinicians, athletic trainers have been taught to think of the inflammatory response in the traditional sense, as a clinical milieu of signs and symptoms. Attempting to reduce or prevent the signs and symptoms of inflammation after tissue trauma has become dogma, even though the role of inflammation in tissue healing and repair is not fully understood.
The inflammatory process can be initiated through a variety of mechanisms, which include the introduction of pathogens as well as challenges to the system through chemical, thermal, and mechanical stresses. Regardless of the inciting factors, the events accompanying inflammation are somewhat consistent. For research purposes, a reproducible injury model of tissue inflammation and repair is required; one such model involves eccentric overload to skeletal muscle. 2–4 Although this approach allows us to specifically discuss inflammation in muscle tissue and provides discrete insight into muscle-vessel communication, it also fulfills a broader purpose. Specifically, it serves as an accepted paradigm to aid in the understanding of the body's generalized inflammatory process and, therefore, of a significant number of athletic injuries.
Ultimately, effective clinical care of stressed or traumatized tissues depends on a thorough understanding of the cellular and molecular events leading to the physiologic response we recognize as classical inflammation. Unfortunately, for many this understanding has been limited to a relatively simple listing of events, such as vasoconstriction, vasodilation, margination, diapedesis, exudate formation, and phagocytosis. These are all key elements of the process, but a clinically useful understanding of inflammation must go further. The cellular processes of inflammation are regulated by a series of specific cell signals that stimulate a variety of cell types, resulting in a cascade of events including white blood cell (WBC) recruitment and activation. The physiologic response to these signals or WBC activity (or both) results in the traditional inflammatory response: the clinically observable milieu of signs and symptoms associated with tissue injury and healing. Current researchers continue to uncover the integrated processes implicating the inflammatory response in injury exacerbation and tissue repair.
In this review, we will examine the roles of neutrophils and macrophages in muscle injury and repair, concentrating on the integration of cellular communication as a controlling signal between the beneficial and perhaps detrimental aspects of inflammation. Specifically, we will discuss 4 major topics: (1) the acute response of neutrophils to exercise, (2) the role of neutrophils in inflammation after muscle injury, (3) the relationships of inflammation and tissue healing with respect to neutrophils and macrophages, and (4) clinical implications. Although the implications for clinical therapeutics are not fully realized at this time, early intervention may prove the most beneficial strategy to minimizing tissue injury and facilitating tissue repair and recovery of function.
ACUTE RESPONSE OF NEUTROPHILS TO EXERCISE
If inflammation is regarded as the proliferation of WBCs after soft tissue injury, then the cellular inflammatory response actually begins at the onset of exercise, when the circulating level of neutrophils increases significantly. 5–8 Neutrophils are the first WBC population to arrive and affect the host inflammatory response during exercise and soft tissue injury ( Table). These cells have both specific and nonspecific defensive mechanisms, some of which are capable of causing additional tissue damage. 15–18 In the past, the early effects of damaging eccentric exercise were proposed to result in increased numbers of circulating neutrophils, as these cells would be required to enter the injury site to initiate phagocytosis or removal of damaged tissues. However, this immediate response has also been observed after both noninjurious passive stretching and isometric exercise, illustrating that the presence of neutrophils does not necessarily always lead to injury. 19
Roles of Selected Immune Cells in Inflammation and Repair.

The mechanism for early neutrophilia postexercise is likely due to a combination of factors. During rest, more than half of the circulating neutrophils are marginated along the endothelial walls of blood vessels. At the onset of exercise, increases in epinephrine, blood flow, and cell-signaling molecules demarginate these neutrophils away from the vessel walls, resulting in their mobilization into the circulation. 5, 20, 21 Demargination allows the neutrophils to enter the circulation and redistribute elsewhere in the body, as needed. The mechanisms by which neutrophils localize in damaged or stressed tissue are just beginning to be understood and may represent key strategies for intervention to limit certain aspects of inflammation.
The movement of a neutrophil from the circulation into the tissue, called diapedesis, is under tight regulatory control of the underlying tissue. In skeletal muscle, diapedesis can occur rapidly during exercise. 22 Neutrophil recruitment is ultimately the responsibility of the muscle fibers (myocytes) together with mast cells from a variety of tissues, including the local connective tissue. If a myocyte is perturbed in some fashion, such as in the case of an active stretch or contusion, it communicates with the endothelial wall of the adjacent blood vessel, initiating a cascade of signaling events and resulting in diapedesis. This intercellular communication is accomplished, in part, by a series of cell-signaling molecules, or cytokines, that are essential to any understanding of immune cell function.
The term cytokine is derived from the Greek root meaning “to put cells into motion.” 17 All nucleated cells in the body produce cytokines and similarly express cytokine receptors on their surface membranes. Cytokines act at the surface of the target cells, principally to alter cell function. 23 Skeletal muscle continually produces cytokines in an effort to maintain homeostasis and to regulate function. Simple perturbations of skeletal muscle, such as an active stretch during eccentric exercise, markedly increase the expression of interleukin-1β (IL-1β) and tumor necrosis factor–α (TNF-α). 24 These proinflammatory cytokines upregulate the expression of endothelial-leukocyte adhesion molecules (E-selectin) within the endothelium of the adjacent blood vessels. 17, 25, 26 Activation of the endothelium is site specific and can result in the release of additional IL-1β, as well as additional proinflammatory cytokines, including IL-6 and IL-8, both of which have been shown to attract neutrophils. 27–31 Thus, endothelial activation serves 2 purposes: encouraging the adhesion of neutrophils at the site of cell stress (margination) and assisting the cell in recruiting additional neutrophils ( Figure 1).
Figure 1. Modified muscle use may result in an increased intracellular calcium concentration, resulting in an increased cell production of proinflammatory cytokines such as TNF-α and IL-1β, which in turn upregulate the expression of endothelial-leukocyte adhesion molecules (E-selectin and P-selectin). The activated endothelium attracts neutrophils to the region and also releases the neutrophil chemoattractants and proinflammatory cytokines IL-6 and IL-8. IL-6 indicates interleukin 6; IL-8, interleukin 8; IL-1β, interleukin 1-beta; TNF-α, tumor necrosis factor–alpha; 1β; TNF-α, tumor necrosis factor–α; and ECM, extracellular matrix.

The temporary adhesion of neutrophils to the endothelium results in their immobilization and, hence, prolonged signaling from the muscle cell. 32 Without margination, no effective communication would be possible between the myocytes and the neutrophils, because these cell types would not be in close proximity for an adequate length of time. This cytokine-mediated communication results in a reorganization of neutrophil 33, 34 and endothelial 35, 36 cell structure, allowing the neutrophils to pass from the endothelium (diapedesis) to the extracellular matrix (ECM) adjacent to the myocytes. The traditional thinking has been that these cytokines were released only by injured or damaged myocytes, resulting in the localization of neutrophils to these injured tissues. This finding has been observed after eccentric contractions 4, 37 and has led to speculation that inflammation is the process responsible for delayed-onset muscle soreness. 19, 20, 38 However, simple muscle activation and passive stretch have recently been shown to be sufficient stimuli for diapedesis to occur, with subsequent localization of neutrophils within the ECM of skeletal muscle. 39, 40 Currently, researchers are focused on the mechanisms for neutrophil recruitment and the function of these neutrophils in otherwise healthy, uninjured muscle. Among the most important questions regarding neutrophils and inflammation is whether the localization of neutrophils in the ECM facilitates healing or tissue destruction. Evidence is beginning to indicate that it is more likely a combination of both repair and further damage. The latter seems somewhat surprising but may even be an important signal for tissue repair.
THE ROLE OF NEUTROPHILS IN INFLAMMATION AFTER MUSCLE INJURY
Neutrophils are the first subpopulation of WBCs to enter traumatized or stressed tissues. 17 The potential for these cells to exacerbate muscle injury has been carefully studied in a variety of experimental models producing tissue damage through eccentric loading. For instance, observable alterations in cell structure after eccentric exercise have included the loss of sarcomeric organization; the loss of intermediate filament proteins, including desmin 41–43; and the infiltration of neutrophils within the muscle cells. 37, 39, 40, 44 This subcellular muscle damage is augmented through repetitive eccentric exercise and further compromises muscle contractility and function. 45, 46 Currently, the role of neutrophils in muscle injury remains controversial. Although neutrophils contribute to preexisting muscle damage, 47 evidence is mounting that they may provide the principal insult to the muscle membrane. 39
Neutrophils as Contributory Factors in Muscle Injury
Membrane disruption has often been considered to be the initial, propagating event in muscle injury. 47–50 This hypothesis has centered on the mechanical disruption of the cell membrane due to the high forces transmitted during eccentric exercise. 18, 48, 51 In this manner, membrane disruption is thought to be the initial, precipitating factor, and the localization of neutrophils to the damaged area is required for removal of damaged tissue through phagocytosis. Several authors have reported an exacerbation of injury after eccentric exercise, 48, 52 including observations that the initial injury is often followed by a secondary loss of muscle force, arguably due to additional but delayed damage to the muscle fibers. 53, 54 This so-called secondary damage has been proposed to be caused by invading neutrophils, 16, 18 potentially due to a second burst of neutrophilia within 24 hours of cessation of exercise. 5 More importantly, this secondary burst appears to mediate damage through the release of cytotoxic compounds. 55–57 This secondary response likely results from bone marrow release of neutrophils in response to elevated blood catecholamine levels. 21 Interestingly, these neutrophils appear to be more oxidatively active than the first group of neutrophils emigrating to the ECM. 5
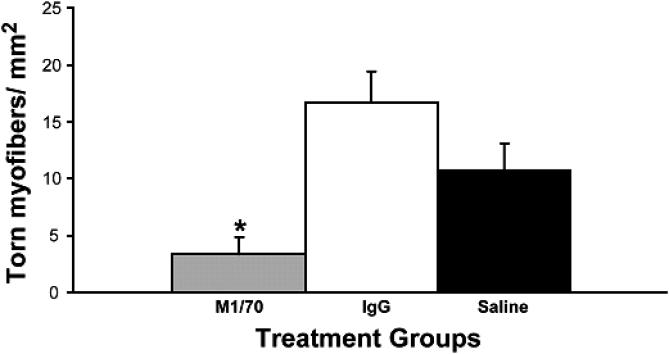
After muscle injury, myocytes and other cells release a number of cytokines, such as IL-1β, IL-6, IL-8, and TNF-α, that cause neutrophils to become activated to produce a host of cytotoxic substances, including reactive oxygen species, such as superoxide anions, hypochloride, and hydrogen peroxide. 2, 3 The cytokines IL-1, IL-6, and TNF-α all stimulate pathways that contribute to activation of the enzyme NADPH-oxidase, which generates a “respiratory burst” and the subsequent release of reactive oxygen species. Recently, when the neutrophil's respiratory burst was blocked with monoclonal antibody M1/70, membrane damage was diminished after a single eccentric stretch protocol ( Figure 2), 58 illustrating a potential role of neutrophils in stretch-induced muscle injury. Conversely, using a series of repetitive eccentric contractions, Lowe et al 59 observed no difference in injury variables between healthy mice and neutropenic mice. These somewhat contradictory results may be explained by the different models (single stretch versus repetitive exercise) used to study this phenomenon. Therefore, evidence in support of a secondary role for neutrophils in muscle fiber damage is still controversial and is certainly not well understood in humans.
Figure 2. Rabbit tibialis anterior muscles were subjected to an injurious eccentric exercise protocol after 1 of 3 treatments. *Note the significant reduction ( P < .01) in torn fibers postexercise in the M1/70 group (blocked respiratory burst from neutrophils) compared with the 2 control groups exposed to saline and immunoglobulin G (IgG), with intact neutrophil function and burst not blocked. Used with permission from the American Physiological Society. 58 .
Understanding the relationship between muscle membrane disruption and the contribution of neutrophil-mediated damage remains a key objective in identifying mechanisms of inflammation and damage. Using an ischemia-reperfusion model, researchers have begun to appreciate the damaging actions of neutrophils in the absence of prior tissue injury. When blood flow is restored to ischemic muscle, neutrophils quickly marginate and rapidly extravasate into the muscle ECM and myocytes. Although tissue reperfusion clearly provides a significant stress to the tissues (ie, metabolic stress), notable in these models is the absence of mechanical tissue injury or trauma. The removal of neutrophils from experimental animal models of ischemia-reperfusion results in less oxidant production and the attenuation of membrane disruption during the reperfusion of ischemic tissue. 60 In this model, the essential step in the inflammatory process is the adhesion of neutrophils to the endothelium, which ultimately leads to neutrophil-mediated cell destruction and necrosis. 61 Therefore, the primary mechanism of tissue destruction is the release of cytotoxic agents by the invading neutrophils, in the absence of any initial mechanical event. The recruitment of neutrophils to the muscle in each of these nonactivated, noncontractile models may assist in elucidating the mechanisms by which neutrophils undergo activation and diapedesis, regardless of the muscle activation or contraction type.
Neutrophils as Principal Factors in Muscle Injury
When subjected to a single active stretch, the tibialis anterior muscles of neutropenic rabbits showed significantly reduced membrane disruption compared with healthy control muscles. 62 The lack of observable membrane disruption in the absence of neutrophils and the attenuation of the degradation of the intermediate filament proteins in the muscle cells 58 imply that much of the observed cellular destruction after eccentric exercise may be explained through an inflammatory cell-mediated response. Such observations call into question the role of mechanical events as a necessary, predisposing factor for membrane disruption and even indicate a principal role of neutrophils in stretch-induced muscle injury. These findings need to be confirmed in humans before any definitive conclusions can be drawn.
Recently, dystrophic muscle has been used as a model to study membrane disruption after eccentric exercise. 63 Dystrophic muscle exhibits a chronic inflammatory response in addition to recurrent membrane disruption and failed cellular repair. Dystrophic muscle has an inherently fragile membrane structure, resulting in frequent membrane disruptions after the slightest mechanical stress or strain. However, it is now becoming clear that chronic inflammation is an important aspect of the cyclic fiber disruption and necrosis observed in these muscles. 64 In fact, membrane disruption does not appear to be the primary point of damage after active stretch. 65 When stretch-activated calcium channels (SACs) are blocked before dystrophic muscle is stretched, the observed membrane disruption does not differ from the membrane disruption in healthy muscle after identical stretch magnitudes. 63 Similar results with healthy muscle have supported this idea. After the SACs in rat skeletal muscle were blocked, torque production was similar, but injury was reduced after a series of eccentric contractions. 66 These findings imply that cellular, ionic, or inflammatory processes in muscle may play a more primary role in fiber membrane disruption than do the previously accepted mechanical factors.
The role of neutrophils, therefore, must be carefully examined in this scenario. In vitro, stretched myotubes in skeletal muscle directly recruit neutrophils, and these neutrophils subsequently damage myotube membranes. 67 Accordingly, a new role for neutrophils as the primary mediators of muscle injury after modified use is emerging. Limited, noninjurious muscle use results in neutrophilia, recruitment, and diapedesis to the muscle ECM. Altering or increasing the magnitude of the stimuli, through altered activation or loading, appears to result in greater recruitment and the subsequent appearance of neutrophils within the fibers themselves after membrane disruption.
Findings such as these are causing researchers to focus on the possible role of neutrophils in the initiation of cell destruction, similar to that observed in ischemia-reperfusion injury, but how do these results apply to an intact physiologic system? If muscle membrane destruction is not required to activate the endothelium of adjacent blood vessels, then what causes margination of neutrophils and subsequent diapedesis into the ECM of uninjured muscle? Possibilities include the expression and release of cytokines that may be related to membrane calcium channels. 68 Stretch-activated calcium channels are abundant on the membranes of muscle fibers. These SACs result in transient increases in intracellular calcium after stretch and may be the intracellular signaling molecule responsible for the release of proinflammatory cytokines from the muscle. Interestingly, dystrophic muscle has a higher membrane permeability to calcium, 69 which may be due to an overabundance or hyperactivity of SACs in the membranes of muscle fibers. 63, 70 This finding may explain the observed differences in membrane injury and inflammation after eccentric contractions using calcium channel blockers, 66, 71 whereby decreased transient intracellular calcium concentrations during repetitive muscle activation result in decreased cytokine production and cell signaling. 72 Although currently unproven, the strong possibility exists of a cause-and-effect relationship between abnormal calcium dynamics and chronic inflammation in dystrophic muscle. Teleologically, by reducing transient intracellular calcium influx, cytokine production could be attenuated. This would lead to decreased endothelial stimulation and reduced neutrophil recruitment and, ultimately, would result in diminished neutrophil-mediated membrane destruction ( Figure 3). Furthermore, this may also have an important effect on tissue repair, as calcium channel blocking has recently been shown to reduce macrophage responsiveness. 68 Clearly, future studies are required to establish the link between calcium influx and inflammatory cell–mediated fiber disruption and repair.
Figure 3. Schematic illustration demonstrating the proposed role of the stretch-activated calcium channels (SACs) and neutrophil recruitment in the inflammatory response of skeletal muscle. Solid lines represent initial events in muscle inflammation, and dotted lines denote the proinflammatory signaling cascade that results. Hypothetically, modified skeletal muscle use results in an increased intracellular calcium concentration. This starts a cascade of cell signaling, endothelial activation, and white blood cell recruitment, the magnitude of which may be stress or strain dependent. ROS indicates reactive oxygen species; ECM, extracellular matrix.

THE RELATIONSHIPS OF INFLAMMATION AND TISSUE HEALING WITH RESPECT TO NEUTROPHILS AND MACROPHAGES
Tissue injury results in a rapid early response by neutrophils, but macrophages soon follow. 38 Although the mechanisms responsible for attracting macrophages remain unknown, injured tissue probably releases a temporally variable series of signaling molecules 12 responsible for recruiting these cells. However, the resulting contributions and interactions of neutrophils and macrophages in tissue repair remain poorly understood. Neutrophils and macrophages coexist at the site of tissue stress, yet evidence indicates that these cells interact to perform disparate functions. Neutrophils, for example, produce destructive oxidizing agents in the absence of other WBCs, such as macrophages, 13 and attempts at blocking neutrophil function have resulted in significantly diminished early tissue destruction. 58 It is important, however, to recognize that some of these findings may have limited importance given that they have only been observed in vitro. 13 Similarly, macrophages do not contribute to membrane disruption during the inflammatory process, 12 but their reparative functions can be inhibited by the absence of neutrophils, as the reduced phagocytosis of cellular debris by neutrophils results in slower cellular regeneration and repair by macrophages. 73 The role of WBC communication and interaction is further complicated by the observation that prolonged neutrophil accumulation results in impaired tissue regeneration, possibly through an inordinate amount of tissue destruction. 74 Therefore, it is becoming evident that some neutrophil function is necessary for tissue repair. Recently, Teixeira et al 75 showed that skeletal muscle injected with snake venom regenerated at a slower rate in the absence of neutrophils than did control muscle. Thus, early phagocytosis of tissue debris may serve as the rate-limiting process in cellular repair and regeneration, and the temporal response and communication between neutrophils and macrophages appears essential to proper regeneration.
Macrophages and Tissue Repair
Although many clinicians mistakenly conceptualize macrophages as being a single type of immune cell, the reality is that 2 cell populations appear to perform multiple functions at various times in the inflammatory process ( Table). The ED1 + macrophages are most prominent within necrotic fibers as early as 1 day after neutrophil invasion and are activated by the proinflammatory cytokines, such as TNF-α and IL-1β. 76 The ED2 + macrophages, however, appear during the latter stages of inflammation and sequester themselves in the ECM. 14 Thus, many tissues can recruit macrophages, including endothelium and neutrophils, but the macrophage response may differ by subpopulation and possibly by function. Once ED1 + macrophages are activated, they also contribute to the exacerbation of inflammation by producing and releasing more than 100 substances, including proinflammatory cytokines such as prostaglandins (PGE 2) and IL-1β. 77 This cell signaling serves to not only magnify the macrophage response in the tissue but may also recruit additional neutrophils, resulting in a positive proinflammatory feedback loop. 77 However, unlike neutrophils, macrophages also release a series of growth factors essential for tissue repair and regeneration. Because ED2 + macrophages do not enter necrotic tissues, their primary role may be in tissue repair through cell signaling and cytokine production. These findings, however, remain to be validated in human studies.
Macrophages, particularly the ED2 + subpopulation, can mediate tissue repair and growth by the release of a number of growth-promoting factors and cytokines. Of particular interest are cell growth regulatory cytokines such as fibroblast growth factor (FGF), insulin-like growth factor (IGF-1), and transforming growth factor–β1 (TGF-β1). In addition, macrophages also have the ability to cleave decorin, an abundant proteoglycan, resulting in the additional release of TGF-β1. Together, these cytokines (FGF, IGF-1, TGF-β1) recruit and activate fibroblasts that will eventually secrete matrix molecules such as collagen to begin the repair process. 78 During tissue repair, fibroblasts continue to secrete their own proinflammatory cytokines, such as IL-6 and IL-1, as well as to recruit additional neutrophils through the production of IL-8. 17 Ultimately, fibroblasts may perpetuate the chronic inflammatory response through their release of PGE 2 in response to cell stress and strain. Although satellite cell proliferation has been shown to be enhanced in the presence of macrophages, 79 the release of excessive quantities of TGF-β1 inhibits satellite cell differentiation, compromising fiber regeneration. 80
CLINICAL IMPLICATIONS
Historically, the acute management of athletic musculoskeletal injury has focused on limiting the cardinal signs of inflammation in an effort to expedite the rehabilitative process and to facilitate an early return to competition. 81 To this end, the use of ice, compression, and elevation for initial management of injuries has flourished. Over the last 25 years, rationales for acute treatment practices have changed, focusing on retarding secondary injury in an effort to minimize total injury. 82, 83 Regardless of the rationale, the practice of using ice, compression, and elevation in managing acute inflammation is well ingrained. Although a potential role for the use of physical agents, such as cryotherapy, in attenuating the neutrophilic response has been demonstrated in the laboratory, 84, 85 the actual clinical evidence supporting the efficacy of these practices is limited.
Similarly, limiting inflammation and enhancing tissue repair through the suppression of neutrophil recruitment and activation may reduce tissue damage postexercise. Such efforts have been established, as nonsteroidal inflammatory drugs (NSAIDs) have been used for centuries in an attempt to limit the inflammatory response. However, the anti-inflammatory effects may be confounded by the analgesic action of these drugs, 86 which has long been the focus of early interventions for muscle injury. 81 It has been suggested that the magnitude of pain after tissue trauma corresponds to the concentration of WBCs within the injured tissue. 87 However, this theory has not been supported in the literature. For example, although tendinitis is a common diagnosis, the absence of WBCs in tissues affected by this condition indicates that this is not a true inflammatory response. 88 Conversely, the mere presence of WBCs does not always coincide with the cardinal signs of inflammation. White blood cells have been observed in the absence of obvious tissue trauma, 39 even though this situation is generally not referred to as an inflammatory process.
A challenge to reducing inflammation through pharmacologic intervention is the multiple cellular pathways by which the inflammatory response can be mediated. Traditional NSAIDs block the cyclooxygenase (COX) pathway that contributes to cell-mediated prostaglandin (PGE 2) production 89 and, arguably, neutrophil recruitment. 90 However, other proinflammatory pathways exist for the cell to recruit neutrophils to damaged or exercised muscle, including the alternative lipoxygenase pathway 91 and the nuclear factor kappa-B (NF-κB)–mediated induction of proinflammatory genes. 92 Although a great deal of information on the efficacy of NSAID use exists in the literature, the immediate and long-term use of NSAIDs to control inflammation remains controversial. This may be due to the multiple proinflammatory pathways, the wide variety of emerging NSAIDs designed to target specific cellular pathways, and their respective effects of muscle repair and regeneration.
The effects of NSAIDs on inflammatory cell accumulation in the muscle and their relationship with muscle repair remain controversial. 44 For instance, inhibition of NF-κB by curcumin has been shown to accelerate muscle regeneration. 93 Although non–NF-κB inhibitors such as the NSAID naproxen have been shown to have no effect on muscle cell regeneration, 94 NS-398 (a COX-2–specific inhibitor) reduced neutrophil and macrophage entry into the muscle, delayed regeneration and healing, and resulted in increased TGF-β1 and increased tissue fibrosis. 89 Evidence is accumulating that NSAIDs may actually interfere with satellite cell proliferation, differentiation, and fusion 89, 95 and, therefore, may adversely affect muscle regeneration and repair. 89, 96, 97 Similarly, inhibited tissue repair after NSAID administration has also been shown after injury in other soft tissues, including ligaments. 98 Ultimately, although NSAID treatment for soft tissue injuries is common in sports medicine settings, no concrete evidence demonstrates that such treatments are justified, even for the analgesic effects. 99
If it is beneficial to limit the neutrophilic response, then the timing and dosage of NSAIDs are likely important. Neutrophils are the dominant immune cells for the first 4 to 24 hours postinjury, after which macrophages predominate. 3 Some potential clearly exists for limiting the neutrophil-mediated damage that appears to accompany mechanical stress to muscle and other tissues. However, whether more is to be gained by combating the secondary neutrophilic damage but potentially interfering with the muscle regeneration process or by accepting the secondary damage in hopes that faster regeneration is stimulated remains unclear. It is important to note that evidence of impeding regeneration 75 was observed in neutrophil-depleted mice. That is, regeneration was impaired in an animal model in which no neutrophils were present, indicating that neutrophils may play a key role in muscle repair. Although this laboratory model is useful, it does not reflect the clinical reality of acute intervention in the injured athlete. Completely abolishing the neutrophilic response using typical clinical cryotherapy or NSAID therapy would be practically impossible. Therefore, we expect that some level of neutrophilic response would be seen, regardless of our acute intervention. No data presently describe whether a partially muted response would be beneficial or harmful.
In skeletal muscle, the propensity for an enhanced inflammatory response and fibroblast proliferation exceeds the muscle's ability to regenerate, particularly in humans, resulting in the formation of a fibrotic scar. Until recently, this fibrotic response was presumed to be a necessary step in the formation of nascent myotubes for muscle fiber repair. However, fibrotic scar formation is not an optimal outcome and may be due to excessive cell signaling 100 and inflammatory response. 101 When the function of fibroblasts and TGF-β1 was inhibited after laceration injuries, skeletal muscle had the inherent ability to regenerate damaged fibers. 102–104 Although this finding has not been studied in models of severe strain injury, the manipulation of cell signaling may provide a glimpse into the possible future of therapeutic agents designed to modify tissue healing.
Clinically, return to activity can result in an exacerbation of the inflammatory response. 105 However, evidence is also accumulating that regular exercise acts as an anti-inflammatory agent. 106 Neutrophil function and cytotoxicity may be modified through exercise, 107 and these modifications may depend on exercise intensity. 21 The production of cytokines by neutrophils as well as the resulting response from all WBCs can be modified with long-term exercise. 15 The exact mechanisms are not known, but the low-level inflammatory response produced through regular exercise may blunt the cells' response to cytokines or inhibit their production and subsequent release. In this regard, regular exercise may suppress the release of proinflammatory cytokines, such as TNF-α. 106 Recently, using isolated chondrocytes, the proinflammatory response was shown to vary depending on the magnitude of the mechanical signal applied to the tissue. 108 Thus, exercise may produce both proinflammatory and anti-inflammatory effects, depending on the magnitude of the stimulus and the corresponding level of cytokine released.
SUMMARY
The inflammatory response of tissue to stress is a culmination of a series of multifaceted cellular events, leading to a recognizable physiologic response. Although clinicians have become cognizant of the many therapeutic interventions available for treating inflammation, the dogmatic approach commonly used has little scientific basis in the current literature. Most research regarding these clinical treatments has been very limited and has focused on clinical signs and symptoms in very small numbers of subjects. There remains a dearth of cellular evidence or significant clinical trials to support the traditional treatments used by athletic trainers to treat soft tissue injury and inflammation. This probably reflects the complicated and integrated cellular response to tissue trauma and stress and the concomitant “good” and “bad” aspects of the inflammatory response. Although it is becoming clear that neutrophils cause cell death and necrosis, their phagocytic actions are still required for proper macrophage function and tissue repair. What is not yet clear is the degree to which clinical attempts to minimize neutrophilic tissue damage may interfere with neutrophilic roles in tissue repair. Elucidating this balance is a key goal of current inflammation research. Novel therapies may focus on modifying the cellular response at both the tissue and immune cell levels, possibly attenuating the detrimental effects of inflammation while facilitating tissue repair. The fact that we are unlikely to completely attenuate the neutrophilic response after musculoskeletal injury indicates that we may be able to strike some balance between the potentially beneficial aspects of our acute treatments and their concomitant interference with repair. At this time, however, no data describe this balance or even indicate if any such balance can be achieved. For now, care remains focused on minimizing the body's physiologic response to tissue stress, as authors of well-controlled clinical studies and parallel basic science investigations attempt to uncover more appropriate therapeutic strategies.
REFERENCES
- Rocha e Silva M. A brief survey of the history of inflammation, 1978. Agents Actions. 1994;43:86–90. doi: 10.1007/BF01986675. [DOI] [PubMed] [Google Scholar]
- Best TM, Fiebig R, Corr DT, Brickson S, Ji L. Free radical activity, antioxidant enzyme, and glutathione changes with muscle stretch injury in rabbits. J Appl Physiol. 1999;87:74–82. doi: 10.1152/jappl.1999.87.1.74. [DOI] [PubMed] [Google Scholar]
- Brickson S, Hollander J, Corr DT, Ji LL, Best TM. Oxidant production and immune response after stretch injury in skeletal muscle. Med Sci Sports Exerc. 2001;33:2010–2015. doi: 10.1097/00005768-200112000-00006. [DOI] [PubMed] [Google Scholar]
- Tsivitse SK, Mylona E, Peterson JM, Gunning WT, Pizza FX. Mechanical loading and injury induce human myotubes to release neutrophil chemoattractants. Am J Physiol Cell Physiol. 2005;288:C721–C729. doi: 10.1152/ajpcell.00237.2004. [DOI] [PubMed] [Google Scholar]
- Quindry JC, Stone WL, King J, Broeder CE. The effects of acute exercise on neutrophils and plasma oxidative stress. Med Sci Sports Exerc. 2003;35:1139–1145. doi: 10.1249/01.MSS.0000074568.82597.0B. [DOI] [PubMed] [Google Scholar]
- Smith LL, Bond JA, Holbert D. Differential white cell count after two bouts of downhill running. Int J Sports Med. 1998;19:432–437. doi: 10.1055/s-2007-971941. et al. [DOI] [PubMed] [Google Scholar]
- MacIntyre DL, Reid WD, Lyster DM, Szasz IJ, McKenzie DC. Presence of WBC, decreased strength, and delayed soreness in muscle after eccentric exercise. J Appl Physiol. 1996;80:1006–1013. doi: 10.1152/jappl.1996.80.3.1006. [DOI] [PubMed] [Google Scholar]
- MacIntyre DL, Reid WD, Lyster DM, McKenzie DC. Different effects of strenuous eccentric exercise on the accumulation of neutrophils in muscle in women and men. Eur J Appl Physiol. 2000;81:47–53. doi: 10.1007/PL00013796. [DOI] [PubMed] [Google Scholar]
- Majno G, Joris I. Cells, Tissues, and Disease: Principles of General Pathology. 2nd ed. Oxford, UK: Oxford University Press; 2004.
- McLennan IS. Degenerating and regenerating skeletal muscles contain several subpopulations of macrophages with distinct spatial and temporal distributions. J Anat. 1996;188:17–28. (pt 1) [PMC free article] [PubMed] [Google Scholar]
- Marsolais D, Cote CH, Frenette J. Neutrophils and macrophages accumulate sequentially following Achilles tendon injury. J Orthop Res. 2001;19:1203–1209. doi: 10.1016/S0736-0266(01)00031-6. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Berchenko E, Frenette J. Macrophage invasion does not contribute to muscle membrane injury during inflammation. J Leukoc Biol. 1999;65:492–498. [PubMed] [Google Scholar]
- Nguyen HX, Tidball JG. Interactions between neutrophils and macrophages promote macrophage killing of rat muscle cells in vitro. J Physiol. 2003;547:125–132. doi: 10.1113/jphysiol.2002.031450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Pierre BA, Tidball JG. Differential response of macrophage subpopulations to soleus muscle reloading after rat hindlimb suspension. J Appl Physiol. 1994;77:290–297. doi: 10.1152/jappl.1994.77.1.290. [DOI] [PubMed] [Google Scholar]
- Peake JM. Exercise-induced alterations in neutrophil degranulation and respiratory burst activity: possible mechanisms of action. Exerc Immunol Rev. 2002;8:49–100. [PubMed] [Google Scholar]
- Cannon JG, Orencole SF, Fielding RA. Acute phase response in exercise: interaction of age and vitamin E on neutrophils and muscle enzyme release. Am J Physiol. 1990;259:R1214–R1219. doi: 10.1152/ajpregu.1990.259.6.R1214. et al. [DOI] [PubMed] [Google Scholar]
- Cannon JG, St Pierre BA. Cytokines in exertion-induced skeletal muscle injury. Mol Cell Biochem. 1998;179:159–167. doi: 10.1023/a:1006828425418. [DOI] [PubMed] [Google Scholar]
- Armstrong RB. Initial events in exercise-induced muscular injury. Med Sci Sports Exerc. 1990;22:429–435. [PubMed] [Google Scholar]
- Smith LL, McCammon M, Smith S, Chamness M, Israel RG, O'Brien KF. White blood cell response to uphill walking and downhill jogging at similar metabolic loads. Eur J Appl Physiol Occup Physiol. 1989;58:833–837. doi: 10.1007/BF02332215. [DOI] [PubMed] [Google Scholar]
- MacIntyre DL, Reid WD, McKenzie DC. Delayed muscle soreness: the inflammatory response to muscle injury and its clinical implications. Sports Med. 1995;20:24–40. doi: 10.2165/00007256-199520010-00003. [DOI] [PubMed] [Google Scholar]
- Pyne DB. Regulation of neutrophil function during exercise. Sports Med. 1994;17:245–258. doi: 10.2165/00007256-199417040-00005. [DOI] [PubMed] [Google Scholar]
- Fielding RA, Manfredi TJ, Ding W, Fiatarone MA, Evans WJ, Cannon JG. Acute phase response in exercise, III: neutrophil and IL-1 beta accumulation in skeletal muscle. Am J Physiol. 1993;265:R166–R172. doi: 10.1152/ajpregu.1993.265.1.R166. [DOI] [PubMed] [Google Scholar]
- Taniguchi T. Cytokine signaling through nonreceptor protein tyrosine kinases. Science. 1995;268:251–255. doi: 10.1126/science.7716517. [DOI] [PubMed] [Google Scholar]
- Malm C, Nyberg P, Engstrom M. Immunological changes in human skeletal muscle and blood after eccentric exercise and multiple biopsies. J Physiol. 2000;529:243–262. doi: 10.1111/j.1469-7793.2000.00243.x. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua MP, Pober JS, Mendrick DL, Cotran RS, Gimbrone MA., Jr. Identification of an inducible endothelial-leukocyte adhesion molecule. Proc Natl Acad Sci U S A. 1987;84:9238–9242. doi: 10.1073/pnas.84.24.9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller A, Isenmann S, Vestweber D. Cloning of the mouse endothelial selectins: expression of both E- and P-selectin is inducible by tumor necrosis factor alpha. J Biol Chem. 1992;267:15176–15183. [PubMed] [Google Scholar]
- Kaplanski G, Farnarier C, Kaplanski S. Interleukin-1 induces interleukin-8 secretion from endothelial cells by a juxtacrine mechanism. Blood. 1994;84:4242–4248. et al. [PubMed] [Google Scholar]
- Detmers PA, Powell DE, Walz A, Clark-Lewis I, Baggiolini M, Cohn ZA. Differential effects of neutrophil-activating peptide 1/IL-8 and its homologues on leukocyte adhesion and phagocytosis. J Immunol. 1991;147:4211–4217. [PubMed] [Google Scholar]
- Detmers PA, Lo SK, Olsen-Egbert E, Walz A, Baggiolini M, Cohn ZA. Neutrophil-activating protein 1/interleukin 8 stimulates the binding activity of the leukocyte adhesion receptor CD11b/CD18 on human neutrophils. J Exp Med. 1990;171:1155–1162. doi: 10.1084/jem.171.4.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackarel AJ, Russell KJ, Brady CS, FitzGerald MX, O'Connor CM. Interleukin-8 and leukotriene-B(4), but not formylmethionyl leucylphenylalanine, stimulate CD18-independent migration of neutrophils across human pulmonary endothelial cells in vitro. Am J Respir Cell Mol Biol. 2000;23:154–161. doi: 10.1165/ajrcmb.23.2.3853. [DOI] [PubMed] [Google Scholar]
- Liu X, Spolarics Z. Methemoglobin is a potent activator of endothelial cells by stimulating IL-6 and IL-8 production and E-selectin membrane expression. Am J Physiol Cell Physiol. 2003;285:C1036–C1046. doi: 10.1152/ajpcell.00164.2003. [DOI] [PubMed] [Google Scholar]
- Rubin BB, Romaschin A, Walker PM, Gute DC, Korthuis RJ. Mechanisms of postischemic injury in skeletal muscle: intervention strategies. J Appl Physiol. 1996;80:369–387. doi: 10.1152/jappl.1996.80.2.369. [DOI] [PubMed] [Google Scholar]
- Yap B, Kamm RD. Cytoskeletal remodeling and cellular activation during deformation of neutrophils into narrow channels. J Appl Physiol. 2005;99:2323–2330. doi: 10.1152/japplphysiol.00503.2005. [DOI] [PubMed] [Google Scholar]
- Yap B, Kamm RD. Mechanical deformation of neutrophils into narrow channels induces pseudopod projection and changes in biomechanical properties. J Appl Physiol. 2005;98:1930–1939. doi: 10.1152/japplphysiol.01226.2004. [DOI] [PubMed] [Google Scholar]
- Lewis RE, Granger HJ. Diapedesis and the permeability of venous microvessels to protein macromolecules: the impact of leukotriene B4 (LTB4) Microvasc Res. 1988;35:27–47. doi: 10.1016/0026-2862(88)90048-9. [DOI] [PubMed] [Google Scholar]
- Paterson IS, Klausner JM, Goldman G. The endothelial cell cytoskeleton modulates extravascular polymorphonuclear leukocyte accumulations in vivo. Microvasc Res. 1989;38:49–56. doi: 10.1016/0026-2862(89)90016-2. et al. [DOI] [PubMed] [Google Scholar]
- Tidball JG. Interactions between muscle and the immune system during modified musculoskeletal loading. Clin Orthop Rel Res. 2002;403:S100–S109. doi: 10.1097/00003086-200210001-00012. (suppl): [DOI] [PubMed] [Google Scholar]
- Smith LL. Acute inflammation: the underlying mechanism in delayed onset muscle soreness? Med Sci Sports Exerc. 1991;23:542–551. [PubMed] [Google Scholar]
- Pizza FX, Koh TJ, McGregor SJ, Brooks SV. Muscle inflammatory cells after passive stretches, isometric contractions, and lengthening contractions. J Appl Physiol. 2002;92:1873–1878. doi: 10.1152/japplphysiol.01055.2001. [DOI] [PubMed] [Google Scholar]
- McLoughlin TJ, Mylona E, Hornberger TA, Esser KA, Pizza FX. Inflammatory cells in rat skeletal muscle are elevated after electrically stimulated contractions. J Appl Physiol. 2003;94:876–882. doi: 10.1152/japplphysiol.00766.2002. [DOI] [PubMed] [Google Scholar]
- Barash IA, Peters D, Friden J, Lutz GJ, Lieber RL. Desmin cytoskeletal modifications after a bout of eccentric exercise in the rat. Am J Physiol Regul Integr Comp Physiol. 2002;283:R958–R963. doi: 10.1152/ajpregu.00185.2002. [DOI] [PubMed] [Google Scholar]
- Peters D, Barash IA, Burdi M. Asynchronous functional, cellular and transcriptional changes after a bout of eccentric exercise in the rat. J Physiol. 2003;553:947–957. doi: 10.1113/jphysiol.2003.048462. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber RL, Shah S, Friden J. Cytoskeletal disruption after eccentric contraction-induced muscle injury. Clin Orthop Rel Res. 2002;403:S90–S99. doi: 10.1097/00003086-200210001-00011. (suppl): [DOI] [PubMed] [Google Scholar]
- Pizza FX, Cavender D, Stockard A, Baylies H, Beighle A. Anti-inflammatory doses of ibuprofen: effect on neutrophils and exercise-induced muscle injury. Int J Sports Med. 1999;20:98–102. doi: 10.1055/s-2007-971100. [DOI] [PubMed] [Google Scholar]
- Butterfield TA, Herzog W. Quantification of muscle fiber strain during in vivo repetitive stretch-shortening cycles. J Appl Physiol. 2005;99:593–602. doi: 10.1152/japplphysiol.01128.2004. [DOI] [PubMed] [Google Scholar]
- Geronilla KB, Miller GR, Mowrey KF. Dynamic force responses of skeletal muscle during stretch-shortening cycles. Eur J Appl Physiol. 2003;90:144–153. doi: 10.1007/s00421-003-0849-8. et al. [DOI] [PubMed] [Google Scholar]
- Frenette J, St-Pierre M, Cote CH, Mylona E, Pizza FX. Muscle impairment occurs rapidly and precedes inflammatory cell accumulation after mechanical loading. Am J Physiol Regul Integr Comp Physiol. 2002;282:R351–R357. doi: 10.1152/ajpregu.00189.2001. [DOI] [PubMed] [Google Scholar]
- Armstrong RB, Ogilvie RW, Schwane JA. Eccentric exercise-induced injury to rat skeletal muscle. J Appl Physiol. 1983;54:80–93. doi: 10.1152/jappl.1983.54.1.80. [DOI] [PubMed] [Google Scholar]
- Warren GL, Hayes DA, Lowe DA, Armstrong RB. Mechanical factors in the initiation of eccentric contraction-induced injury in rat soleus muscle. J Physiol. 1993;464:457–475. doi: 10.1113/jphysiol.1993.sp019645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil PL, Khakee R. Disruptions of muscle fiber plasma membranes: role in exercise-induced damage. Am J Pathol. 1992;140:1097–1109. [PMC free article] [PubMed] [Google Scholar]
- Schwane JA, Armstrong RB. Effect of training on skeletal muscle injury from downhill running in rats. J Appl Physiol. 1983;55:969–975. doi: 10.1152/jappl.1983.55.3.969. [DOI] [PubMed] [Google Scholar]
- McCully KK, Faulkner JA. Injury to skeletal muscle fibers of mice following lengthening contractions. J Appl Physiol. 1985;59:119–126. doi: 10.1152/jappl.1985.59.1.119. [DOI] [PubMed] [Google Scholar]
- Frenette J, Cai B, Tidball JG. Complement activation promotes muscle inflammation during modified muscle use. Am J Pathol. 2000;156:2103–2110. doi: 10.1016/S0002-9440(10)65081-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizza FX, Hernandez IJ, Tidball JG. Nitric oxide synthase inhibition reduces muscle inflammation and necrosis in modified muscle use. J Leukoc Biol. 1998;64:427–433. [PubMed] [Google Scholar]
- Stauber WT, Fritz VK, Vogelbach DW, Dahlmann B. Characterization of muscles injured by forced lengthening, I: cellular infiltrates. Med Sci Sports Exerc. 1988;20:345–353. doi: 10.1249/00005768-198808000-00004. [DOI] [PubMed] [Google Scholar]
- Kasperek GJ, Snider RD. Increased protein degradation after eccentric exercise. Eur J Appl Physiol Occup Physiol. 1985;54:30–34. doi: 10.1007/BF00426294. [DOI] [PubMed] [Google Scholar]
- Cannon JG, Meydani SN, Fielding RA. Acute phase response in exercise: II, associations between vitamin E, cytokines, and muscle proteolysis. Am J Physiol. 1991;260:R1235–R1240. doi: 10.1152/ajpregu.1991.260.6.R1235. et al. [DOI] [PubMed] [Google Scholar]
- Brickson S, Ji LL, Schell K, Olabisi R, St Pierre Schneider B, Best TM. M1/70 attenuates blood-borne neutrophil oxidants, activation, and myofiber damage following stretch injury. J Appl Physiol. 2003;95:969–976. doi: 10.1152/japplphysiol.00005.2003. [DOI] [PubMed] [Google Scholar]
- Lowe DA, Warren GL, Ingalls CP, Boorstein DB, Armstrong RB. Muscle function and protein metabolism after initiation of eccentric contraction-induced injury. J Appl Physiol. 1995;79:1260–1270. doi: 10.1152/jappl.1995.79.4.1260. [DOI] [PubMed] [Google Scholar]
- Crinnion JN, Homer-Vanniasinkam S, Hatton R, Parkin SM, Gough MJ. Role of neutrophil depletion and elastase inhibition in modifying skeletal muscle reperfusion injury. Cardiovasc Surg. 1994;2:749–753. doi: 10.1177/096721099400200614. [DOI] [PubMed] [Google Scholar]
- Rosen GM, Pou S, Ramos CL, Cohen MS, Britigan BE. Free radicals and phagocytic cells. FASEB J. 1995;9:200–209. doi: 10.1096/fasebj.9.2.7540156. [DOI] [PubMed] [Google Scholar]
- Toumi H, Best TM. The role of neutrophils in injury and repair following muscle stretch. 2006;208:459–470. doi: 10.1111/j.1469-7580.2006.00543.x. J Anat. (4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung EW, Allen DG. Stretch-activated channels in stretch-induced muscle damage: role in muscular dystrophy. Clin Exp Pharmacol Physiol. 2004;31:551–556. doi: 10.1111/j.1440-1681.2004.04027.x. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Wehling-Henricks M. Damage and inflammation in muscular dystrophy: potential implications and relationships with autoimmune myositis. Curr Opin Rheumatol. 2005;17:707–713. doi: 10.1097/01.bor.0000179948.65895.1a. [DOI] [PubMed] [Google Scholar]
- Consolino CM, Brooks SV. Susceptibility to sarcomere injury induced by single stretches of maximally activated muscles of mdx mice. J Appl Physiol. 2004;96:633–638. doi: 10.1152/japplphysiol.00587.2003. [DOI] [PubMed] [Google Scholar]
- Willems ME, Stauber WT. Streptomycin and EDTA decrease the number of desmin-negative fibers following stretch injury. Muscle Nerve. 2005;32:310–315. doi: 10.1002/mus.20370. [DOI] [PubMed] [Google Scholar]
- Pizza FX, McLoughlin TJ, McGregor SJ, Calomeni EP, Gunning WT. Neutrophils injure cultured skeletal myotubes. Am J Physiol Cell Physiol. 2001;281:C335–C341. doi: 10.1152/ajpcell.2001.281.1.C335. [DOI] [PubMed] [Google Scholar]
- Cuschieri J, Gourlay D, Garcia I, Jelacic S, Maier RV. Slow channel calcium inhibition blocks proinflammatory gene signaling and reduces macrophage responsiveness. J Trauma. 2002;52:434–442. doi: 10.1097/00005373-200203000-00004. [DOI] [PubMed] [Google Scholar]
- Tutdibi O, Brinkmeier H, Rudel R, Fohr KJ. Increased calcium entry into dystrophin-deficient muscle fibres of MDX and ADR-MDX mice is reduced by ion channel blockers. J Physiol. 1999;515:859–868. doi: 10.1111/j.1469-7793.1999.859ab.x. (Pt 3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen DG. Skeletal muscle function: role of ionic changes in fatigue, damage and disease. Clin Exp Pharmacol Physiol. 2004;31:485–493. doi: 10.1111/j.1440-1681.2004.04032.x. [DOI] [PubMed] [Google Scholar]
- Yeung EW, Whitehead NP, Suchyna TM, Gottlieb PA, Sachs F, Allen DG. Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J Physiol. 2005;562:367–380. doi: 10.1113/jphysiol.2004.075275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin ER. Role of Ca2+/calmodulin-dependent kinases in skeletal muscle plasticity. J Appl Physiol. 2005;99:414–423. doi: 10.1152/japplphysiol.00015.2005. [DOI] [PubMed] [Google Scholar]
- Grounds MD. Phagocytosis of necrotic muscle in muscle isografts is influenced by the strain, age, and sex of host mice. J Pathol. 1987;153:71–82. doi: 10.1002/path.1711530110. [DOI] [PubMed] [Google Scholar]
- Pizza FX, Peterson JM, Baas JH, Koh TJ. Neutrophils contribute to muscle injury and impair its resolution after lengthening contractions in mice. J Physiol. 2005;562:899–913. doi: 10.1113/jphysiol.2004.073965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira CF, Zamuner SR, Zuliani JP. Neutrophils do not contribute to local tissue damage, but play a key role in skeletal muscle regeneration, in mice injected with Bothrops asper snake venom. Muscle Nerve. 2003;28:449–459. doi: 10.1002/mus.10453. et al. [DOI] [PubMed] [Google Scholar]
- Hirani N, Antonicelli F, Strieter RM. The regulation of interleukin-8 by hypoxia in human macrophages—a potential role in the pathogenesis of the acute respiratory distress syndrome (ARDS) Mol Med. 2001;7:685–697. et al. [PMC free article] [PubMed] [Google Scholar]
- Scott A, Khan KM, Roberts CR, Cook JL, Duronio V. What do we mean by the term “inflammation”? A contemporary basic science update for sports medicine. Br J Sports Med. 2004;38:372–380. doi: 10.1136/bjsm.2004.011312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs EJ, DiPietro LA. Fibrogenic cytokines and connective tissue production. FASEB J. 1994;8:854–861. doi: 10.1096/fasebj.8.11.7520879. [DOI] [PubMed] [Google Scholar]
- Merly F, Lescaudron L, Rouaud T, Crossin F, Gardahaut MF. Macrophages enhance muscle satellite cell proliferation and delay their differentiation. Muscle Nerve. 1999;22:724–732. doi: 10.1002/(sici)1097-4598(199906)22:6<724::aid-mus9>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Allen RE, Boxhorn LK. Inhibition of skeletal muscle satellite cell differentiation by transforming growth factor-beta. J Cell Physiol. 1987;133:567–572. doi: 10.1002/jcp.1041330319. [DOI] [PubMed] [Google Scholar]
- Almekinders LC. Anti-inflammatory treatment of muscular injuries in sport: an update of recent studies. Sports Med. 1999;28:383–388. doi: 10.2165/00007256-199928060-00001. [DOI] [PubMed] [Google Scholar]
- Merrick MA. Secondary injury after musculoskeletal trauma: a review and update. J Athl Train. 2002;37:209–217. [PMC free article] [PubMed] [Google Scholar]
- Knight KL. Effects of hypothermia on inflammation and swelling. Athl Train J Natl Athl Train Assoc. 1976;11:7–10. [Google Scholar]
- Mowlavi A, Neumeister MW, Wilhelmi BJ, Song YH, Suchy H, Russell RC. Local hypothermia during early reperfusion protects skeletal muscle from ischemia-reperfusion injury. Plast Reconstr Surg. 2003;111:242–250. doi: 10.1097/01.PRS.0000034936.25458.98. [DOI] [PubMed] [Google Scholar]
- Scumpia PO, Sarcia PJ, Kelly KM, DeMarco VG, Skimming JW. Hypothermia induces anti-inflammatory cytokines and inhibits nitric oxide and myeloperoxidase-mediated damage in the hearts of endotoxemic rats. Chest. 2004;125:1483–1491. doi: 10.1378/chest.125.4.1483. [DOI] [PubMed] [Google Scholar]
- Hertel J. The role of nonsteroidal anti-inflammatory drugs in the treatment of acute soft tissue injuries. J Athl Train. 1997;32:350–358. [PMC free article] [PubMed] [Google Scholar]
- Curl WW. Clinical relevance of sports-induced inflammation. In: Leadbetter WB, Buckwalter J, Gordon SL, eds. Sport-Induced Inflammation. Park Ridge, IL: American Academy of Orthopaedic Surgeons; 1989:149– 154 .
- Wilson JJ, Best TM. Common overuse tendon problems: a review and recommendations for treatment. Am Fam Physician. 2005;72:811–818. [PubMed] [Google Scholar]
- Shen W, Li Y, Tang Y, Cummins J, Huard J. NS-398, a cyclooxygenase-2–specific inhibitor, delays skeletal muscle healing by decreasing regeneration and promoting fibrosis. Am J Pathol. 2005;167:1105–1117. doi: 10.1016/S0002-9440(10)61199-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronstein BN, Van de Stouwe M, Druska L, Levin RI, Weissmann G. Nonsteroidal antiinflammatory agents inhibit stimulated neutrophil adhesion to endothelium: adenosine dependent and independent mechanisms. Inflammation. 1994;18:323–335. doi: 10.1007/BF01534273. [DOI] [PubMed] [Google Scholar]
- Ali H, Haribabu B, Richardson RM, Snyderman R. Mechanisms of inflammation and leukocyte activation. Med Clin North Am. 1997;81:1–28. doi: 10.1016/s0025-7125(05)70503-4. [DOI] [PubMed] [Google Scholar]
- Kumar A, Boriek AM. Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: a possible role in Duchenne muscular dystrophy. FASEB J. 2003;17:386–396. doi: 10.1096/fj.02-0542com. [DOI] [PubMed] [Google Scholar]
- Thaloor D, Miller KJ, Gephart J, Mitchell PO, Pavlath GK. Systemic administration of the NF-kappaB inhibitor curcumin stimulates muscle regeneration after traumatic injury. Am J Physiol. 1999;277:C320–C329. doi: 10.1152/ajpcell.1999.277.2.C320. [DOI] [PubMed] [Google Scholar]
- Thorsson O, Rantanen J, Hurme T, Kalimo H. Effects of nonsteroidal antiinflammatory medication on satellite cell proliferation during muscle regeneration. Am J Sports Med. 1998;26:172–176. doi: 10.1177/03635465980260020401. [DOI] [PubMed] [Google Scholar]
- Mendias CL, Tatsumi R, Allen RE. Role of cyclooxygenase-1 and -2 in satellite cell proliferation, differentiation, and fusion. Muscle Nerve. 2004;30:497–500. doi: 10.1002/mus.20102. [DOI] [PubMed] [Google Scholar]
- Almekinders LC, Gilbert JA. Healing of experimental muscle strains and the effects of nonsteroidal antiinflammatory medication. Am J Sports Med. 1986;14:303–308. doi: 10.1177/036354658601400411. [DOI] [PubMed] [Google Scholar]
- Mishra DK, Friden J, Schmitz MC, Lieber RL. Anti-inflammatory medication after muscle injury: a treatment resulting in short-term improvement but subsequent loss of muscle function. J Bone Joint Surg Am. 1995;77:1510–1519. doi: 10.2106/00004623-199510000-00005. [DOI] [PubMed] [Google Scholar]
- Elder CL, Dahners LE, Weinhold PS. A cyclooxygenase-2 inhibitor impairs ligament healing in the rat. Am J Sports Med. 2001;29:801–805. doi: 10.1177/03635465010290062101. [DOI] [PubMed] [Google Scholar]
- Reynolds JF, Noakes TD, Schwellnus MP, Windt A, Bowerbank P. Non-steroidal anti-inflammatory drugs fail to enhance healing of acute hamstring injuries treated with physiotherapy. S Afr Med J. 1995;85:517–522. [PubMed] [Google Scholar]
- McCroskery S, Thomas M, Platt L. Improved muscle healing through enhanced regeneration and reduced fibrosis in myostatin-null mice. J Cell Sci. 2005;118:3531–3541. doi: 10.1242/jcs.02482. et al. [DOI] [PubMed] [Google Scholar]
- Szpaderska AM, DiPietro LA. Inflammation in surgical wound healing: friend or foe? Surgery. 2005;137:571–573. doi: 10.1016/j.surg.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Chan YS, Li Y, Foster W. Antifibrotic effects of suramin in injured skeletal muscle after laceration. J Appl Physiol. 2003;95:771–780. doi: 10.1152/japplphysiol.00915.2002. et al. [DOI] [PubMed] [Google Scholar]
- Foster W, Li Y, Usas A, Somogyi G, Huard J. Gamma interferon as an antifibrosis agent in skeletal muscle. J Orthop Res. 2003;21:798–804. doi: 10.1016/S0736-0266(03)00059-7. [DOI] [PubMed] [Google Scholar]
- Fukushima K, Badlani N, Usas A, Riano F, Fu F, Huard J. The use of an antifibrosis agent to improve muscle recovery after laceration. Am J Sports Med. 2001;29:394–402. doi: 10.1177/03635465010290040201. [DOI] [PubMed] [Google Scholar]
- Peake J, Nosaka K, Suzuki K. Characterization of inflammatory responses to eccentric exercise in humans. Exerc Immunol Rev. 2005;11:64–85. [PubMed] [Google Scholar]
- Petersen AM, Pedersen BK. The anti-inflammatory effect of exercise. J Appl Physiol. 2005;98:1154–1162. doi: 10.1152/japplphysiol.00164.2004. [DOI] [PubMed] [Google Scholar]
- Smith JA, Pyne DB. Exercise, training, and neutrophil function. Exerc Immunol Rev. 1997;3:96–116. [PubMed] [Google Scholar]
- Agarwal S, Deschner J, Long P. Role of NF-kappaB transcription factors in antiinflammatory and proinflammatory actions of mechanical signals. Arthritis Rheum. 2004;50:3541–3548. doi: 10.1002/art.20601. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]