

Abstract
Granulomatous experimental autoimmune thyroiditis (G-EAT) is induced in DBA/1 mice by adoptive transfer of mouse thyroglobulin (MTg)-primed spleen cells. TNFα is an important proinflammatory cytokine and apoptotic molecule involved in many autoimmune diseases. To study its role in G-EAT, anti-TNFα mAb was given to recipient mice. Disease severity was comparable between mice with or without anti-TNFα treatment at day 19–21, the time of maximal severity of G-EAT, suggesting TNFα is not essential for development of thyroid inflammation. However, thyroid lesions resolved at day 48 in anti-TNFα-treated mice, while thyroids of rat Ig-treated controls had fibrosis. These results suggested that reducing TNFα contributed to resolution of inflammation and inhibited fibrosis. Gene and protein expression of inflammatory molecules was examined by RT-PCR and immunostaining, and apoptosis was detected using TUNEL staining and an apoptosis kit. Thyroids of anti-TNFα-treated controls had reduced proinflammatory and profibrotic molecules, e.g. IFNγ, IL-1β, IL-17, iNOS and MCP-1, at day 19 compared to thyroids of rat Ig-treated mice. There were more apoptotic thyrocytes in rat Ig-treated controls than in anti-TNFα-treated mice. The site of expression of the anti-apoptotic molecule FLIP also differed between rat Ig-treated and anti-TNFα-treated mice. FLIP was predominantly expressed by inflammatory cells of rat Ig-treated mice and by thyrocytes of anti-TNFα-treated mice. These results suggest that anti-TNFα may regulate expression of proinflammatory cytokines and apoptosis in thyroids, resulting in less inflammation, earlier resolution and reduced fibrosis.
Keywords: Rodent, autoimmunity, cytokines
Introduction
Granulomatous experimental autoimmune thyroiditis (G-EAT) is an organ-specific autoimmune disease that can be induced in genetically susceptible mice by injection of MTg and adjuvant [1–5] or by adoptive transfer of spleen cells from MTg-primed donors activated in vitro with MTg and IL-12 [6–8]. G-EAT is characterized by proliferation of thyroid epithelial cells, granuloma formation, and destruction of the thyroid by T lymphocytes, large numbers of histiocytes, multinucleated giant cells, and variable numbers of neutrophils [6–8]. CD4+ T cells are the primary effector cells [6,7], while CD8+ T cells promote resolution of G-EAT [9]. Although the mechanisms by which CD4+ T cells cause thyroid destruction are not well understood, cytokines produced by activated CD4+ T cells are known to play an important role in the pathogenesis of EAT [6–8]. TNFα and other proinflammatory cytokines are upregulated in thyroids of mice with G-EAT [10]. Moreover, early resolution of G-EAT was observed in IFNγ −/− recipients, and reduction of several cytokines including TNFα may contribute to G-EAT resolution in IFNγ −/− mice [10].
TNFα is a proinflammatory cytokine that plays a critical role in diverse cellular events. The binding of TNFα to TNFα receptors triggers a series of intracellular events that ultimately result in production of inflammatory cytokines via NF-kB activation or apoptosis [11,12]. Thus, TNFα is a major mediator of apoptosis as well as inflammation and immunity. TNFα is expressed in virtually all inflammatory autoimmune diseases and has been implicated in the pathogenesis of a wide spectrum of human autoimmune diseases, including rheumatoid arthritis, diabetes, multiple sclerosis, and inflammatory bowel disease [11–15]. However, the pathophysiological effects of TNFα in autoimmune diseases are still incompletely understood, and research with other animal models may further clarify its functions [14]. TNFα is also a profibrotic cytokine, but its role in fibrosis and its mechanism of action are not well defined [16–19].
This study was undertaken to define the role of TNFα in autoimmune thyroiditis and to determine if anti-TNFα antibodies might be useful for decreasing autoimmune inflammation and fibrosis. TNFα neutralization significantly promoted resolution of inflammation and reduced development of fibrosis in G-EAT. By detecting expression of proinflammatory cytokines, fibrotic and apoptotic molecules, the mechanisms by which TNFα contributes to G-EAT pathology were determined.
Materials and Methods
Mice
DBA/1 mice were bred in our animal facilities in accordance with the University of Missouri institutional guidelines for animal care. Both male and female mice (8–12 wk old) were used for these experiments.
Induction of G-EAT
G-EAT was induced as previously described [4]. Briefly, mice were injected i.v. twice at 10-day intervals with 150 μg MTg prepared as previous described [6] and 15 μg LPS (Escherichia coli 011:B4; Sigma Chemical Co., St. Louis, MO). Seven days later, donor spleen cells were restimulated in vitro with 25 μg/ml MTg and 5 ng/ml IL-12 [1, 4]. Cells were harvested after 72 h, washed twice, and 3.5 × 107 cells were transferred i.v. to 500-Rad irradiated syngeneic recipients. Recipient thyroids were removed at day 19–21 (peak of disease) or 38–56 days (resolution) after cell transfer [6–8].
Anti-TNFα treatment
Rat anti-mouse TNFα (ATCC HB-10649) mAb was purified from culture supernatant using protein G-Sepharose. Recipient mice were given 0.3mg rat anti-mouse TNFα mAb or normal rat IgG (Jackson Immunoreserch) 1–2 days after cell transfer and every 3–4 days until termination of the experiment. The dosage of anti-TNFα mAb was chosen based on use of anti-TNFα mAb in other models [20,21].
Evaluation of thyroiditis
Thyroids were collected, fixed in formalin, sectioned and stained with hematoxylin and eosin (H & E) as previously described [4]. Thyroids were scored quantitatively for EAT severity (the extent of thyroid follicle destruction) using a scale of 1+ to 5+, as described previously [6–8]. 1+ thyroiditis is defined as an infiltrate of at least 125 cells in one or several foci; 2+ is 10–20 foci of cellular infiltration involving up to 25% of the gland; 3+ indicates that 25–50% of the gland is infiltrated; 4+ indicates that >50% of the gland is destroyed by infiltrating inflammatory cells; and 5+ indicates virtually complete destruction of the thyroid with few or no remaining follicles. Thyroid lesions were also evaluated qualitatively. Thyroid lesions designated as granulomatous had enlargement and proliferation of thyroid follicular cells, with numerous histiocytes, multinucleated giant cells, and increased numbers of neutrophils in addition to the mononuclear cell infiltration. The more severely inflamed granulomatous thyroids (4–5+ severity scores) also had microabscess formation, necrosis, and focal fibrosis, and inflammation extended beyond the thyroid to involve adjacent muscle and connective tissue [7]. For evaluation of collagen deposition, some thyroid sections were stained using Masson’s Trichrome.
Serum thyroxine (T4) assay
Serum T4 levels were determined using a T4 enzyme immunoassay kit (Biotecx Labs, Houston, TX) according to the manufacturer’s instructions. Results are expressed as μg T4/dL of serum. Using this kit, values >3 μg T4/dl of serum are considered normal [22].
Immunohistochemistry and confocal analysis
Myofibroblasts, important cells involved in development of fibrosis, were recognized by staining using a mouse anti-α-smooth muscle actin (α SMA) (clone 1A4, Sigma) on paraffin sections of thyroids. Staining of TNFα, active TGFβ, iNOS, MCP-1, FLIP and caspase-3 was performed using the immunoperoxidase method as previously described [10,22,23]. For IL-17 staining, rabbit anti-IL-17 [H-132; Santa Cruz Biotechnology, Santa Cruz, CA) was used. Following incubation with a secondary biotinylated goat anti-rabbit antibody (1:500) (Jackson ImmunoResearch, West Grove, PA), immunoreactivity was demonstrated using the avidin-biotin complex immunoperoxidase system (Vector ABC peroxidase kit; Vector Laboratories, Burlingame, CA) with 3,3-diaminobenzidine tetrahydrochloride (DAB), Vector VIP (Vector Laboratories) or Vector NovaRED (Vector Laboratories) as the chromogen. Slides were counterstained with hematoxylin. Negative controls used non-immune rat, rabbit or goat Ig at a protein concentration equivalent to the respective primary antibodies. These controls were always negative.
Dual staining and confocal microscopy were used to detect apoptotic thyrocytes. Thyrocytes were identified by cytokeratin staining, and apoptosis was detected using an in-situ cell death kit [23]. To more specifically detect apoptosis of thyrocytes, dual immunofluorescence and confocal laser scanning microscopy was done. Prior to staining, tissue sections were pretreated by microwave irradiation for antigen retrieval [23], and thyroid follicular cells were detected by pan-cytokeratin staining using FITC-labeled PCK-26 (Sigma, St. Louis). Apoptosis was detected using an in-situ cell death kit (Roche, Indianapolis, IN). Slides were observed with a BioRad Radiance 2000 confocal system coupled to an Olympus IX70 inverted microscope.
Reverse transcription-PCR (RT-PCR) amplification and real-time quantitative PCR (Q-PCR)
RT-PCR was performed as previously described [10] using specific primers [22,24]. To determine the relative initial amounts of target cDNA, each cDNA sample was serially diluted 1:5 and 1:25, and amplified with specific primers. Hypoxanthine phosphoribosyltransferase (HPRT) was used as a housekeeping gene to verify that the same amount of RNA was amplified. To compare relative levels of mRNA transcripts between different groups, samples were reverse transcribed and amplified at the same time using aliquots of reagent from the same master mix. The PCR products were analyzed using a digital imaging system (Life Sciences, St. Louis, MO). Samples within the linear-relationship between input cDNA and final PCR products (usually 1/25 cDNA dilution) were collected, and empirically determined concentrations of first-strand cDNA were used in RT-PCR to ensure linear amplification of sequences. The densitometric units for each cytokine band were normalized to those for the corresponding HPRT band. Most cytokine gene primers used in this study have been described previously [22,24]. Primer sequences for IL-17 were: sense: GGTCAACCTCAAAGTCTTTAACTC, anti-sense: TTAAAA ATGCAAGTAAGTTTGCTG.
Real-time PCR was performed using an ABI Prism 7000 Sequence Detection System using SYBR Green PCR Master Mix (ABgene, Surrey, UK) and following manufacturer’s protocols. Primers used were: IFN sense: TCAAGTGGCATAGATGTG GAAGAA, IFN anti-sense: TGGCTCTGCAGGATTTTC ATG. MCP-1 forward: GTTGGCTCAGCCAGATGCA, MCP-1 reverse: AGCCTACTCATTGG GATCATCTT G. IL-17F sense: CCCATGGGATTACAACATCACTC, IL-17F anti-sense: CACTGGG CCTCAGCGATC. IL-10 forward: CAGCCTTG GAGAAAAGAGAG, IL-10 reverse: GGAAGTGGGTGGTGTTATTG. Each sample was amplified in triplicate. Values represent relative expression levels normalized to HPRT.
Statistical analysis
All experiments were repeated at least three times. Statistical significance was evaluated using the Mann-Whitney test. Values with a p value <0.05 were considered significant and are designated by * in the figure legends.
Results
Inhibition of TNFα has no effect on development of G-EAT, but decreases fibrosis and promotes resolution of inflammation
All recipients given anti-TNFα developed G-EAT that reached maximal severity 19 days after cell transfer (Fig. 1). Rat Ig-treated control recipients always had very severe (4–5+) G-EAT 19 days after cell transfer, and anti-TNFα had no significant effect on G-EAT severity at day 19 (P=0.06). However, anti-TNFα significantly reduced G-EAT severity 38–56 days after cell transfer (P<0.05) (Fig. 1). Thyroids of most rat Ig-treated mice maintained disease severity scores of 5+ 38–56 days after cell transfer, while thyroid lesions of 8 of 15 anti-TNFα-treated mice were resolving, with a severity score of 1–3+. Thus, anti-TNFα promoted resolution of inflammation in G-EAT.
Figure 1.

Effect of anti-TNFα on induction and outcome of G-EAT. G-EAT severity scores were evaluated 19 and 38–56 days after cell transfer on a scale from 0–5+. Each symbol represents G-EAT severity scores of individual mice. A significant difference in the severity scores of anti-TNFα compared with rat Ig-treated mice at day 38–56 is indicated by an asterisk (p < 0.05). Results are pooled from 3 separate experiments.
G-EAT lesions were characterized by infiltration of inflammatory cells and destruction of thyroid follicles (Fig. 2A–H). There were few or no thyroid follicles at day 19 in thyroids of mice given rat IgG (Fig 2A), or anti- TNFα with 5+ severity scores (Fig 2E), but there were some residual thyroid follicles in thyroids with 4+ severity scores (Fig 2B and F, arrowhead). Inflammation persisted in rat-Ig-treated mice, with 5+ severity scores, and thyroids were atrophic (Fig. 2C, arrows) with few or no thyroid follicles. Thyroids of rat-Ig-treated mice with 4–5+ severity scores had a few remaining follicles and persistent inflammation at day 40 (Fig. 2D). At day 40, thyroids of anti-TNFα-treated mice with 4–5+ severity scores were less atrophic, and lesions had started to resolve in most mice (Fig. 2G), while thyroids of anti-TNFα treated mice with 2–3+ severity scores were resolving with many thyroid follicles and less inflammation (Fig. 2H).
Figure 2.
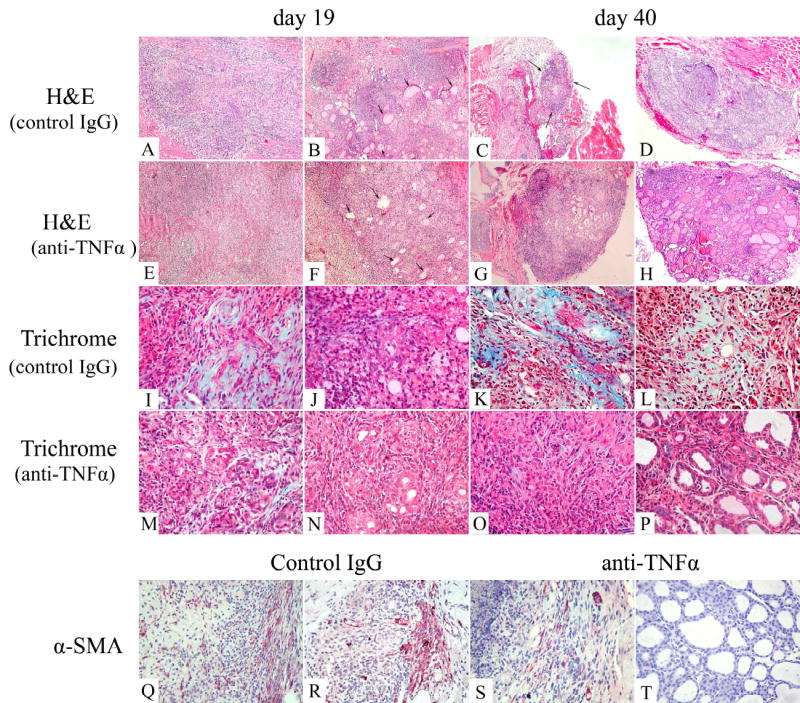
Histopathology of G-EAT and fibrosis in thyroids of rat Ig-treated and anti-TNFα-treated mice. A–H: H&E staining of thyroids from rat Ig-treated and anti-TNFα- treated mice at day 19 or day 40. G-EAT reached maximal severity in both control (A, B) and anti-TNFα-treated (E, F) mice with a score of 5+ (A, E) or 4+ (B, F) severity at day 19. Inflammation was sustained at day 40 with a score of 5+ (C) or 4–5+ severity (D) in rat Ig-treated mice (C, D), and thyroids with 5+ severity scores were atrophic (C). In anti-TNFα-treated mice, thyroids with 4–5+ severity scores were not atrophic (G), and inflammation was resolving at day 40 in thyroids with 2–3+ severity scores (H). I–P: Trichrome staining for collagen in rat Ig-treated and anti-TNFα-treated mice. Collagen was evident in thyroids of rat Ig-treated mice with 5+ (I) or 4+ (J) severity at day 19 (I, J: blue), and was more prominent in thyroids of rat Ig-treated mice with 5+ (K) or 4+ (L) severity at day 40. Collagen was also present in thyroids of anti-TNFα-treated mice with 5+ severity scores (M) but was minimal in thyroids with 4+ severity scores (N) at day 19. Fibrosis was minimal in thyroids of anti-TNFα-treated mice with 5+ severity scores (O) and was barely discernible in 3+ G-EAT thyroids (P) at day 40. Q–T: Myofibroblasts were identified by -SMA staining in thyroids of both rat Ig-treated and anti-TNFα-treated mice at day 19 with 5+ severity scores. There were more myofibroblasts in thyroids of rat Ig-treated mice (Q and R) than in thyroids of anti-TNFα-treated mice at day 19 (S and T). Original magnification: A–H: 100×; I–T: 400×. Photos are representative of 3 experiments for rat Ig-treated and anti-TNFα treated mice.
The hallmark of fibrosis is the deposition of collagen and myofibroblasts, which can be detected by Trichrome (Fig. 2I–P) and α-smooth muscle actin staining (Fig. 2Q–T), respectively. Fibrosis developed at day 19 in thyroids of rat Ig-treated mice with 5+ (Fig 2I) or 4+ (Fig. 2J) severity scores as shown by the blue collagen staining. Collagen deposition increased considerably at day 40 in thyroids of rat Ig-treated mice with 5+ (Fig. 2K) or 4+ (Fig. 2L) severity scores. There was less collagen deposition in thyroids of anti-TNFα-treated mice at day 19 (Fig. 2, M and N) or day 40 (Fig. 2O and P), and little or no collagen was present in thyroids of anti-TNFα-treated mice with 3+ severity scores at day 40 (Fig. 2P). Myofibroblasts represent activated fibroblasts and are prominent inducers of collagen [25,26]. Infiltration of myofibroblasts was more prominent at day 19 (Fig. 2Q) and day 40 (Fig. 2R) in thyroids of rat Ig-treated mice than in thyroids of anti-TNFα-treated mice at day 19 (Fig. 2S) or day 40 (Fig. 2T). Serum T4 level which considered normal with a value >3 μg/dl is an indicator of thyroid function, and will be decreased during development of fibrosis. All mice with very severe thyroid destruction and fibrosis had 5+ severity scores and had low serum T4 (Fig. 3). Serum T4 levels were similar for rat Ig-treated and anti-TNFα-treated mice at day 19 (P=0.169) (Fig. 3). However, at days 38–56, T4 levels for thyroids with 4–5+ severity scores were significantly lower in rat Ig-treated mice than in anti-TNFα-treated mice (P<0.05) (Fig. 3). Serum T4 levels in mice with 1–3+ G-EAT severity scores were all normal (Fig. 3), and serum T4 was normal when lesions resolved in anti-TNFα-treated mice.
Figure 3.
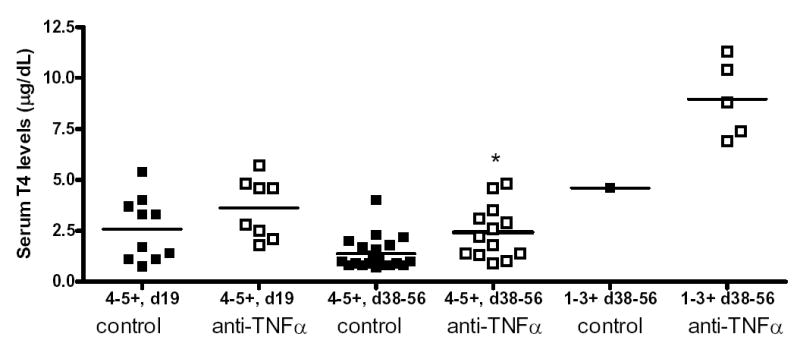
Serum T4 levels in rat Ig-treated and anti-TNFα-treated mice. Serum T4 levels were determined at day 19 or day 38–56 using a T4 enzyme immunoassay kit as described in Methods. Results are expressed as μg T4/dL of serum. * indicates that serum T4 levels were higher at day 38–56 in anti-TNFα-treated compared to rat Ig-treated mice (P<0.05).
The reduced thyroid destruction and fibrosis in thyroids of anti-TNFα-treated mice is solely due to inhibition of TNFα and is not a nonspecific effect of rat IgG, since repeated and prolonged injection of rat IgG or no antibody both resulted in comparable severity of G-EAT at day 19 with atrophy and fibrosis at day 35–60 [7,10,22, 23].
In conclusion, anti-TNFα did not affect incidence, onset or severity of G-EAT, suggesting that endogenous TNFα is not essential for development of G-EAT induced by sensitized cells. However, resolution of G-EAT lesions was promoted and fibrosis was reduced after TNFα neutralization.
Effect of anti-TNFα on expression of pro- and anti- inflammatory molecules
To determine whether anti-TNF modulated expression of cytokines in thyroids, expression of TNFα, IFNγ, IL-1β, iNOS, IL-17, IL4, IL-13, IL-5 and IL-10 were analyzed by RT-PCR. None of the cytokines were detected in normal thyroids, but mRNA was upregulated in thyroids of rat Ig-treated and anti-TNFα-treated mice at day 19, and declined at day 48 (Fig. 4A). Compared to rat-Ig treated controls, thyroids of anti-TNFα-treated mice had significantly lower expression of proinflammatory cytokines such as TNFα, IFNγ, IL-1β, iNOS and IL-17 at day 19 (Fig. 4A). Th2 cytokines IL-4, IL13, IL-5 and IL-10 were also upregulated in thyroids of both rat Ig-treated and anti-TNFα-treated mice at day 19 (Fig. 4B), and anti-TNFα had no effect on expression of IL-4, IL-13, IL-5 or IL-10 at day 19 (Fig. 4B). These results suggest that IL-4 and IL-13 are not involved in development of fibrosis in G-EAT although IL-4 and IL-13 are implicated in development of fibrosis in other models [27–29]. Expression of all cytokines was very low at day 48 and only TNFα was significantly decreased in anti-TNFα-treated mice compared to rat Ig-treated mice (Fig. 4).
Figure 4.
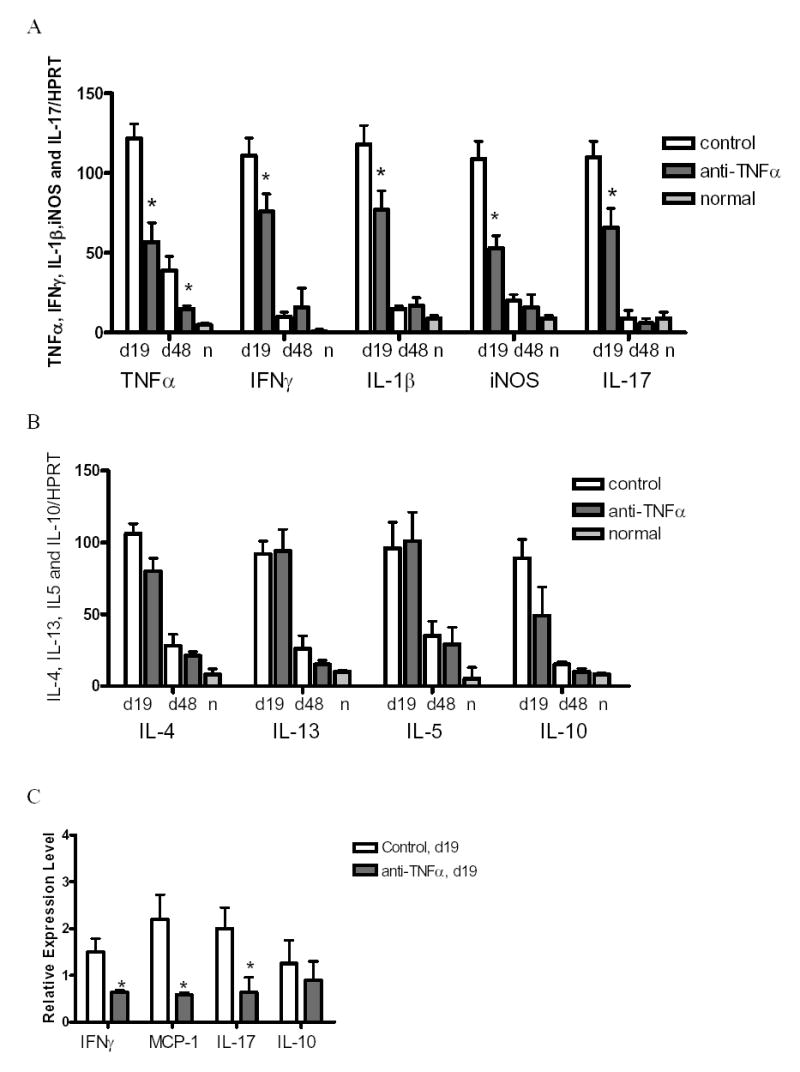
Comparison of cytokine expression in rat Ig-treated and anti-TNFα-treated mice. mRNA expression levels of TNFα, IFNγ, IL-1β, iNOS and IL-17 (A), IL-4, IL13, IL-5 and IL-10 (B) in thyroids of rat Ig-treated and anti-TNFα-treated mice 19 or 48 days after cell transfer. Bars represent mean results for thyroids of five individual mice ± SD. Results are expressed as the mean ratio of cytokine densitometric U/HPRT ± SD (×100), and are representative of two independent experiments. C: Real-time PCR analysis of IFNγ, MCP-1, IL-17 and IL-10 mRNA expression in thyroids of rat Ig-treated and anti-TNFα-treated mice at day 19. The results are the mean ± SD from two independent experiments. Data are presented as relative expression levels normalized against the housekeeping gene HPRT. A significant difference between rat Ig-treated and anti-TNFα-treated groups is indicated by * (P<0.05). N in A and B represents normal thyroids. In A, B and C: G-EAT severity was 4–5+ at day 19 in thyroids of both rat Ig-treated and anti-TNFα-treated mice. Thyroids of rat Ig-treated mice had 4–5+ severity scores and fibrosis at day 48, while G-EAT was resolving with a score of 0–4+ in thyroids of anti-TNFα-treated mice.
Real-time quantitative PCR indicated a significant reduction in the expression of mRNA for IFNγ, MCP-1 and IL-17 in thyroids of anti-TNFα-treated mice compared to rat-Ig treated mice (Fig. 4C). However, anti-TNFα had no effect on mRNA for IL-10 (Fig. 4C).
To determine if protein expression of proinflammatory and profibrotic cytokines was also reduced in anti-TNFα-treated mice, immunostaining was used to examine expression of TNFα, IL-17, iNOS, MCP-1 and active TGFβ1 in G-EAT thyroids. The results confirmed that the protein expression of TNFα (Fig 5A and B), IL-17 (Fig. 5C and D), iNOS (Fig. 5E and F) and MCP-1 (Fig. 5G and H) at day 19–21 correlated with expression of their transcripts, being reduced in thyroids of anti-TNFα-treated mice (Fig. 5, B, D, F, H) compared to rat-Ig treated mice (Fig. 5, A, C, E, G) although G-EAT severity (4–5+) at day 19 was comparable in both groups. Active TGFβ which contributes to development of fibrosis was highly expressed in thyroids of rat Ig-treated mice with 4–5+ severity scores at day 19 (Fig. 5I), whereas active TGFβ1 appeared to be reduced in thyroids of anti-TNFα-treated mice with 4–5+ G-EAT (Fig. 5J). Confocal analysis showed that IL-17 was expressed by infiltrating CD4+ T cells, and expression of IL-17 by CD4+ T cells was reduced in anti-TNFα-treated mice compared to rat-Ig treated mice (data not shown).
Figure 5.
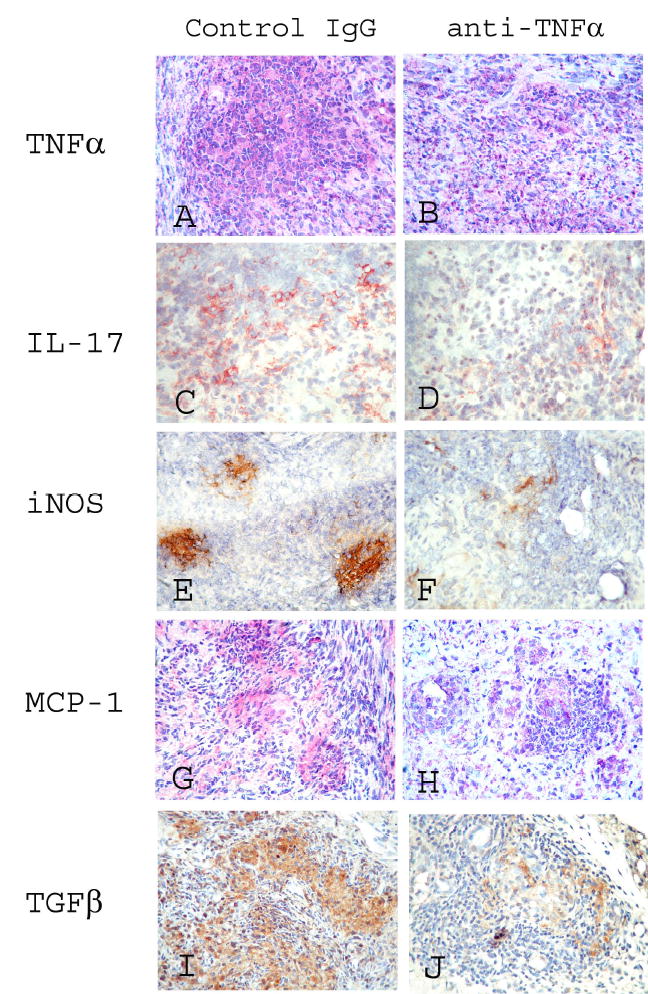
Protein expression of TNFα, IL-17, iNOS, MCP-1 and TGFβ in thyroids of rat Ig-treated and anti-TNFα-treated mice. A-H: representative areas of photomicrographs demonstrating TNFα (A, B: red), IL-17 (C, D: red), iNOS (E, F: brown), MCP-1 (G, H: red), and active TGFβ (I, J: brown) at day 19 in thyroids of rat Ig-treated mice (A, C, E, G, I) and anti-TNFα-treated mice (B, D, F, H, J). G-EAT severity was 4–5+ at day 19 in thyroids of both rat Ig-treated and anti-TNFα-treated mice. Thyroids of mice given rat Ig-G had 4–5+ severity scores and fibrosis at day 40, but G-EAT lesions were resolving with little fibrosis at day 40 in thyroids of anti-TNFα-treated mice. Magnification: A–H: 400×.
Together, these results indicated that anti-TNFα modulated expression of proinflammatory and profibrotic cytokines, but had no effect on expression of Th2 cytokines. Reduced protein expression of the profibrotic cytokines MCP-1 and active TGFβ in thyroids of anti-TNFα-treated mice may contribute to inhibition of fibrosis.
Effect of anti-TNFα on apoptosis and expression of anti-apoptotic molecules
TNFα is also a major mediator of apoptosis [11,12,30]. To determine if anti-TNFα might regulate expression of apoptosis-related molecules in G-EAT thyroids, different methods were used to examine the effect of anti-TNFα on pro and anti-apoptotic molecules and apoptosis in thyroids. TUNEL staining showed apoptotic cells in thyroids of both rat Ig-treated (Fig. 6A) and anti-TNFα-treated mice (Fig. 6B). Apoptotic cells were predominant in thyroid follicular cells in thyroids of rat Ig-treated mice (Fig. 6A, arrows), but predominant in inflammatory cells in thyroids of anti-TNFα-treated mice (Fig. 6B, arrows). TNFα signaling triggers apoptosis through cleavage of caspase-3, and results in production of active caspase-3, which represents cells undergoing apoptosis [31]. Using a monoclonal antibody that recognizes the active form of caspase-3, expression of active caspase-3 was predominant in thyroid follicular cells in rat Ig-treated mice (Fig. 6C, arrows), but was predominant in inflammatory cells in anti-TNFα-treated mice (Fig. 6D, arrow). Confocal analysis confirmed that there were many apoptotic thyrocytes in rat Ig-treated mice (Fig. 6E), while there were fewer apoptotic thyrocytes in anti-TNFα-treated mice (Fig. 6F). These results suggest TNFα may contribute to apoptotic destruction of thyrocytes.
Figure 6.
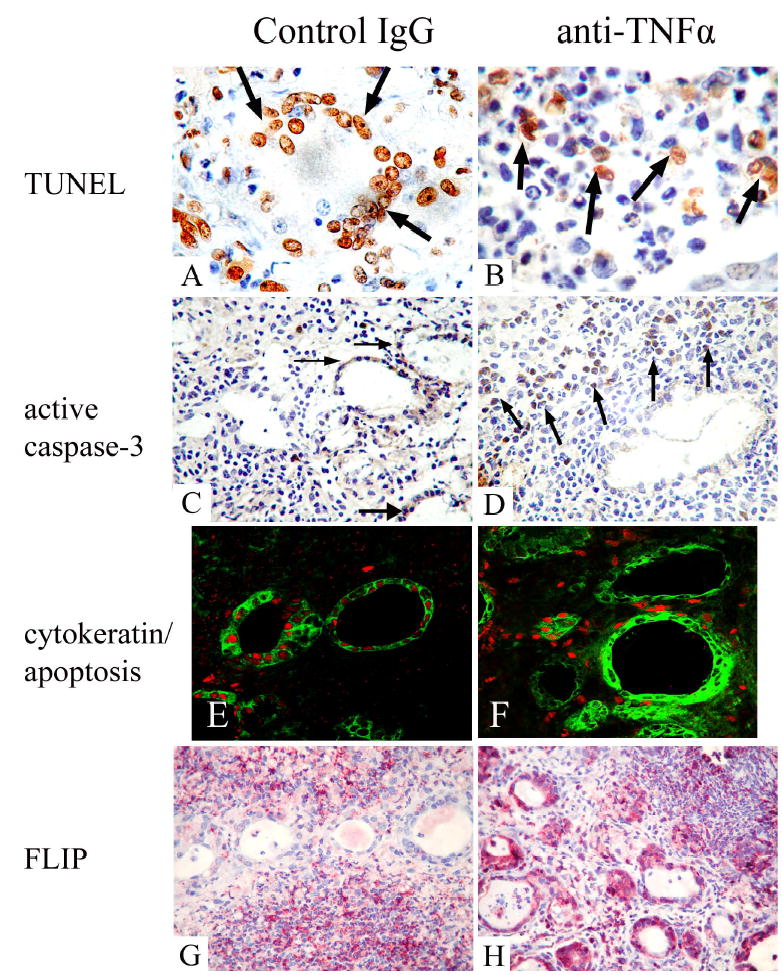
Expression of apoptotic and anti-apoptotic molecules in thyroids of mice given rat IgG and anti-TNFα 19 days after cell transfer. A, B: TUNEL staining (brown) in thyroids of rat Ig-treated (A) and anti-TNFα treated mice (B). C, D: Expression of active caspase-3 (reddish brown) in thyroids of rat Ig-treated (C) and anti-TNFα-treated mice (D). E, F: Confocal analysis demonstrates apoptosis of cytokeratin+ thyrocytes in thyroids mice given rat Ig-G (E) and anti-TNFα (F). Thyrocytes were identified by cytokeratin (green, cytoplasmic staining in E and F), and apoptosis (red, nuclear staining in E, F) was detected using an in situ cell death kit. G, H: Expression of FLIP (red) in thyroids of rat Ig-treated (G) and anti-TNFα-treated mice (H). All thyroids had 4–5+ severity scores. Original magnification: A, B: 1000×; C, D, G and H: 400×; E and F, 600×.
FLIP is an important anti-apoptotic molecule in TNFα and Fas signaling pathways [12,31], and has been implicated in resolution of G-EAT [10]. Expression of FLIP was predominant on infiltrating inflammatory cells in thyroids of rat Ig-treated mice (Fig. 6G), and predominant on thyroid follicular cells in thyroids of anti-TNFα-treated mice (Fig. 6H). The expression pattern of FLIP in thyroids of anti-TNFα-treated mice was similar to that in thyroids of IFNγ −/− mice in which G-EAT lesions spontaneously resolved [10], suggesting FLIP may be an important molecule protecting against apoptotic destruction of thyroids in G-EAT.
Discussion
The G-EAT model established in our laboratory is an excellent model for studying mechanisms that determine whether an autoimmune response will resolve or progress to fibrosis. G-EAT lesions reach maximal severity 19–21 days after cell transfer, and inflammation continues with development of fibrosis in DBA/1 mice (Figs. 1 and 2). This study was undertaken to understand the role of TNFα in development of thyroid inflammation and fibrosis. Our results indicate that TNFα is not essential for the development of G-EAT, but sustained expression of TNFα is important for maintenance of inflammation and for promoting fibrosis. Furthermore, neutralization of TNFα contributes to resolution of inflammation and inhibits fibrosis.
TNFα is not essential for development of G-EAT induced by sensitized cells since anti-TNFα did not reduce disease severity at day 19. Anti-TNFα also did not reduce disease severity in collagen-induced arthritis or experimental autoimmune encephalomyelitis [11,32,33], and TNFα-deficient mice develop arthritis [14,34]. Our results together with these data are consistent with the notion that TNFα is generally not critical for the induction of organ-specific autoimmune disease [14,35]. By using knockout of different cytokines on the DBA/1 background, e.g. IFNγ −/−, IL-12−/− and IL-4−/− mice, we found that these individual cytokines were also not required for induction of severe G-EAT [6,10,36]. Our results also suggest that it is not an individual cytokine but the combined effects of several proinflammatory cytokines that contribute to the pathogenesis of G-EAT. TNFα may act together with other proinflammatory cytokines to induce thyroid destruction.
However, TNFα contributes to the maintenance of inflammation in G-EAT since thyroid lesions resolved earlier in mice given anti-TNFα. One mechanism by which anti-TNFα promotes G-EAT resolution may due to its ability to modulate production of other cytokines. TNFα plays a key role in the inflammatory process by inducing transcription of several proinflammatory cytokines [11]. Many proinflammatory cytokines such as TNFα, IL-1β, IFNγ, iNOS and IL-17 are expressed in thyroids of mice with G-EAT. Inhibition of TNFα reduced expression of proinflammatory cytokines at day 19 in the thyroid and had no significant effect on expression of Th2 cytokines such as IL-4, IL-13 and IL-10. Thyroids with 4–5+ G-EAT severity scores at day 35–60 are very small and atrophic, and those in which inflammation has largely resolved have very few inflammatory cells remaining. Therefore, expression of cytokines is very low in both of these groups of thyroids regardless of G-EAT severity. The fact that TNFα, IL-1β, IFNγ, iNOS and IL-17 were more highly expressed at day 19 in thyroids of rat Ig-treated mice compared to anti-TNFα-treated mice, suggests that TNFα has a proinflammatory role in thyroiditis [5,37]. TNFα may upregulate production of IL-1β, IFNγ, IL-17 and iNOS, which coordinate with TNFα to maintain autoimmune inflammation in G-EAT. IL-17 contributes to the pathogenesis of many autoimmune and inflammatory diseases by acting as a proinflammatory mediator [38–42]. The decreased expression of IL-17 in thyroids of anti-TNFα-treated mice suggested that TNFα may act directly or indirectly to modulate expresson of IL-17. Proinflammatory cytokines including TNFα and iNOS were also reduced in thyroids of IFNγ −/− mice in which G-EAT lesions spontaneously resolved [10], suggesting that these cytokines may mutually stimulate each other’s production, further amplify inflammation and contribute to sustained inflammation in G-EAT. Our study suggests that a complex interplay among cytokines produced by activated CD4+ T cells is likely to control the outcome of an autoimmune disease.
TNFα, IFNγ and IL-1β can all affect apoptosis [11,12,31,37,43–45]. Therefore, through modulation of proinflammatory cytokines, TNFα may modulate the apoptosis of different cell types. Apoptosis of inflammatory cells was increased whereas apoptotic thyrocytes were decreased in anti-TNFα-treated mice (Fig. 6B, D, F). Indeed, anti-TNFα treatment induced apoptosis of inflammatory cells in patients with arthritis and in animal models of Crohn's disease [30,46]. FasL expression by thyrocytes and CD8+ T cells play an important role in mediating the killing of Fas+ inflammatory cells, contributing to G-EAT resolution [47,48]. TNFα and other proinflammatory cytokines IFNγ, IL-1β and iNOS could induce apoptosis in tissues cells such as pancreatic islet cells and thyrocytes [10,37,44,45,49]. The current study supports a role for TNFα, IFNγ, IL-1β and iNOS in autoimmune thyroiditis both for perpetuation of the autoimmune disease and induction of tissue damage via apoptosis of thyrocytes [10,37,44]. Expression of FLIP was shown to protect cells against TNFα- and/or Fas- induced apoptosis [31,48,50]. High expression of FLIP by thyrocytes of anti-TNFα-treated mice correlated with reduced apoptosis of thyrocytes (Fig. 6H), and may protect thyrocytes in anti-TNFα-treated mice from apoptosis.
The results of this study also support a role for TNFα in development of fibrosis [16–18, 51–56]. The fact that profibrotic cytokines were reduced in thyroids of anti-TNFα-treated mice suggests that TNFα contributes to the development of fibrosis. Thyroids from rat IgG-treated control mice highly expressed TNFα, myofibroblasts and collagen. Our finding is consistent with other observations indicating that increased levels of TNFα correlate with increased collagen deposition and development of fibrosis in other animal models and also in humans [16–18,51–56]. Thyroids of rat Ig-treated mice had extensive inflammation, and serum T4 levels began to decrease at day 19 and further decreased by day 38–56 as a result of thyroid fibrosis. In contrast, G-EAT lesions began to resolve by day 38–56 in anti-TNFα-treated mice, fibrosis was reduced, and serum T4 levels were normal in anti-TNFα-treated mice when lesions resolved. In the present study, reduced fibrosis in thyroids of mice given anti-TNFα correlated with reduced expression of active TGFβ and MCP-1 and decreased infiltration of myfbs in thyroids. Anti-TNFα also markedly suppressed bleomycin- induced lung fibrosis [18,54] and decreased expression of TGFβ and MCP-1 in the lung [18]. Anti-TNFα may inhibit thyroid fibrosis through down-regulation of profibrotic cytokines such as TGFβ and MCP-1 [22,57]. Consistent with the results presented here, TNFα has been shown to be profibrotic in several other different animal models and in human fibrotic disorders [16–18, 51–56], but TNFα was also shown to inhibit fibrosis in some studies [19,58]. The effect of TNFα can differ depending on many factors, e.g. whether TNFα is constitutively expressed or temporally blocked and what kind of cells expressed TNFα [11,14,15,56,58]. Fibrosis is a common response to injury and can be the outcome of perturbation in the function of any tissue. Fibrosis occurs as a result of autoimmune inflammation in systemic sclerosis and idiopathic pulmonary fibrosis. However, fibrotic diseases respond poorly to currently available therapy. Our results suggest that inhibition of TNFα could provide an alterative therapeutic intervention for fibrosis due to autoimmune inflammation.
In conclusion, unregulated TNFα production characterizes many autoimmune diseases. Our results showed that although development of G-EAT was not suppressed by blocking TNFα, blocking TNFα led to beneficial outcomes in G-EAT with earlier resolution of inflammation and inhibition of fibrosis. Expression of IFNγ, IL-1β, IL-17 and iNOS was also decreased, suggesting that blocking TNFα decreased production of proinflammatory cytokines, but had no effect on Th2 cytokines. TNFα could also contribute to destruction of thyroids through its effect on apoptosis or through regulation of expression of pro- and/or anti-apoptotic molecules. Our study supports a proinflammatory and/or disease-promoting role for TNFα in G-EAT, and indicates an important role for TNFα in the pathogenesis of G-EAT and development of fibrosis that develops after sustained autoimmune inflammation.
Acknowledgments
This work was supported by NIH Grant DK35527 and by the Arthritis National Research Foundation. We thank Patti Mierzwa for excellent technical assistance. We also thank Dr. Charles Brown for real-time PCR primers for MCP-1.
Footnotes
K.C and. Y.W. contributed equally to this work.
Abbreviations used in this paper: G-EAT, granulomatous experimental autoimmune thyroiditis; MTg, mouse thyroglobulin.
References
- 1.Rose NR, Kong YC, Okayasu I, Giraldo AA, Beisel K, Sundick RS. T-cell regulation in autoimmune thyroiditis. Immunol Rev. 1981;55:299–314. doi: 10.1111/j.1600-065x.1981.tb00346.x. [DOI] [PubMed] [Google Scholar]
- 2.Charreire J. Immune mechanisms in autoimmune thyroiditis. Adv Immunol. 1989;46:263–334. doi: 10.1016/s0065-2776(08)60656-2. [DOI] [PubMed] [Google Scholar]
- 3.Kong Y, Audibert CF, Giraldo AA, Rose NR, Chedid L. Effects of natural or synthetic microbial adjuvants on induction of autoimmune thyroiditis. Infect Immun. 1985;49:40–45. doi: 10.1128/iai.49.1.40-45.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lira SA, Martin AP, Marinkovic T, Furtado GC. Mechanisms regulating lymphocytic infiltration of the thyroid in murine models of thyroiditis. Crit Rev Immunol. 2005;25:251–262. doi: 10.1615/critrevimmunol.v25.i4.10. [DOI] [PubMed] [Google Scholar]
- 5.Zaccone P, Fehervari Z, Cooke A. Tumour necrosis factor-alpha is a fundamental cytokine in autoimmune thyroid disease induced by thyroglobulin and lipopolysaccharide in interleukin-12 p40 deficient C57BL/6 mice. Immunology. 2003;108:50–54. doi: 10.1046/j.1365-2567.2003.01547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braley-Mullen H, Johnson M, Sharp GC, Kyriakos M. Induction of experimental autoimmune thyroiditis in mice with in vitro activated splenic T cells. Cell Immunol. 1985;93:132–143. doi: 10.1016/0008-8749(85)90394-6. [DOI] [PubMed] [Google Scholar]
- 7.Braley-Mullen H, Sharp GC, Tang H, Chen K, Kyriakos M, Bickel JT. Interleukin-12 promotes activation of effector cells that induce a severe destructive granulomatous form of murine experimental autoimmune thyroiditis. Am J Pathol. 1998;152:1347–1358. [PMC free article] [PubMed] [Google Scholar]
- 8.Braley-Mullen H, Sharp GC. Adoptive transfer murine model of granulomatous experimental autoimmune thyroiditis. Int Rev Immunol. 2000;19:535–555. doi: 10.3109/08830180009088511. [DOI] [PubMed] [Google Scholar]
- 9.Braley-Mullen H, McMurray RW, Sharp GC, Kyriakos M. Regulation of the induction and resolution of granulomatous experimental autoimmune thyroiditis in mice by CD8+ T cells. Cell Immunol. 1994;153:492–504. doi: 10.1006/cimm.1994.1045. [DOI] [PubMed] [Google Scholar]
- 10.Chen K, Wei Y, Sharp GC, Braley-Mullen H. Mechanisms of spontaneous resolution versus fibrosis in granulomatous experimental autoimmune thyroiditis. J Immunol. 2003;171:6236–6243. doi: 10.4049/jimmunol.171.11.6236. [DOI] [PubMed] [Google Scholar]
- 11.Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 12.Muppidi JR, Tschopp J, Siegel RM. Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity. 2004;21:461–465. doi: 10.1016/j.immuni.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Yang XD, Tisch RS, Singer M, Cao ZA, Liblau RS, Schreiber RD, McDevitt HO. Effect of tumor necrosis factor alpha on insulin-dependent diabetes mellitus in NOD mice. I The early development of autoimmunity and the diabetogenic process. J Exp Med. 1994;180:995–1004. doi: 10.1084/jem.180.3.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kassiotis G, Kollias G. TNF and receptors in organ-specific autoimmune disease: multi-layered functioning mirrored in animal models. J Clin Invest. 2001;107:1507–1508. doi: 10.1172/JCI13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green EA, Flavell RA. The temporal importance of TNF[alpha] expression in the development of diabetes. Immunity. 2000;12:459–469. doi: 10.1016/s1074-7613(00)80198-3. [DOI] [PubMed] [Google Scholar]
- 16.Booth M, Mwatha JK, Joseph S, Jones FM, Kadzo H, Ireri E, Kazibwe F, Kemijumbi J, Kariuki C, Kimani G, Ouma JH, Kabatereine NB, Vennervald BJ, Dunne DW. Periportal fibrosis in human Schistosoma mansoni infection is associated with low IL-10, low IFN-gamma, high TNF-alpha, or low RANTES, depending on age and gender. J Immunol. 2004;172:1295–1303. doi: 10.4049/jimmunol.172.2.1295. [DOI] [PubMed] [Google Scholar]
- 17.Chou DH, Lee W, McCulloch CA. TNF-alpha inactivation of collagen receptors: implications for fibroblast function and fibrosis. J Immunol. 1996;156:4354–4362. [PubMed] [Google Scholar]
- 18.Zhang K, Gharaee-Kermani B, McGarry M, Remick D, Phan SH. TNF-alpha-mediated lung cytokine networking and eosinophil recruitment in pulmonary fibrosis. J Immunol. 1997;158:954–959. [PubMed] [Google Scholar]
- 19.Fujita M, Shannon JM, Morikawa O, Gauldie J, Hara N, Mason RJ. Overexpression of tumor necrosis factor-{alpha} diminishes pulmonary fibrosis induced by bleomycin or transforming growth factor-β. Am J Respir Cell Mol Biol. 2003;29:669–676. doi: 10.1165/rcmb.2002-0046OC. [DOI] [PubMed] [Google Scholar]
- 20.Marinova-Mutafchieva L, Williams RO, Funa K, Maini RN, Zvaifler NJ. Inflammation is preceded by tumor necrosis factor-dependent infiltration of mesenchymal cells in experimental arthritis. Arthritis Rheum. 2002;46:507–513. doi: 10.1002/art.10126. [DOI] [PubMed] [Google Scholar]
- 21.Butler DM, Malfait AM, Maini RN, Brennan FM, Feldmann M. Anti-IL-12 and anti-TNF antibodies synergistically suppress the progression of murine collagen-induced arthritis. Eur J Immunol. 1999;29:2205–2212. doi: 10.1002/(SICI)1521-4141(199907)29:07<2205::AID-IMMU2205>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 22.Chen K, Wei Y, Sharp GC, Braley-Mullen H. Inhibition of TGFβ1 by anti-TGFβ1 antibody or lisinopril reduces thyroid fibrosis in granulomatous experimental autoimmune thyroiditis. J Immunol. 2002;169:6530–6538. doi: 10.4049/jimmunol.169.11.6530. [DOI] [PubMed] [Google Scholar]
- 23.Chen K, Wei Y, Sharp GC, Braley-Mullen H. Balance of proliferation and cell death between thyrocytes and myofibroblasts regulates thyroid fibrosis in granulomatous experimental autoimmune thyroiditis (G-EAT) J Leukoc Biol. 2005;77:166–172. doi: 10.1189/jlb.0904538. [DOI] [PubMed] [Google Scholar]
- 24.Tang H, Sharp GC, Chen K, Braley-Mullen H. The kinetics of cytokine gene expression in the thyroids of mice developing granulomatous experimental autoimmune thyroiditis. J Autoimmun. 1998;11:581–589. doi: 10.1006/jaut.1998.0247. [DOI] [PubMed] [Google Scholar]
- 25.Zhang K, Rekhter MD, Gordon D, Phan SH. Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis. A combined immunohistochemical and in situ hybridization study. Am J Pathol. 1994;145:114–125. [PMC free article] [PubMed] [Google Scholar]
- 26.Phan SH. The myofibroblast in pulmonary fibrosis. Chest. 2002;122:286S–289. doi: 10.1378/chest.122.6_suppl.286s. [DOI] [PubMed] [Google Scholar]
- 27.Liu T, Jin H, Ullenbruch M, Hu B, Hashimoto N, Moore B, McKenzie A, Lukacs NW, Phan SH. Regulation of Found in inflammatory zone 1 expression in bleomycin-induced lung fibrosis: role of IL-4/IL-13 and mediation via STAT-6. J Immunol. 2004;173:3425–3431. doi: 10.4049/jimmunol.173.5.3425. [DOI] [PubMed] [Google Scholar]
- 28.Sempowski GD, Beckmann MP, Derdak S, Phipps RP. Subsets of murine lung fibroblasts express membrane-bound and soluble IL-4 receptors. Role of IL-4 in enhancing fibroblast proliferation and collagen synthesis. J Immunol. 1994;152:3606–3614. [PubMed] [Google Scholar]
- 29.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Catrina AI, Trollmo C, af KE, Engstrom M, Lampa J, Hermansson Y, Klareskog L, Ulfgren AK. Evidence that anti-tumor necrosis factor therapy with both etanercept and infliximab induces apoptosis in macrophages, but not lymphocytes, in rheumatoid arthritis joints: extended report. Arthritis Rheum. 2005;52:61–72. doi: 10.1002/art.20764. [DOI] [PubMed] [Google Scholar]
- 31.Thome M, Tschopp J. Regulation of lymphocyte proliferation and death by FLIP. Nat Rev Immunol. 2001;1:50–58. doi: 10.1038/35095508. [DOI] [PubMed] [Google Scholar]
- 32.Suryaprasad AG, Prindiville T. The biology of TNF blockade. Autoimmun Rev. 2003;2:346–357. doi: 10.1016/s1568-9972(03)00048-x. [DOI] [PubMed] [Google Scholar]
- 33.Liu J, Marino M, Wong WG, Grail D, Dunn A, Bettadapura J, Slavin AJ, Old L, Bernard CC. TNF is a potent anti-inflammatory cytokine in autoimmune-mediated demyelination. Nat Med. 1998;4:78–83. doi: 10.1038/nm0198-078. [DOI] [PubMed] [Google Scholar]
- 34.Campbell IK, O'Donnell K, Lawlor KE, Wicks IP. Severe inflammatory arthritis and lymphadenopathy in the absence of TNF. J Clin Invest. 2001;107:1519–1527. doi: 10.1172/JCI12724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cantor J, Haskins K. Effector function of diabetogenic CD4 Th1 T cell clones: A central role for TNF-α. J Immunol. 2005;175:7738–7745. doi: 10.4049/jimmunol.175.11.7738. [DOI] [PubMed] [Google Scholar]
- 36.Tang H, Sharp GC, Peterson KE, Braley-Mullen H. Induction of granulomatous experimental autoimmune thyroiditis in IL-4 gene-disrupted mice. J Immunol. 1998;160:155–162. [PubMed] [Google Scholar]
- 37.Wang SH, Bretz JD, Phelps E, Mezosi E, Arscott PL, Utsugi S, Baker JR., Jr A unique combination of inflammatory cytokines enhances apoptosis of thyroid follicular cells and transforms nondestructive to destructive thyroiditis in experimental autoimmune thyroiditis. J Immunol. 2002;168:2470–2474. doi: 10.4049/jimmunol.168.5.2470. [DOI] [PubMed] [Google Scholar]
- 38.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wynn TA. T(H)-17: a giant step from T(H)1 and T(H)2. Nat Immunol. 2005;6:1069–1070. doi: 10.1038/ni1105-1069. [DOI] [PubMed] [Google Scholar]
- 40.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 41.Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005;22:285–294. doi: 10.1016/j.immuni.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 42.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 43.McLoughlin RM, Witowski J, Robson RL, Wilkinson TS, Hurst SM, Williams AS, Williams JD, Rose-John S, Jones SA, Topley N. Interplay between IFN-γ and IL-6 signaling governs neutrophil trafficking and apoptosis during acute inflammation. J Clin Invest. 2003;112:598–607. doi: 10.1172/JCI17129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bretz JD, Mezosi E, Giordano TJ, Gauger PG, Thompson NW, Baker JR., Jr Inflammatory cytokine regulation of TRAIL-mediated apoptosis in thyroid epithelial cells. Cell Death Differ. 2002;9:274–286. doi: 10.1038/sj.cdd.4400965. [DOI] [PubMed] [Google Scholar]
- 45.Taverne J, Rayner DC, Van der Meide PH, Lydyard PM, Bidey SP, Cooke A. Cytotoxicity of tumor necrosis factor for thyroid epithelial cells and its regulation by interferon-gamma. Eur J Immunol. 1987;17:1855–1858. doi: 10.1002/eji.1830171229. [DOI] [PubMed] [Google Scholar]
- 46.Van Den Brande JM, Peppelenbosch MP, Van Deventer SJ. Treating Crohn's disease by inducing T lymphocyte apoptosis. Ann N Y Acad Sci. 2002;973:166–180. doi: 10.1111/j.1749-6632.2002.tb04628.x. [DOI] [PubMed] [Google Scholar]
- 47.Wei Y, Chen K, Sharp GC, Yagita H, Braley-Mullen H. Expression and regulation of Fas and Fas ligand on thyrocytes and infiltrating cells during induction and resolution of granulomatous experimental autoimmune thyroiditis. J Immunol. 2001;167:6678–6686. doi: 10.4049/jimmunol.167.11.6678. [DOI] [PubMed] [Google Scholar]
- 48.Wei Y, Chen K, Sharp GC, Braley-Mullen H. Fas Ligand Is Required for Resolution of Granulomatous Experimental Autoimmune Thyroiditis. J Immunol. 2004;173:7615–7621. doi: 10.4049/jimmunol.173.12.7615. [DOI] [PubMed] [Google Scholar]
- 49.Suk K, Kim S, Kim YH, Kim KA, Chang I, Yagita H, Shong M, Lee MS. IFN-γ/TNF-α synergism as the final effector in autoimmune diabetes: A key role for STAT1/IFN regulatory factor-1 pathway in pancreatic β cell death. J Immunol. 2001;166:4481–4489. doi: 10.4049/jimmunol.166.7.4481. [DOI] [PubMed] [Google Scholar]
- 50.Guiet C, Silvestri E, De SE, Franzoso G, Vito P. c-FLIP efficiently rescues TRAF-2−/− cells from TNF-induced apoptosis. Cell Death Differ. 2002;9:138–144. doi: 10.1038/sj.cdd.4400947. [DOI] [PubMed] [Google Scholar]
- 51.Miyazaki Y, Araki K, Vesin C, Garcia I, Kapanci Y, Whitsett JA, Piguet PF, Vassalli P. Expression of a tumor necrosis factor-alpha transgene in murine lung causes lymphocytic and fibrosing alveolitis. A mouse model of progressive pulmonary fibrosis. J Clin Invest. 1995;96:250–259. doi: 10.1172/JCI118029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hasegawa M, Fujimoto M, Kikuchi K, Takehara K. Elevated serum tumor necrosis factor-alpha levels in patients with systemic sclerosis: association with pulmonary fibrosis. J Rheumatol. 1997;24:663–665. [PubMed] [Google Scholar]
- 53.Peng J, Gurantz D, Tran V, Cowling RT, Greenberg BH. Tumor necrosis factor-alpha-induced AT1 receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis. Circ Res. 2002;91:1119–1126. doi: 10.1161/01.res.0000047090.08299.d5. [DOI] [PubMed] [Google Scholar]
- 54.Piguet PF, Collart MA, Grau GE, Kapanci Y, Vassalli P. Tumor necrosis factor/cachectin plays a key role in bleomycin-induced pneumopathy and fibrosis. J Exp Med. 1989;170:655–663. doi: 10.1084/jem.170.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vassallo R, Matteson E, Thomas CF., Jr Clinical response of rheumatoid arthritis-associated pulmonary fibrosis to tumor necrosis factor-alpha inhibition. Chest. 2002;122:1093–1096. doi: 10.1378/chest.122.3.1093. [DOI] [PubMed] [Google Scholar]
- 56.Lundblad LK, Thompson-Figueroa J, Leclair T, Sullivan MJ, Poynter ME, Irvin CG. Tumor necrosis factor-alpha overexpression in lung disease: a single cause behind a complex phenotype. Am J Respir Crit Care Med. 2005;171:1363–1370. doi: 10.1164/rccm.200410-1349OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lloyd CM, Minto AW, Dorf ME, Proudfoot A, Wells TN, Salant DJ, Gutierrez-Ramos JC. RANTES and monocyte chemoattractant protein-1 (MCP-1) play an important role in the inflammatory phase of crescentic nephritis, but only MCP-1 is involved in crescent formation and interstitial fibrosis. J Exp Med. 1997;185:1371–1380. doi: 10.1084/jem.185.7.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saika S, Ikeda K, Yamanaka O, Flanders KC, Okada Y, Miyamoto T, Kitano A, Ooshima A, Nakajima Y, Ohnishi Y, Kao WW. Loss of tumor necrosis factor alpha potentiates transforming growth factor beta-mediated pathogenic tissue response during wound healing. Am J Pathol. 2006;168:1848–1860. doi: 10.2353/ajpath.2006.050980. [DOI] [PMC free article] [PubMed] [Google Scholar]