

SUMMARY
Voltage-gated calcium channels play a central role in regulating the electrical and biochemical properties of neurons and muscle cells. One of the ways in which calcium channels regulate long-lasting neuronal properties is by activating signaling pathways that control gene expression, but the mechanisms that link calcium channels to the nucleus are not well understood. We report that a C-terminal fragment of CaV1.2, an L-type voltage-gated calcium channel (LTC), translocates to the nucleus and regulates transcription. We show that this calcium channel associated transcription regulator (CCAT) binds to a nuclear protein, associates with an endogenous promoter, and regulates the expression of a wide variety of endogenous genes important for neuronal signaling and excitability. The nuclear localization of CCAT is regulated both developmentally and by changes in intracellular calcium. These findings provide evidence that voltage-gated calcium channels can directly activate transcription and suggest a mechanism linking voltage-gated channels to the function and differentiation of excitable cells.
INTRODUCTION
Changes in intracellular calcium regulate many cellular events, including synaptic transmission, cell division, survival, and differentiation. Voltage-gated calcium channels are an important route of calcium entry and are essential for converting electrical activity into biochemical events in excitable cells (Catterall et al., 2005). Among the ten different types of voltage gated calcium channels, L-type channels (LTC), encoded by the CaV1.2 and CaV1.3 pore-forming subunits, are particularly effective at inducing changes in gene expression that underlie plasticity and adaptive neuronal responses (Bading et al., 1993). Calcium influx through LTCs activates transcription factors such as CREB, MEF, and NFAT (Graef et al., 1999; Mao et al., 1999; Sheng et al., 1990) that lead to the expression of genes such as c-fos and BDNF (Morgan and Curran, 1986; Murphy et al., 1991; Zafra et al., 1990). Two mechanisms link LTCs, particularly CaV1.2, to the activation of transcription factors such as CREB. Calcium entering through the channels can diffuse to the nucleus and activate nuclear calcium-dependent enzymes, such as CaMKIV, that regulate the activity of transcription factors and coregulators (Hardingham et al., 2001). In addition, calcium entering cells through LTCs can activate calcium-dependent signaling proteins around the mouth of the channel, which propagate the signal to the nucleus (Deisseroth et al., 1998; Dolmetsch et al., 2001).
In this study we have identified a new mechanism by which calcium channels control gene expression. We report that neurons produce a C-terminal fragment of CaV1.2 that can regulate transcription and that we call calcium channel associated transcriptional regulator or CCAT. CCAT is located in the nucleus of many neurons in the developing and adult brain, and its production and nuclear localization are regulated developmentally. In addition, calcium influx through LTCs and NMDA receptors causes CCAT export from the nucleus. In the nucleus, CCAT interacts with the transcriptional regulator p54(nrb)/NonO and can activate transcription of both reporter and endogenous genes. Using microarrays and real-time PCR, we show that CCAT affects the transcription of many neuronal genes, including a gap junction, an NMDA-receptor subunit, and the sodium-calcium exchanger. CCAT binds to the enhancer of the Connexin 31.1 gene (Cx31.1) and directly regulates both the expression of a Cx31.1 reporter gene and the expression of the endogenous gene. Finally, we show that CCAT expression can cause an increase in neurite extension in primary neurons. This is the first example of a calcium channel having a dual function as an ion pore and a transcription factor.
RESULTS
CCAT Is Found in the Nucleus of Neurons in the Brain
Experiments in neurons and cardiac myocytes have suggested that the C terminus of CaV1.2 is proteolytically cleaved, yielding a truncated channel and a cytoplasmic C-terminal fragment (De Jongh et al., 1994; Gerhardstein et al., 2000). To investigate the function of the C-terminal fragment, we developed an antibody to a 14-amino acid peptide in the C terminus of CaV1.2 (aa 2106–2120) and used it to probe HEK 293T cells expressing CaV1.2. The C-terminal antibody (anti-CCAT) recognizes both the intact channel and a short cleavage product that corresponds to the C-terminal fragment. In contrast, an antibody recognizing an epitope in the II-III cytoplasmic loop of CaV1.2 (anti-II-III loop) detects full-length and C-terminally truncated channels only (Figure 1A).
Figure 1.
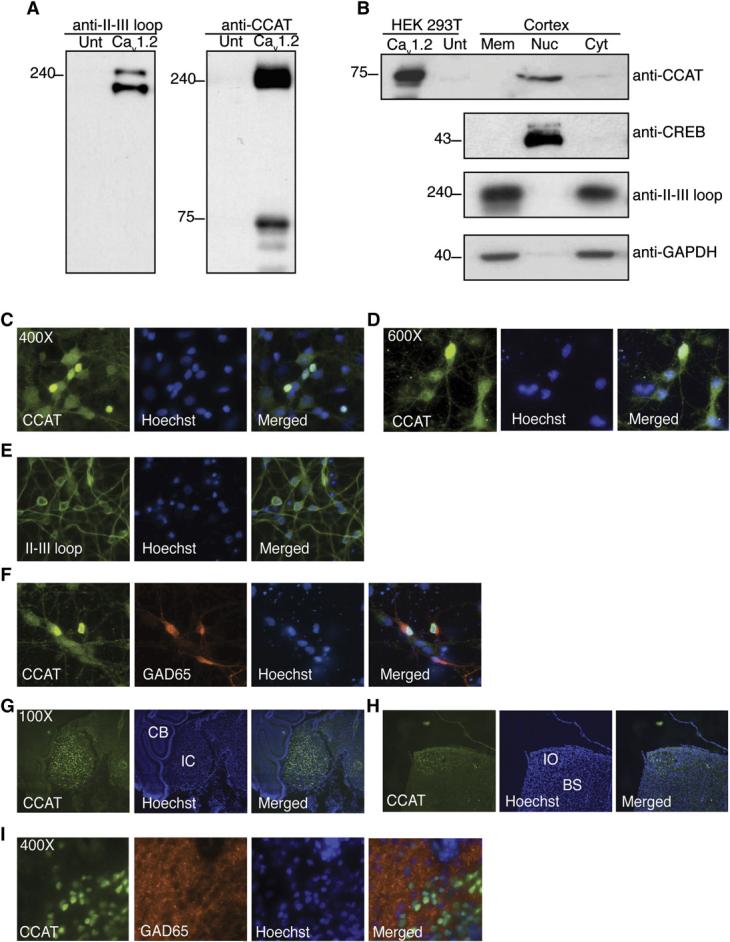
- (A) Western blot of HEK 293T cells expressing Cav1.2 probed with anti-II-III loop antibody (left gel) or anti-CCAT (right gel). The first lane of each gel contains lysate from untransfected cells (Unt), and the second contains lysate from cells expressing CaV1.2.
- (B) Western blots of membrane (Mem), nuclear (Nuc), and cytoplasmic (Cyt) extracts from the cortex of P7 rats analyzed with the anti-CCAT (upper gel), anti-CREB (second gel), anti-Cav1.2 II-III loop (third gel), and anti-GAPDH (bottom gel) antibodies. The first two lanes contain extracts from HEK 293T cells expressing Cav1.2. CREB, and CCAT immunoreactivity are detected only in the nuclear fraction, Cav1.2 probed with anti-II-III loop antibody and GAPDH are found in the membrane and cytoplasmic fractions confirming the efficacy of the fractionation.
- (C) Immunocytochemistry of cortical neurons grown 6 days in vitro. Anti-CCAT staining is shown in green and nuclei is shown in blue.
- (D) High-power image shows strong anti-CCAT staining (green) of nuclei (blue) and lighter staining of dendrites.
- (E) Staining with anti-CaV1.2 II-III loop antibody (green) reveals strong membrane and ER staining but little nuclear staining (blue).
- (F) Costaining with anti-CCAT antibody (green) and anti-GAD65 antibody (red) reveals that CCAT is strongly nuclear in a subpopulation of GAD65-positive neurons.
- (G and H) Immunohistochemistry of P30 rat-brain sagittal sections reveals strong nuclear staining with the anti-CCAT antibody (green) in the inferior colliculus (G:IC) and inferior olivary nucleus (H:IO). The cerebellum is labeled C and the brain stem is labeled BS.
- (I) High-power images of anti-CCAT (green), anti-GAD65 (red), and nuclear (blue) staining of rat olfactory lobe neurons shows that only a subpopulation of neurons have nuclear CCAT and that many of the cells are positive for GAD65.
To determine where CCAT is localized in cells in the brain, we purified nuclear, cytoplasmic, and membrane fractions of postnatal day 7 (P7) rat brain cortex and used western blotting to probe them with the anti-CCAT antibody (Figure 1B). Surprisingly, we found that the nuclear extracts contained high levels of CCAT, suggesting that the C terminus of CaV1.2 is localized in the nucleus of cells in the brain. In contrast, the N-terminal portion of the channel was localized in the membrane and cytoplasmic fractions, as expected for an ion channel. To provide further evidence that CCAT is indeed nuclear in neurons or glial cells, we examined its localization by immunostaining primary cortical cultures. The anti-CCAT antibody stained the cell body and dendrites of neurons weakly (Figure 1D), suggesting that the anti-CCAT antibody recognizes some intact CaV1.2 channels. Importantly, however, CCAT staining was observed in the nucleus of many neurons and was particularly pronounced in 10% of the cells (Figure 1C). In contrast, the II-III loop antibody stained the cell bodies and dendrites of neurons but was excluded from the nucleus, suggesting that the full-length channel is not nuclear (Figure 1E).
To investigate which types of neurons have nuclear CCAT, we costained neurons with anti-CCAT and with antibodies that stain precursor cells (nestin), glial cells (GFAP), excitatory neurons (VGLUT), or inhibitory neurons (GAD65) in the cortex. We found that cells that have strong nuclear CCAT also expressed glutamic acid decarboxylase (GAD65), suggesting that CCAT is strongly nuclear in inhibitory neurons that produce GABA (Figure 1F). To determine if CCAT is also in the nucleus of neurons in vivo, we used the anti-CCAT antibody to stain P30 rat brain sections. A subset of cells in the thalamus (data not shown), inferior colliculus (Figure 1G), inferior olivary nucleus (Figure 1H), and in the olfactory bulb (Figure 1I) displayed prominent nuclear CCAT staining. In the cortex and the hippocampus, CCAT was nuclear in a small number of neurons, consistent with its localization in a subset of GAD65-positive neurons in cortical cultures (data not shown). Taken together, these experiments indicate that CCAT is localized in the nucleus of inhibitory neurons, in culture and in restricted regions of the brain in vivo.
To provide further evidence that CCAT can translocate to the nucleus, we fused yellow fluorescent protein (YFP) to the C terminus of full-length CaV1.2 (CaV1.2-YFP). We observed cytoplasmic and nuclear fluorescence when CaV1.2-YFP was expressed in neurons (Figure 2A), cardiac myocytes (data not shown), or Neuro2A glioblastoma cells (Figure S2). In contrast, in neurons expressing CaV1.2 tagged at its N terminus with YFP, the channel was localized in the membrane and in the endoplasmic reticulum (Figure 2B). We did not observe nuclear fluorescence in HEK 293T cells expressing CaV1.2-YFP, consistent with previous reports that in HEK 293T cells the C terminus of CaV1.2 remains associated with the plasma membrane following cleavage (Gao et al., 2001; Gerhardstein et al., 2000; Hulme et al., 2005). However, a fusion of YFP and the last 503 amino acids of CaV1.2 was nuclear and formed distinct nuclear punctae in neurons, myocytes, and HEK 293T cells (c503, Figures 2C and 2E). Interestingly, this punctate pattern did not seem to be the result of overexpression, as it was also observed in some neurons by confocal imaging of endogenous CCAT staining (Figure 2D) and it was enhanced by incubation in low calcium media (Figure 3B). These experiments provide further evidence that CCAT is indeed nuclear and suggest that formation of punctae by endogenous CCAT is modulated by signaling events in the cell.
Figure 2.
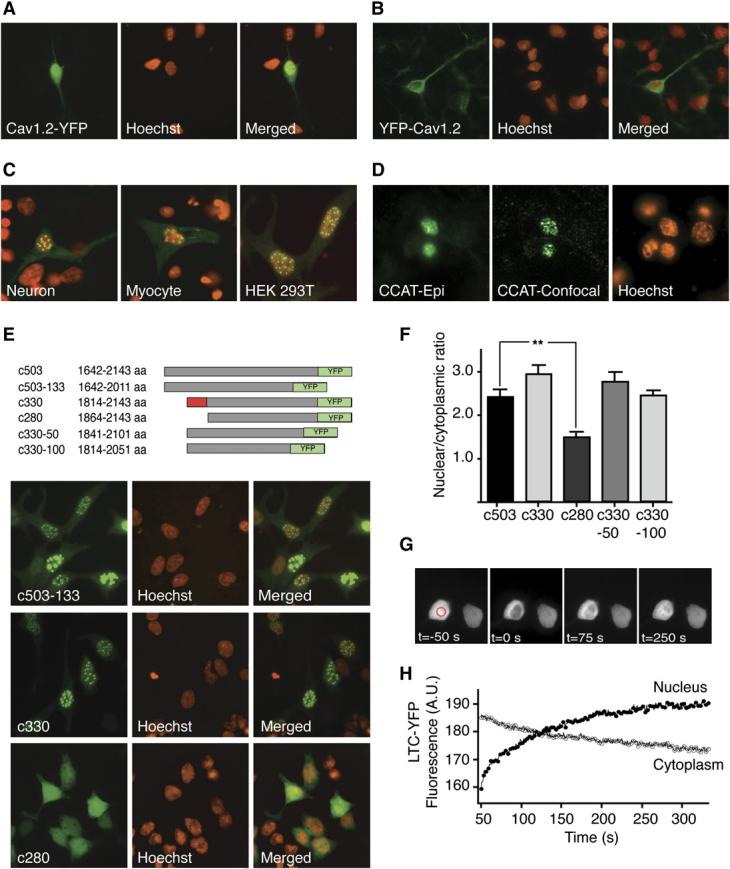
- Neurons expressing Cav1.2 tagged at the C terminus with YFP (Cav1.2-YFP) show pronounced nuclear and cytoplasmic fluorescence. Nuclei are shown in red in all panels.
- Neurons expressing the Cav1.2 tagged at the N terminus with YFP (YFP-Cav1.2) show membrane and ER fluorescence but little nuclear fluorescence.
- Full-length CCAT-YFP (c503-YFP) is nuclear when expressed in neurons, cardiac myocytes, and HEK 293T cells and forms prominent punctae.
- Epifluorescence (left panel) and confocal (right panel) image of neurons stained with anti-CCAT antibody showing endogenous nuclear punctae.
- Schematic representation of YFP-tagged CCAT-deletion mutants (top). Top panel and second panels show nuclear punctae in HEK 293T cells expressing CCAT containing a deletion of 133 aa from the carboxyl terminus (c503–133) and a truncated CCAT lacking aa 1642–1814 of Cav1.2 (c330) respectively. The third panel shows cells expressing a truncated CCAT lacking aa 1814–1864 (c280). The domain important for nuclear localization is highlighted in red in the schematic.
- Mean nuclear to cytoplasmic fluorescence ratios for all the CCAT deletions constructs schematized in part (E) (means ± SEM; n > 75). ** p < 0.001 versus c503.
- Time-lapse images showing fluorescence recovery after photobleaching (FRAP) of Cav1.2-YFP in the nucleus of Neuro2A cells. The area outlined in the red circle was bleached for 300 ms with a high-intensity pulsed 488 nM laser beam. Images were collected every 300 ms.
- Time course of recovery of nuclear fluorescence (solid symbols) and time course of loss of cytoplasmic fluorescence (empty symbols) following bleaching. Nuclear fluorescence recovered with a time course faster than 300 ms in cells expressing YFP alone. Shown is a representative example of eleven experiments.
Figure 3.
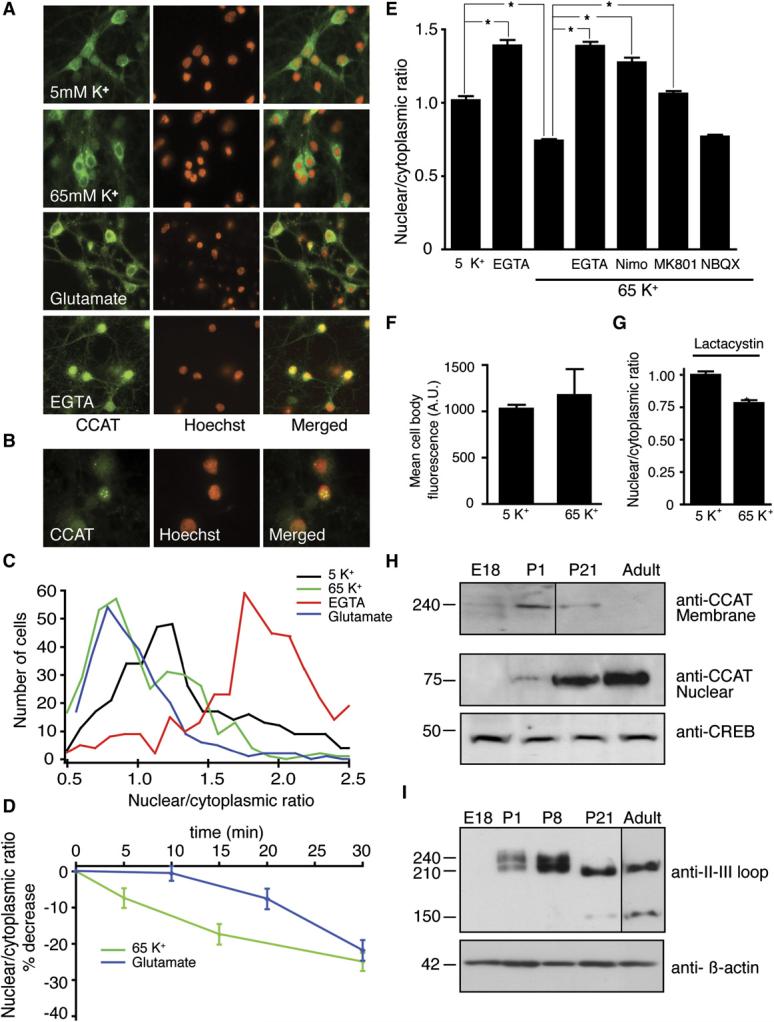
- Cortical neurons stained with anti-CCAT antibody (green) and Hoechst (red) treated with 5 mM K+ (1 hr), 65 mM K+ (1 hr), 100 μM glutamate (30 min), or 2.5 mM EGTA (1 hr).
- Higher magnification image of neurons treated with 2.5 mM EGTA reveals strong and punctate anti-CCAT staining.
- Histograms of the ratio of nuclear to cytoplasmic anti-CCAT fluorescence in neurons treated with 65 mM K+, glutamate, or EGTA. Treatment with 100 mM glutamate (blue) and 65 mM K+ (green) reduce the amount of CCAT in the nucleus of neurons, while EGTA (red) increases the amount of CCAT in the nucleus. Control cells in 5 mM K+ are shown in black (n = 375 per condition).
- Time course of the decrease in anti-CCAT nuclear fluorescence following stimulation with 65 mM K+ (green line; n = 200) or 100 μM glutamate (blue line; n = 200).
- Mean nuclear to cytoplasmic fluorescence ratio of CCAT in neurons treated for 1 hr with 2.5 mM EGTA (bar 2), 65 mM K+ (bar 3), and 65 mM K+ in the presence of 2.5 mM EGTA, 10 μM nimodipine, 10 μM MK-801 or 10 μM NBQX (bars 5–8; n = 200). (means ± SEM) * p < 0.001
- Mean anti-CCAT cell body fluorescence in neurons treated with 5mM or 65mM K+ (n=50).
- Mean nuclear to cytoplasmic fluorescence ratio of CCAT in neurons treated with 5mM and 65mM K+ in the presence of 5μM lactacystin (n=50).
- Western blot analysis of membrane (top) and nuclear fractions (middle) obtained from E18, P1, P21, and adult-brain cortex probed with the anti-CCAT antibody. CREB was used as a loading control (lower gel).
- Western blot analysis of cortical membrane extracts obtained from E18, P1, P8, P21, and adult rats analyzed with anti-CaV1.2 II-III loop antibody (top gel) and anti-β-actin (bottom) as a loading control. Adult lane exposed 4X relative to other lanes.
Nuclear CCAT does not contain a canonical nuclear localization sequence, suggesting that it enters the nucleus via an alternative pathway, perhaps as has been described for Stat1 protein, where nuclear import is mediated by direct interaction with nucleoporins (Marg et al., 2004). To identify the regions of CCAT that are necessary for its nuclear localization, we made truncations of the 503-YFP protein and introduced them in HEK 293T cells. Deletion of the carboxyl end of CCAT and of amino acids 1642–1814 of CaV1.2 (c330) had little effect on the protein's localization. In contrast, deletion of amino acids 1814–1864 (c280) decreased nuclear retention and abolished punctae formation (Figures 2E and 2F). Comparison of the CaV1.2 sequence from other vertebrates indicates that this nuclear retention domain is conserved evolutionarily (Figure S1A), suggesting that it plays an important role in the function of CaV1.2 and CCAT proteins.
Endogenous CCAT is predicted to be a 75 kDa protein; therefore, nuclear translocation of CCAT is likely to involve an active process rather than passive diffusion across nuclear pores. To estimate the rate of CCAT import into the nucleus, we used fluorescence recovery after photo-bleaching (FRAP) and time-lapse microscopy of Neuro2A cells expressing CaV1.2-YFP. After photobleaching of nuclear CCAT, nuclear fluorescence recovered over the course of 300 s with a single exponential time course (t = 48 ±16 s; n = 11), while cytoplasmic fluorescence declined over the same time period (Figures 2G-2H). In control cells expressing YFP alone, we observed an almost instantaneous recovery of nuclear fluorescence after photobleaching, concomitant with a decrease in cytoplasmic fluorescence, consistent with the observation that YFP diffuses rapidly through nuclear pores. The slow rate of recovery of CCAT-YFP nuclear fluorescence suggests that this protein is actively imported into the nucleus at a rate similar to that of NFAT, another transcription factor that translocates to the nucleus (Shibasaki et al., 1996). Measurements of CCAT export by bleaching cytoplasmic fluorescence indicate that CCAT returns to the cytoplasm with a time course of approximately 400 s (t = 62 ±21 s; n = 5) (data not shown). These results are consistent with the idea that CCAT is constitutively transported into the nucleus and that CCAT shuttles between the cytoplasm and the nucleus of unstimulated cells.
The Concentration of Nuclear CCAT Is Regulated by Intracellular Calcium
To determine whether the nuclear localization of CCAT is regulated by changes in intracellular calcium, we assessed the distribution of CCAT by immunocytochemistry in cortical neurons following treatment with agents that affect intracellular calcium levels. Decreasing free extra-cellular calcium using 2.5 mM EGTA caused a robust increase in nuclear CCAT fluorescence (Figures 3A and 3C) and caused CCAT to aggregate into punctae in the nucleus of many neurons (Figure 3B). Conversely, treatment with 65 mM KCl, which mimics tonic electrical activity by increasing the activity of VGCCs, and treatment with 100 μM glutamate caused a significant decrease in the nuclear fluorescence (Figures 3A and 3C). The decrease in nuclear CCAT could be reliably detected after 5 min and reached a maximum after 30 min of stimulation with either depolarization or glutamate, although depolarization had a more pronounced effect at earlier time points (Figure 3D). The nuclear fluorescence of Neuro2A cells expressing the CaV1.2-YFP also declined with tonic depolarization, providing further evidence that electrical activity leads to a net decrease of CCAT from the nucleus (Figure S2A–S2B). The decrease in nuclear CCAT triggered by depolarization was blocked by removing extracellular calcium or by treating cells with the CaV1.2 blocker nimodipine. Application of NMDA-receptor blocker MK-801 partially blocked the activity-induced decrease in nuclear CCAT, but treatment with the AMPA-receptor inhibitor NBQX had no effect (Figure 3E), suggesting that NMDA but not AMPA receptors can also influence the export of CCAT from the nucleus of cortical neurons.
The decrease in nuclear CCAT observed in response to high intracellular calcium could be due to a net export from the nucleus or to selective degradation of CCAT in the nucleus. To determine if CCAT is degraded following a rise in intracellular calcium, we measured total CCAT immunoreactivity before and after depolarization. We found that depolarization had no effect on the total CCAT staining in neurons or on the levels of CCAT-YFP expressed in Neuro2A cells (Figure 3F). Furthermore, addition of the proteosome inhibitor lactacystin failed to block the depolarization-induced decrease in nuclear CCAT (Figure 3G). The lack of a decrease in total CCAT levels in depolarized neurons and Neuro2A cells and the lack of effect of lactacystin on CCAT nuclear localization argue that the decrease in CCAT following depolarization is not due to protein degradation.
Nuclear CCAT Is Regulated Developmentally
The levels of nuclear CCAT vary considerably among neurons in the developing brain (Figures 1G-1I). Since neurons in the central nervous system differentiate at different rates, we considered whether the levels of CCAT in the nucleus could be regulated developmentally. To investigate this possibility, we assessed the levels of nuclear and total CCAT found in brains taken from embryonic day 18 (E18), postnatal day 1 (P1), 3-week-old (P21), and adult rats. The levels of CCAT immunoreactivity in the nuclear fractions increased substantially with age (Figure 3H, middle panel), whereas the amount of CCAT-containing channel at the membrane appeared to decrease (Figure 3H, upper panel). This is consistent with increasing cleavage of CaV1.2 during development. The total levels of CaV1.2, as determined by immunoreactivity of the CaV1.2 internal-loop antibody, were also regulated developmentally. CaV1.2 levels were low at E18 and increased through P8 before declining in P21 and adult brains (Figure 3I, upper panel). Interestingly, early in development a long and a short form of CaV1.2 could be detected, whereas only the short form of the channel and a new, 150 kDa band were observed in both P21 and adult brains, suggesting that there is increasing cleavage and possibly different cleavage events in older brains. Together, these results indicate that the levels of CCAT in the nucleus, the cleavage of CaV1.2, and the levels of CaV1.2 are regulated independently to yield a complex pattern of channel- and transcription-factor expression.
CCAT Binds to a Nuclear Protein
To get an indication of CCAT's function, we looked for proteins that interact with CCAT in the nucleus. We expressed CCAT or a mutant form lacking the nuclear localization domain in Neuro2A cells, immunoprecipitated them via epitope tags, and identified interacting proteins by mass spectrometry. One of the proteins that coimmunoprecipitated with full length CCAT was p54(nrb)/NonO, a nuclear protein that plays a role in regulating transcription downstream of the neuronal Wiscott Aldrich Protein (Wu et al., 2006), the retinoic acid receptor, and the thyroid hormone receptor (Mathur et al., 2001). We verified the interaction of p54(nrb)/NonO with CCAT by coimmunoprecipitation followed by Western blotting against endogenous p54(nrb)/NonO (Figure 4A). These results indicate that CCAT is associated with a nuclear protein that participates in transcriptional regulation and regulates mRNA splicing, and suggest a role for the C terminus of CaV1.2 in the nucleus.
Figure 4.
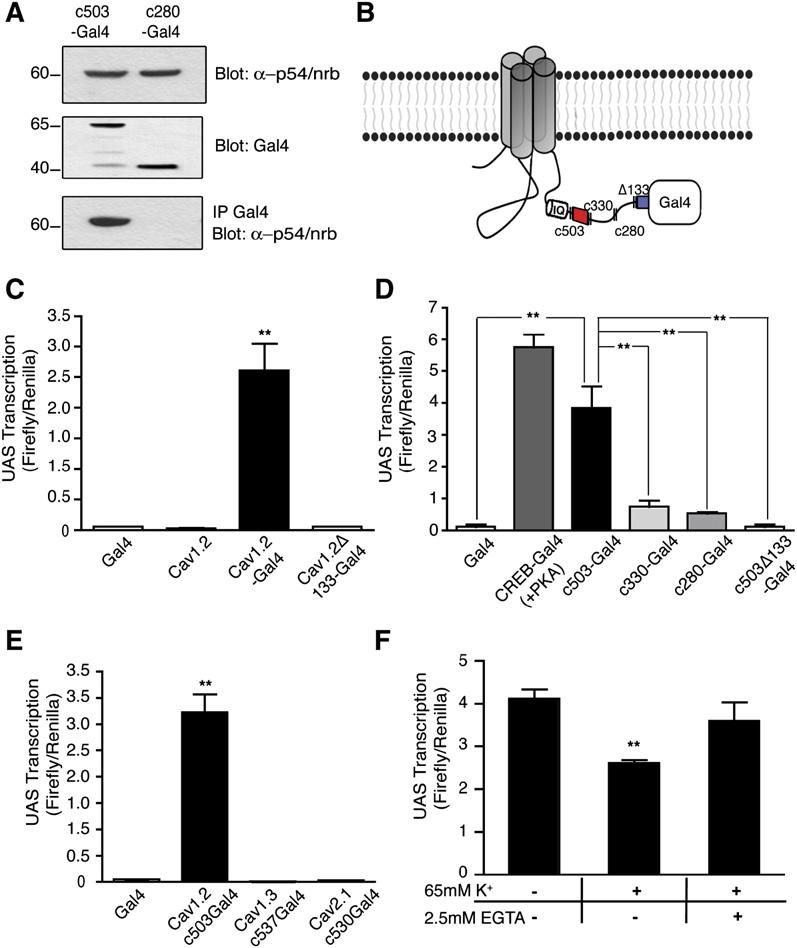
- CCAT immunoprecipitates endogenous p54(nrb)/NonO. Upper panel shows p54nrb/ NonO levels in the lysates. The middle panel shows immunoprecipitated c503-Gal4 and c280-Gal4 that lacks a nuclear localization domain. The lower panel shows endogenous p54(nrb)/NonO coimmunoprecipitated by c503-Gal4 but not by c280-Gal4.
- Schematic representation of the CaV1.2-Gal4 fusion and CCAT deletion proteins. c503 CCAT is the full-length C terminus of CaV1.2 downstream of the IQ motif. c330 CCAT lacks the N-terminal transcriptional activation domain (shown in red). c280 lacks the nuclear retention domain of CCAT and c503D133 lacks the C-terminal transactivation domain (shown in blue).
- Reporter gene activity of Neuro2A cells expressing a UAS-luciferase-reporter plasmid along with either the Gal4-DNA binding domain, full-length CaV1.2, full-length CaV1.2-Gal4, or CaV1.2-Gal4 channel lacking 133 amino acids from the carboxyl terminus. Cells were cotransfected with a Renilla driven by the thymidine kinase promoter to control for cell number and transfection efficiency. The results are given as the ratio of Firefly to Renilla-luciferase activity. (means ± SD) ** p < 0.0001 versus Gal4.
- Luciferase activity of neurons transfected with the UAS-luciferase-reporter gene and either Gal4 alone, CREB-Gal4, or four different CaV1.2 C-terminal fragments c503, c330, or c280 and C503D133 fused to Gal4. The two domains identified as important for transcriptional activation are highlighted in B. (means ± SD; ** p < 0.005)
- Luciferase activity of neurons transfected with Gal4-DNA binding domain alone or Gal4 fused to the C terminus of CaV1.2, CaV1.3, or CaV2.1. (means ± SD) ** p < 0.0001 versus Gal4.
- Luciferase activity of Neuro2A cells expressing CaV1.2-Gal4 treated with 5 mM, 65 mM, and 65 mM K+/2.5 mM EGTA. (means ± SD; ** p < 0.005 versus untreated).
CCAT Activates Transcription
Based on its nuclear localization and its binding to p54(nrb)/NonO, we hypothesized that CCAT might regulate transcription. To investigate whether CCAT can activate transcription when recruited to a promoter by a heterologous DNA binding domain, we made a C-terminal fusion of the intact channel and the Gal4-DNA binding domain from yeast (CaV1.2-Gal4, Figure 4B). The Gal4-DNA binding domain recognizes the UAS-DNA sequence but requires a transcriptional activation domain to activate transcription. We introduced CaV1.2-Gal4 into Neuro2A cells along with a UAS-luciferase-reporter gene and measured luciferase expression. We found that CaV1.2-Gal4 activated transcription approximately 80 times better than Gal4 alone or than the channel lacking the Gal4-DNA binding domain (Figure 4C). These results suggest that the C terminus of CaV1.2 is produced as a soluble protein in cells, that it translocates to the nucleus, and that it activates transcription when recruited to a heterologous gene.
To identify the domains of CaV1.2 that are required for transcriptional activation, we made a family of proteins containing fragments of the C terminus of CaV1.2 fused to Gal4 and tested them in primary neurons for their ability to activate the expression of a UAS-luciferase-reporter gene. A fragment containing 503 amino acids of the CaV1.2 C terminus fused to Gal4-activated transcription almost as well as a CREB-Gal4 fusion protein and about 130 times better than the Gal4-DNA binding domain alone (Figure 4D). Deleting 170 amino acids from the N terminus of this C-terminal CaV1.2 fragment (c330-Gal4) reduced but did not completely abolish the ability of the C terminus to activate transcription. In contrast, deletion of a second domain consisting of the most C-terminal 133 amino acids (c503Δ133-Gal4) completely eliminated the ability of CCAT to activate transcription (Figure 4D). Deletion of these final 133 amino acids in the full-length CaV1.2-Gal4 also produced a channel unable to activate transcription (Figure 4C, bar 4), suggesting that this domain is required for transcriptional regulation by the intact channel. These experiments suggest that CCAT has two domains that are necessary to activate transcription: (1) an N-terminal domain that modulates transcriptional activation and (2) a C-terminal domain that is essential for transcription (red and blue boxes; Figure 4B). Significantly, both transactivation domains are highly conserved in vertebrates (Figure S1B and S1D) and the N-terminal transactivation domain has 42% similarity and 27% identity to a conserved transactivation domain of the transcription factor GATA4, suggesting that it has a bona fide role in transcriptional regulation (Figure S1C).
Because recruiting proteins to DNA via Gal4-DNA binding domains can produce ectopic transcriptional regulators, we also fused various other calcium channel C-terminal domains to Gal4 and expressed these with the UAS-reporter gene. We found that the C termini of CaV1.3 and CaV2.1 when fused to Gal4 had no effect on transcription, suggesting that CaV1.2's C-terminal domain is specific in its ability to activate transcription in neurons (Figure 4E).
In earlier experiments, we observed that the amount of CCAT in the nucleus decreased in response to tonic electrical activity. To determine whether this activity-induced decrease in nuclear CCAT has functional relevance, we depolarized cells expressing CaV1.2-Gal4 and measured activation of the UAS-luciferase reporter (Figure 4F). Prolonged depolarization led to a 30% decline in transcription from the reporter gene, and this effect was blocked by removing extracellular calcium. These results provide evidence that the nuclear localization of CCAT is important for its activation of transcription and are consistent with the observation that nuclear-CCAT concentration is regulated by electrical activity.
CCAT Regulates Transcription of Endogenous Genes
To determine whether CCAT regulates transcription of endogenous genes, we used oligonucleotide microarrays to identify mRNAs that are transcriptionally regulated by CCAT overexpression. We built two plasmids that encode either full-length CCAT or a CCATΔTA that lacks the N-terminal transcriptional-activation domain. Both plasmids also contain a GFP gene driven by a separate promoter that was used to identify transfected cells. We introduced these plasmids into Neuro2A cells and used fluorescence-activated cell sorting (FACS) to select transfected cells. We then compared the mRNA-expression profile of cells expressing full-length CCAT to cells expressing either CCATΔTA or GFP alone, using Agilent mouse whole-genome arrays. In three independent experiments, we found 23 mRNAs that were upregulated more than 2-fold (p < 0.005) in cells expressing CCAT relative to cells expressing CCATΔTA and 22 genes that were down-regulated more than 2-fold by CCAT relative to CCATΔTA (Table S1). Because we subsequently discovered the CCATΔTA still activates transcription albeit at a much lower level than full-length CCAT (see Figure 4D), we also compared mRNA-expression profiles of cells expressing CCAT and GFP to cells expressing GFP alone. In three additional experiments, we found 66 mRNAs upregulated more than 1.8-fold (p < 0.005) in cells expressing CCAT relative to those expressing GFP (Figure S1 and Table S2). The genes that were upregulated by CCAT include the genes for the gap junction protein Cx31.1, the axon guidance factor Netrin4, the regulator of G-protein signaling RGS5, the tight junction protein claudin19 and a broad array of other genes (Figure 5A). Approximately 206 genes were repressed more than 0.55-fold (p < 0.005) by CCAT, including the sodium-calcium exchanger, the cation channel TRPV4, the potassium channel Kcnn3, and the transcription factor GATA6 (Figures 5A and S3 and Table S3; raw data available at http://ncbi.nlm.nih.gov/geo; series #: GSE4180). Combining the results from all six of our microarray experiments (CCAT versus CCATΔTA and CCAT versus GFP) revealed that 16 mRNAs were significantly upregulated (Table S4) and 31 genes were significantly downregulated by CCAT. These results suggest that CCAT can both increase and decrease the expression of a wide set of genes that regulate neuronal differentiation, connectivity, and function.
Figure 5.

- A subset of mRNAs identified in microarray experiments that were upregulated (red bars) or downregulated (green bars) by overexpression of CCAT relative to CCATΔTA or GFP. For a full list of genes, see Tables S1–S4.
- RT-PCR analysis of mRNA levels from Neuro2A cells overexpressing CCAT confirming changes in gene expression for a subset of these genes identified in A. Bars represent mean fold changes relative to CCATΔTA or GFP and were normalized to b-actin and GAPDH levels. Data are means ± SD of three independent experiments performed in triplicate.
- Luciferase activity of neurons expressing a cx31.1-luciferase-reporter gene along with empty vector, full-length CCAT, or CCAT lacking the C-terminal transactivation domain (CCATΔTA) (means ± SD; ** p < 0.0001).
- 4OHT-induced nuclear translocation of a CCAT-ER fusion. Immunocytochemistry of Neuro2A cells expressing myc-CCAT-ER (green) before (top panels) and after (bottom panels) addition of 5 μM 4OHT for 1 hr. Nuclei are shown in blue.
- Transcription of a cx31.1-luciferase-reporter gene in Neuro2A cells expressing a cx31.1-luciferase reporter along with ER alone, CCAT-ER, or CCATΔTA-ER before and after nuclear translocation by addition of 5 μM 4OHT for 6 hr.
- Mean luciferase activity (mean ± SEM; n = 3) of Neuro2A cells expressing CCAT and deleted forms of the cx31.1 promoter. The 125 bp 3′ region of the promoter that binds CCAT is shown in red.
- Representative ChIP assay showing that CCAT immunoprecipitates the endogenous cx31.1 promoter. Agarose gel electrophoresis of PCR products amplified from either input DNA (I) or from DNA that was immunoprecipitated by GST-CCAT (IP) or by GST alone (C). The upper gel shows the PCR products using primers that recognize two regions in the cx31.1 promoter (5′ promoter and 3′ promoter). The lower gel shows PCR products using primers that recognize the 3′ region of the cx31.1 gene several KB from the start site (n = 3).
To verify the results of the microarray experiments, we measured changes in mRNA expression due to CCAT expression using RT-PCR (Figure 5B). CCAT changed the expression of all seven mRNAs tested, in accordance with the results from the array experiments. As normalizing controls we used β-actin and GAPDH, which showed no detectable change in response to overexpression of CCAT. These data provide independent evidence that CCAT regulates expression of endogenous genes, some of which are important for the function of excitable cells.
CCAT Binds and Regulates the Promoter of cx31.1
The microarray and RT-PCR experiments suggested that cx31.1 was strongly regulated by CCAT in cells. To study the regulation of cx31.1 by CCAT in more detail, we constructed a reporter gene consisting of the 2 Kb promoter/ enhancer region of cx31.1 in front of the firefly-luciferase-coding sequence. We introduced this cx31.1-luciferase-reporter gene into neurons along with either the full-length CCAT or CCATΔDTA, a version of CCAT lacking the C-terminal transcriptional activation domain. Full-length CCAT increased the expression of the cx31.1 reporter by 3.4 ± 0.4-fold (n = 12) relative to a control vector or to CCATΔTA (Figure 5C) providing additional evidence that CCAT regulates the expression of cx31.1.
CCAT could affect the transcription of cx31.1 either by regulating the transcriptional machinery in the nucleus directly or by modifying signaling proteins in the cytoplasm of cells that lead to changes in transcription. To determine if CCAT acts in the nucleus, we fused CCAT to the ligand binding domain of a modified estrogen receptor (ER) that binds 4-hydroxytamoxifen (4OHT) but not endogenous estrogen (Littlewood et al., 1995). When expressed in Neuro2A cells, ER-CCAT is largely excluded from the nucleus but brief treatment with 4OHT causes ER-CCAT to move into the nucleus (Figure 5D). Treatment of cells expressing ER-CCAT with 4OHT caused a 50-fold increase in the transcription of cx31.1 relative to untreated cells (Figure 5E). 4OHT had no effect on cells expressing ER alone and caused a much smaller effect in cells expressing ER-CCATΔTA. These results provide compelling evidence that CCAT regulates the transcription of cx31.1 when it is in the nucleus of cells.
To identify regions of the cx31.1 promoter that are important for its regulation by CCAT, we made a series of deletions of the cx31.1 promoter and placed them upstream of the firefly-luciferase gene (Figure 5F). We introduced this library of deletion mutants of the cx31.1 promoter into Neuro2A cells along with full-length CCAT and measured luciferase activity in these cells. CCAT regulation of the cx31.1 promoter was critically dependent on 148 base pairs at the 3′ end of the cx31.1 promoter. Deletion of this domain eliminated the ability of CCAT to activate transcription of the cx31.1-reporter gene, and this domain alone was sufficient to confer CCAT regulation on to a reporter gene (Figure 5F). Together, these data suggest that CCAT regulates the expression of cx31.1 in a sequence-specific manner and that the CCAT-recognition element lies in the final 148 base pairs of the cx31.1-promoter sequence.
In the nucleus, CCAT could affect transcription directly by binding to a complex of proteins on the promoter of genes, or indirectly by binding to other proteins in the transcriptional-activation pathway. We used chromatin immunoprecipitation (ChIP) to determine whether CCAT binds to the promoter of cx31.1 directly. We introduced an epitope-tagged CCAT into cells, crosslinked the protein to the DNA, and immunoprecipitated CCAT from these cells and used PCR to determine if any region in the promoter of the cx31.1 gene was coimmunoprecipitated by CCAT. We found that CCAT reproducibly immunoprecipitate two fragments of the endogenous cx31.1 promoter but not a 3 region of the gene (Figure 5G). These results suggest that CCAT regulates transcription by binding, either directly or through protein-protein interactions, to the promoter of cx31.1, providing further evidence that CCAT is a transcriptional regulator.
Endogenous CaV1.2 and CCAT Regulate Transcription of cx31.1
We have provided evidence that exogenous expression of CaV1.2 leads to the production of CCAT, which in turn affects transcription. To determine whether endogenous CaV1.2 regulates transcription by generating CCAT, we asked whether reducing the levels of endogenous CCAT in the nucleus by depolarization had an effect on expression of the cx31.1-reporter gene or of the endogenous Cx 31.1 gene. Depolarization of cortical neurons reduced activation of the cx31.1-reporter gene by 2.12 ± 0.12-fold (Figure 6A) and caused a 2.4-fold decrease in the expression of the cx31.1-mRNA levels as measured by RT-PCR (Figure 6B). The effects of depolarization on cx31.1-mRNA levels were also apparent in Neuro2As and in cultured thalamic neurons, suggesting that CCAT regulates the expression of cx31.1 in multiple cell types (Figure 6B). These results support the conclusion that CCAT-dependent transcription of the cx31.1 gene requires nuclear localization of CCAT.
Figure 6.
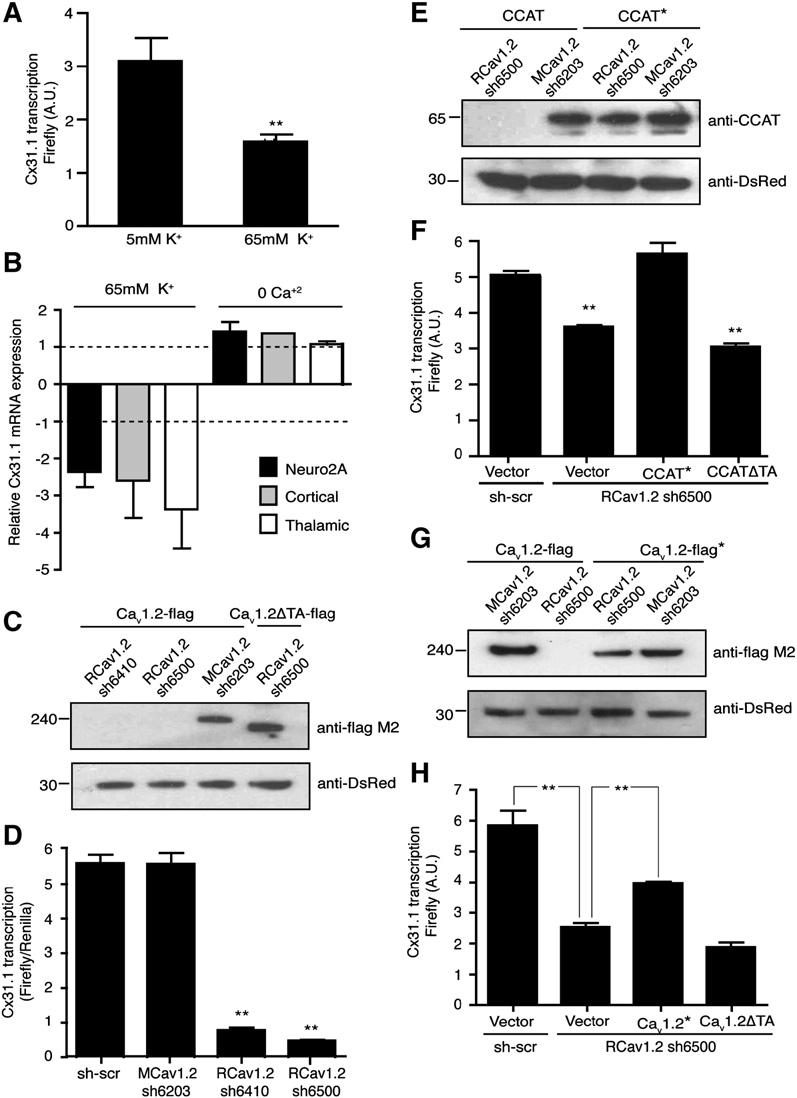
- Depolarization of neurons with 65 mM KCl decreases the activity of the cx31.1 reporter relative to unstimulated cells (5 mM KCl; mean ± SD; n > 3; ** p < 0.0001).
- RT-PCR analysis of endogenous cx31.1 mRNA in Neuro2A cells and cortical and thalamic neurons treated with 65 mM KCl or 0 calcium showing that depolarization causes a pronounced decrease in cx31.1 expression. mRNA levels are normalized to mRNA levels in unstimulated cells (bars represent mean ± SEM; n = 5).
- Western blots showing reduced expression of FLAG-tagged rat CaV1.2 expressed in Neuro2A cells by expression of two rat shRNAs (RCaV1.2 sh6410 and RCaV1.2 sh6500; lanes 1 and 2, top panel) but not by a mouse shRNA (MCaV1.2 sh6203) that differs by two base pairs (lane 3, top panel). CaV1.2ΔTA is resistant to RCaV1.2 sh6500 (lane 4), which targets the TA domain of CaV1.2. Ds-Red, expressed from the shRNA vector, was used as a loading control (bottom panel).
- Mean luciferase activity (± SD) of rat neurons transfected with the cx31.1-luciferase-reporter gene and either a scrambled control shRNA (sh-scr), shRNA constructs targeting the mouse CaV1.2 (MCaV1.2 sh-6203), or the rat CaV1.2 mRNAS (shRNA RCaV1.2 sh-6500, RCaV1.2 sh-6410; ** p < 0.001 versus scrambled (shscr) or MCaV1.2 sh-6203).
- CCAT* is resistant to knockdown by an shRNA targeting CaV1.2 (CaV1.2 sh6500). Western blot analysis of lysates from Neuro2A cells expressing CCAT or an RNAi-resistant CCAT* along with the RCaV1.2 sh-6500 shRNA vector that targets rat CaV1.2 (upper gel). Ds-Red, expressed from the shRNA vector, was used as a loading control (lower gel).
- Expression of RNAi-resistant CCAT* reversed the effect of CaV1.2 sh6500 on cx31.1 expression (bar 3), but CCATΔTA, which lacks the C-terminal transactivation domain, does not (bar 4; means ± SD; n = 3; ** p < 0.001 versus sh-scr).
- CaV1.2-FLAG* is resistant to knockdown by RCav1.2 sh6500. Western blot analysis of lysates from Neuro2A cells expressing CaV1.2-FLAG or RNAi-resistant CaV1.2-FLAG*, and RCaV1.2 sh6500 or MCaV1.2 sh-6203 (upper gel). Ds-Red, expressed from the shRNA vector, was used as a loading control (lower).
- Expression of RNAi-resistant CaV1.2-Flag* partially reversed the effect of RCaV1.2 sh6500 on cx31.1 expression (bar 3), but CaV1.2-ΔTA*, which lacks the C-terminal transactivation domain, did not (bar 4; means ± SD; n = 3). ** p < 0.0001 versus sh-scr.
Because CCAT is derived from CaV1.2, we also asked whether cx31.1 expression depends on the expression of endogenous CaV1.2. We designed several short hairpin RNAs (shRNAs) and asked whether introducing these shRNAs into neurons reduced the expression of cx31.1. Two shRNAs targeting the rat CaV1.2 (RCav1.2 sh6410 and RCaV1.2 sh6500) reduced the expression of rat CaV1.2 expressed in Neuro2A cells, whereas an shRNA targeting the mouse CaV1.2 sequence had no effect on the expression of the rat channel (Figure 6C, lanes 1–3). The shRNAs targeting the rat CaV1.2 also reduced CaV1.2-dependent signaling to CREB in rat-cortical neurons, suggesting that these shRNAs reduce the expression of endogenous CaV1.2 and prevent activation of CREB-dependent transcription (Bading et al., 1993; Dolmetsch et al., 2001; Murphy et al., 1991) (Figure S4A). We next introduced the shRNAs targeting the rat CaV1.2 into cortical neurons and measured the activation of the cx31.1 reporter. Both rat shRNAs decreased the expression of cx31.1 by approximately 6-fold, indicating that CaV1.2 regulates the expression of cx31.1 in neurons (Figure 6D). CaV1.2 knockdown had no effect on Renilla-luciferase expression from the control vector, suggesting that the decrease in cx31.1-reporter activity was not due to decreased viability. To assess whether the effect of the shRNAs targeting CaV1.2 on the transcription of cx31.1 was the result of the loss of calcium influx through the channel, we tested whether L-type calcium channel blockers affected cx31.1 transcription. Twenty-four hr treatment of neurons with 10 mM nimodipine had no effect on the expression of cx31.1 in the presence or absence of CCAT, suggesting that cx31.1 is not regulated by calcium influx through CaV1.2 in unstimulated cells (Figure S4B). To determine if the inhibitory effects of CaV1.2 shRNAs on the cx31.1 promoter are due to reduction of CCAT, we constructed a version of CCAT that is insensitive to the rat CaV1.2 shRNA (CCAT*, Figure 6E) and expressed it in cells along with the shRNA targeting rat CaV1.2. Expression of CCAT* rescued the effect of knocking down the endogenous CaV1.2 on the expression of the cx31.1 gene (Figure 6F, n = 6). In contrast, CCATΔTA that lacked the transcriptional-activation domain did not rescue the effects of the CaV1.2 shRNA on Cx 31.1 expression. This suggests that CCAT alone can restore expression of Cx 31.1 in cells in which CaV1.2 has been reduced by an shRNA and that this effect depends on the transcriptional-activation domain of CCAT. We also made a version of CaV1.2 that is insensitive to the rat CaV1.2 shRNA (CaV1.2*) (Figure 6G) and asked whether this channel can rescue cx31.1 expression in cells lacking endogenous CaV1.2 (Figure 6H). Expression of CaV1.2* in neurons partially rescued the effect of the CaV1.2 shRNA on cx31.1 expression, while a form of CaV1.2* that lacks the C-terminal transcriptional-activation domain did not restore the effects of CaV1.2 knockdown on cx31.1 expression. Together these results support the conclusion that endogenous CaV1.2 modulates transcription of the cx31.1 gene and that this transcriptional regulation depends on the production of CCAT from the C terminus of CaV1.2.
CCAT Expression Promotes Neurite Growth
Our microarray and RT-PCR experiments suggested that CCAT regulates the transcription of a number of genes important in neuronal function and excitability. To explore the cell biological functions of CCAT, we measured the effect of expressing CCAT on the morphology and survival of cerebellar granule neurons. We selected these cells because they are a largely homogenous population of neurons that have low basal levels of CCAT and that have well-characterized survival and dendritic arborization patterns. Expression of CCAT or CCATΔTA did not significantly affect granule cell survival, but it did cause a dramatic change in the length of neurites (Figures 7A and 7B). Full-length CCAT doubled the average length of neu-rites to 10 μm(Figures 7C [bottom panel] and 7D) whereas the CCATΔTA decreased the average length of neurites to approximately 2.7 um (Figures 7C [top panel] and 7D). There was also a small but statistically significant effect of CCAT on the number of neurites, suggesting that under some circumstances CCAT could affect the growth and formation of new dendrites (Figure 7E). Interestingly, expressing CCAT in other cell types such as Neuro2As also caused a change in the morphology of the cells, causing an increase in the production of filopodial extensions (data not shown). This data suggests that CCAT-dependent transcription can lead to rearrangement of the cyto-skeleton and may contribute to changes in the connectivity of neurons during development.
Figure 7.
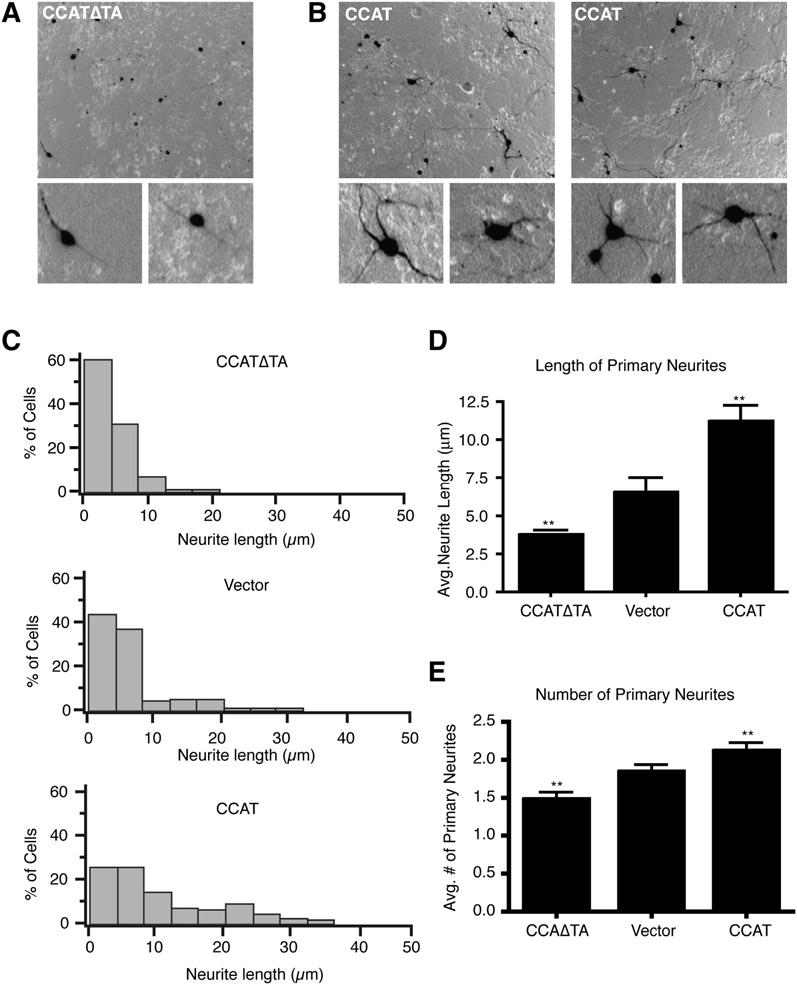
- (A and B) Representative low- and high-magnification images of cerebellar granule cells grown in vitro for 5 days transfected with a vector expressing GFP and either CCATΔTA (A) or CCAT (B).
- (C) Histograms of mean neurite length for granule neurons expressing CCATΔTA (top graph), a vector with GFP alone (middle graph), or CCAT (bottom graph).
- (D) Average neurite length for cells expressing CCATΔTA, GFP alone, or CCAT (means ± SEM, n = 200; ** p < 0.005 versus vector control).
- (E) Average number of primary dendrites for granule neurons expressing CCATΔTA, empty vector, or CCAT.
DISCUSSION
Neurons and myocytes generate characteristic patterns of electrical activity and intracellular calcium that are essential for cell function. The reliability of the calcium signal requires a delicate balance of proteins that import and export calcium from the cytoplasm—proteins whose individual expression is regulated independently in response to cellular function. The expression of voltage-gated calcium channels is closely coordinated with the expression of other ion channels, pumps, and signaling proteins that regulate membrane excitability and calcium homeostasis. In this paper we describe a novel mechanism by which cells coordinate the expression of voltage-gated calcium channels with the expression of other molecules. LTCs generate a transcription factor that integrates information both about the number of calcium channels and the electrical activity of a cell. CCAT is generated from the L-type channel, and its nuclear localization is negatively regulated by the electrical activity of the cell; it is therefore in a privileged position to integrate information about the number of channels with information about the calcium history of a cell.
Several laboratories have reported that LTCs are cleaved at their C terminus, and the site of cleavage of Cav1.1, the homologous LTC in skeletal muscle, was recently identified (Hulme et al., 2005). The cleaved channel carries more calcium, so channel cleavage could have profound effects on the electrical properties of a neuron by changing the properties of the LTC. The proteolytically processed C-terminal domain is also thought to bind to truncated channels, where it exerts an inhibitory effect on channel function (Hulme et al., 2006). This hypothesis does not preclude the idea that the C terminus of CaV1.2 also acts as a transcription factor. By analogy with the potassium-channel binding protein KChip/DREAM, which is also a calcium-sensitive transcriptional repressor, we propose that CCAT both regulates transcription and reduces calcium influx through CaV1.2 (An et al., 2000; Carrion et al., 1999). This hypothesis is appealing in light of the observation that CCAT is exported from the nucleus by elevations in intracellular calcium, suggesting that under conditions of tonically elevated calcium, CCAT would both alter the transcription of specific genes and inhibit the activity of CaV1.2. Thus CCAT may be an important part of a negative-feedback pathway regulating both gene expression and calcium influx in the neurons.
In addition to CaV1.2, it has also been reported that CaV1.3 (Hell et al., 1993), CaV2.1 (Kubodera et al., 2003), and CaV2.2 (Westenbroek et al., 1992) are cleaved in neurons. In the case of CaV2.1, the cleavage product is also approximately 75 kDa and has been localized to the nucleus of Purkinje neurons in the cerebellum (Kordasiewicz et al., 2006). This suggests that C-terminal cleavage is a general feature of CaV channels and that other members of this family may also be transcriptional regulators. In our studies, we did not find that the C-terminal domains of CaV1.3 or CaV2.1 activated transcription in cortical neurons, but it is possible that the C-terminal domains of other channels may act in other types of neurons or may be transcriptional repressors or regulators of chromatin structure. This would be consistent with our finding that in addition to activating transcription CCAT also represses the transcription of many genes.
Despite more than a decade of experiments, the stimuli and mechanisms that lead to cleavage of CaV1.2 remain enigmatic. It has been reported that cleavage of CaV1.2 is triggered by NMDA stimulation in hippocampal slices (Hell et al., 1996), and CaV1.2 cleavage has also been reported to occur in response to sex-hormone stimulation of uterine muscle (Helguera et al., 2002). We did not observe any obvious increase in CCAT following stimulation of neurons in culture with NMDA or potassium chloride; however, it is possible that CaV1.2 cleavage only occurs in the context of hippocampal slices. In cortical neurons, cerebellar granule cells, cardiac myocytes, Neuro2A cells, and PC12 cells exogenous CaV1.2 appears to be cleaved constitutively to yield nuclear and cytoplasmic CCAT. While the production of CCAT did not appear to be regulated, its nuclear localization and its transcriptional effects on the Cx31.1 gene were strongly regulated by changes in cytoplasmic calcium. Therefore, we favor the idea that CCAT is produced in proportion to the number of CaV1.2 channels in cells and that cytoplasmic calcium levels regulate its nuclear localization and transcriptional activity. In addition to being regulated by calcium, nuclear CCAT levels were also regulated in a cell-specific manner, and its appearance in brain nuclear fractions increased substantially over the course of postnatal development. In cultured neurons, CCAT levels were highest in GABAergic inhibitory neurons, while in brain slices CCAT staining was particularly strong in the inferior colliculus, inferior olive, and thalamus. These data suggest that CCAT may play an important role in the development of neurons and in regulation of neuronal properties in specific cell types.
Our studies have identified many interesting genes regulated by CCAT, and these genes offer clues to understanding CCAT's physiologic function. CCAT regulates the expression of several gap-junction proteins, a gluta-mate receptor, several potassium channels, a sodium-calcium exchanger, and signaling proteins such as RGS5, Formin, and Nitric Oxide Synthase. One of the main targets of CCAT in the nucleus is the gap-junction protein Cx31.1. Cx31.1 is expressed in the retina (Guldenagel et al., 2000), in developing embryos (Davies et al., 1996), and in GABAergic striatal-output neurons of the thalamus (Venance et al., 2004). Our array and RT-PCR studies suggest that Cx31.1 is also well expressed in neuroblastoma cells and in thalamic neurons. Transcription of the Cx31.1 gene correlates well with the amount of endogenous CCAT in the nucleus and depolarization, which reduces the amount of nuclear CCAT and also decreases the amount of Cx31.1 transcript, suggesting that these two are correlated. Finally CCAT binds to the promoter of Cx31.1, providing compelling evidence that CCAT is a regulator of Cx31.1 expression in neurons. Connexins play a key role in forming electrical connections between developing neurons and form conduits for signaling molecules that can regulate a developing tissue. The expression of Cx31.1 during development in response to changes in CCAT could thus play an important role in regulating the electrical coupling of neurons and the overall excitability of the brain.
We have found that CCAT expression in neurons increases dendritic length. This effect is blocked by CCAT lacking a transcriptional activation domain. There are many possible mechanisms for this effect of CCAT on neuronal morphology. The observation that CCAT upregulates Cx31.1, formin, claudin 19, procolagen type XI, and an α-catenin-like protein suggests that it might promote the formation of adhesion complexes or junctional contacts between neurons and the extracellular matrix. Alternatively, since CCAT increases the production of Netrin4 and of two chemokines that regulate axonal and dendritic growth, it could lead to increases in neurite length via these mechanisms (Adler and Rogers, 2005; Barallobre et al., 2005). Finally, by downregulating a potassium channel and a sodium-calcium exchanger, CCAT could increase the excitability of neurons and thus regulate their morphology indirectly. Understanding how CCAT modulates dendritic length might help uncover the mechanisms by which L-type calcium channels regulate neuronal morphology.
We provide strong evidence that CaV1.2 encodes a transcription factor that can regulate expression of a variety of genes that are important for the function of neurons and muscle cells. This finding reveals an entirely unsuspected function for a well-characterized calcium channel that plays an essential role in electrical tissues. This new function of CaV1.2 will be a rich area for future study in ion channel physiology and neurobiology.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
HEK 293T cells and Neuro2A and PC12 cells were cultured in Dulbecco's Minimal Essential Media (DMEM) containing 10% fetal bovine serum (FBS; 15% for PC12s), penicillin, streptomycin (P/S), and L-gluta-mine (LQ). Cortical neurons were dissociated from E17-19 Sprague Dawley rats as described (Xia et al., 1996) and maintained for 6–14 days in culture in Basal Medium Eagle with 5% FBS, P/S, LQ, and 1% glucose or in Neurobasal medium containing B27 supplement (Invitrogen). Cardiac myocytes were cultured from P0–P1 rats using a neonatal myocyte isolation kit (Cellutron Life Technology) and maintained in DMEM with 10% FBS, P/S, LQ, and 0.1 mM BRDU for 3–4 days. Cerebellar granule cells were cultured from P5 Sprague Dawley rats and grown as described elsewhere (Dudek et al., 1997). For description of thalamic neuron cultures see Supplemental Experimental Procedures.
HEK 293T (24 hr) cells, cortical and granule neurons (72 hr) were transfected using a standard calcium-phosphate method at a concentration of 2 mg of DNA/106 cells. Neuro2As (24 hr), cortical neurons (96 hr), cardiac myocytes (24 hr), and PC12s (24 hr) were transfected using lipofectamine 2000 according to manufacturer's instructions. For luciferase reporter gene experiments see Supplemental Experimental Procedures.
Plasmid Construction
See Supplemental Experimental Procedures.
Antibody Generation
Peptides of the following sequence DPGQDRAVVPEDES were synthesized (Covance), coupled to KLH (Pierce), injected into rabbits, and affinity purified as previously described (Datta et al., 1997).
Subcellular Fractionation and Western Blotting
The brain was rapidly removed and homogenized in 320 mM sucrose and 20 mM HEPES homogenization buffer, pH 7.2, containing 1 mM EDTA, 1 mM dithiothreitol, Complete protease inhibitors (Roche Applied Science), and calpain inhibitors (A.G. Scientific). The homogenate was centrifuged for 10 min at 1000 g to obtain the nuclear fraction. The supernatant was then centrifuged for 30 min at 100,000 g at 4°C to obtain the cytoplasmic and membrane fractions. The nuclear pellet was extracted using the Dignam method (Dignam et al., 1983). For membrane bound channel visualization, proteins were extracted as described previously (Haase et al., 2000). Western blotting was conducted using standard protocols. Antibodies and dilutions are included in Supplemental Experimental Procedures. Protein concentration was measured by the BCA method (Pierce).
Immunofluorescence
6 day-old cortical cultures cells were fixed in 4% paraformaldehyde/2% sucrose, permeabilized, and blocked with 3% bovine serum albumin (BSA) in phosphate-buffered saline (PBS). Neurons were stained with either rabbit anti-CCAT or rabbit anti-Cav1.2 II-III loop (each diluted 1:100) and anti-GAD65 followed by 1:500 dilutions of Alexa 594-conjugated anti-mouse and Alexa 488-conjugated anti-rabbit antibodies (Molecular Probes). Nuclei were stained using Hoechst 33258 (Molecular Probes). Neuro2A cells expressing CCAT-ER fusions were stained with mouse anti-myc tag (Upstate). P30 rat-brain sections were a gift from Dr. Ben Barres. Sections were blocked and permeabilized 30 min using 10% goat serum, 0.25% Triton X-100 in PBS. Primary and secondary antibody incubations were done as described above. Slides were visualized by conventional epifluorescence micro-copy using a cooled CCD camera (Hamamatsu) coupled to an inverted Nikon Eclipse E2000-U microscope. Confocal images were obtained using the Volocity grid confocal microscope (Improvision, Inc.).
Quantitative Image Analysis
Images were analyzed using OpenLab 4.0.4 software (Improvision, Inc.). For measurements of nuclear and cytoplasmic fluorescence, nuclear and whole-cell regions of interest (ROI) were generated by density slicing the Hoechst and anti-CCAT images, respectively, and cytoplasmic ROIs were obtained by subtraction. Fluorescence measurements were analyzed using Igor Pro software (Wavemetrics).
Fluorescence Recovery after Photobleaching (FRAP)
FRAP experiments were conducted at 37°C using a Zeiss Axiovert 200 M inverted microscope coupled to a Coolsnap cooled CCD camera controlled by Slidebook software. Bleaching was achieved with a 100 ms long 488 nm laser pulse. Images were captured every 400 ms.
Luciferase Assays
Most luciferase assays were performed 24 hr after transfection using the Dual-Glo luciferase assay kit from Promega. For shRNA experiments, assays were performed 72 hr posttransfection. A Veritas 96-well luminometer (Turner Biosystems) was used to measure light emission. CREB-Gal4, constitutively active PKA, and PFA-CMV constructs were obtained as part of the PathDetect transreporting system from Strategene. Data sets were analyzed using Igor Pro and Prism4 software. Two-paired t tests were performed between relevant conditions.
Immunoprecipitation and Mass Spectrometry
HEK 293T cells were transfected with c503-Gal4 or c280-Gal4. Twenty-four hours after transfection, immunoprecipitations were carried out using the ProFound Coimmunoprecipitation Kit (Pierce) and mouse or rabbit anti-Gal4 antibodies (Santa Cruz Biotechnology). SDS-PAGE gels were silver stained using the SilverQuest system from Invitrogen. Individual bands were analyzed by Stanford Mass Spectrometry facility using LC-MS/MS as previously described (Shevchenko et al., 1996).
RNA Isolation and Oligonucleotide Microarrays
Neuro 2A cells were transfected using the CCAT-PA1, CCATΔTA-PA1, or PA1 control vectors. Twenty-four hours after transfection, cells were trypsinized and resuspended in fresh media without phenol red, and GFP-positive cells were sorted using FACS. RNA was isolated from 2 3 106 cells using an RNAeasy kit from Qiagen. RNA was hybridized to Agilent whole-mouse oligomicroarrrays by MOgene, Inc. (St. Louis, MO). Expression data was analyzed using GeneSpring GX 7.3 software (Agilent).
RT-PCR
First-strand synthesis was conducted using the first-strand cDNA synthesis kit from Invitrogen. Five-hundred nanograms of cDNA was used as a template for RT-PCRs performed using an Mx3000P Real-Time System (Stratagene), and the reactions were carried out using Quantitec SYBR green PCR master mix (Qiagen). For a list of primers see Supplemental Experimental Procedures.
Cycling parameters were 95°C for 10 min, followed by 45 cycles of 95°C for 30 s, 55°C for 1 min, 72°C for 30 s. Fluorescence intensities were analyzed using the manufacturer's software, and relative amounts were obtained using the 2−ΔΔCt method (Livak and Schmittgen, 2001).
ChIP
ChIP was carried out as described in the Upstate Biotechnology protocol. Briefly 107 cells were transfected with CCAT-GST or CCATGal4, or GST or Gal4 alone as controls. Proteins were crosslinked to DNA with 1% paraformaldehyde for 10 min, and the nuclei were isolated by centrifugation. The DNA was sheared by sonication to generate DNA fragments between 200 and 1000 bp. The C-terminal fusion proteins were immunoprecipitated, washed, and uncrosslinked by adding high salt and incubating at 65° overnight. DNA was recovered by phenol/chloroform extraction and ethanol precipitation and used as a template for PCR. For primer sequences see Supplemental Experimental Procedures. Reaction products were visualized by agarose gel electrophoresis.
Dendritic Arborization Assays
Cerebellar granules cells were imaged 24–48 hr posttransfection using the ImageXpress 500A system (Molecular Devices). Dendrites were analyzed employing ImageJ and NeuroJ programs.
Supplementary Material
Footnotes
Supplemental Data
Supplemental Data include four figures, four tables, experimental procedures and can be found with this article online at http://www.cell.com/cgi/content/full/127/3/591/DC1/.
REFERENCES
- Adler MW, Rogers TJ. Are chemokines the third major system in the brain? J. Leukoc. Biol. 2005;78:1204–1209. doi: 10.1189/jlb.0405222. [DOI] [PubMed] [Google Scholar]
- An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403:553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- Barallobre MJ, Pascual M, Del Rio JA, Soriano E. The Netrin family of guidance factors: emphasis on Netrin-1 signalling. Brain Res. Brain Res. Rev. 2005;49:22–47. doi: 10.1016/j.brainresrev.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Carrion AM, Link WA, Ledo F, Mellstrom B, Naranjo JR. DREAM is a Ca2+-regulated transcriptional repressor. Nature. 1999;398:80–84. doi: 10.1038/18044. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 2005;57:411–425. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Davies TC, Barr KJ, Jones DH, Zhu D, Kidder GM. Multiple members of the connexin gene family participate in preim-plantation development of the mouse. Dev. Genet. 1996;18:234–243. doi: 10.1002/(SICI)1520-6408(1996)18:3<234::AID-DVG4>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- De Jongh KS, Colvin AA, Wang KK, Catterall WA. Differential proteolysis of the full-length form of the L-type calcium channel alpha 1 subunit by calpain. J. Neurochem. 1994;63:1558–1564. doi: 10.1046/j.1471-4159.1994.63041558.x. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippo-campal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Gao T, Cuadra AE, Ma H, Bunemann M, Gerhardstein BL, Cheng T, Eick RT, Hosey MM. C-terminal fragments of the alpha 1C (CaV1.2) subunit associate with and regulate L-type calcium channels containing C-terminal-truncated alpha 1C subunits. J. Biol. Chem. 2001;276:21089–21097. doi: 10.1074/jbc.M008000200. [DOI] [PubMed] [Google Scholar]
- Gerhardstein BL, Gao T, Bunemann M, Puri TS, Adair A, Ma H, Hosey MM. Proteolytic processing of the C terminus of the alpha(1C) subunit of L-type calcium channels and the role of a proline-rich domain in membrane tethering of proteolytic fragments. J. Biol. Chem. 2000;275:8556–8563. doi: 10.1074/jbc.275.12.8556. [DOI] [PubMed] [Google Scholar]
- Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature. 1999;401:703–708. doi: 10.1038/44378. [DOI] [PubMed] [Google Scholar]
- Guldenagel M, Sohl G, Plum A, Traub O, Teubner B, Weiler R, Willecke K. Expression patterns of connexin genes in mouse retina. J. Comp. Neurol. 2000;425:193–201. [PubMed] [Google Scholar]
- Haase H, Pfitzmaier B, Morano I. Expression of Ca2+ channel subunits during cardiac ontogeny in mice and rats: identification of fetal alpha 1C and beta subunit isoforms. J. Cell. Biochem. 2000;76:695–730. doi: 10.1002/(sici)1097-4644(20000315)76:4<695::aid-jcb17>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat. Neurosci. 2001;4:261–267. doi: 10.1038/85109. [DOI] [PubMed] [Google Scholar]
- Helguera G, Olcese R, Song M, Toro L, Stefani E. Tissue-specific regulation of Ca(2+) channel protein expression by sex hormones. Biochim. Biophys. Acta. 2002;1569:59–66. doi: 10.1016/s0304-4165(01)00234-3. [DOI] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J. Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Breeze LJ, Wang KK, Chavkin C, Catterall WA. N-methyl-D-aspartate receptor-induced proteolytic conversion of postsynaptic class C L-type calcium channels in hippocampal neurons. Proc. Natl. Acad. Sci. USA. 1996;93:3362–3367. doi: 10.1073/pnas.93.8.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme JT, Konoki K, Lin TW, Gritsenko MA, Camp DG, II, Bigelow DJ, Catterall WA. Sites of proteolytic processing and noncovalent association of the distal C-terminal domain of CaV1.1 channels in skeletal muscle. Proc. Natl. Acad. Sci. USA. 2005;102:5274–5279. doi: 10.1073/pnas.0409885102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme JT, Yarov-Yarovoy V, Lin TW, Scheuer T, Catterall WA. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J. Physiol. 2006;576:87–102. doi: 10.1113/jphysiol.2006.111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordasiewicz HB, Thompson RM, Clark HB, Gomez CM. C-termini of P/Q-type Ca2+ channel alpha1A subunits translocate to nuclei and promote polyglutamine-mediated toxicity. Hum. Mol. Genet. 2006;15:1587–1599. doi: 10.1093/hmg/ddl080. [DOI] [PubMed] [Google Scholar]
- Kubodera T, Yokota T, Ohwada K, Ishikawa K, Miura H, Matsuoka T, Mizusawa H. Proteolytic cleavage and cellular toxicity of the human alpha1A calcium channel in spinocerebellar ataxia type 6. Neurosci. Lett. 2003;341:74–78. doi: 10.1016/s0304-3940(03)00156-3. [DOI] [PubMed] [Google Scholar]
- Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999;286:785–790. doi: 10.1126/science.286.5440.785. [DOI] [PubMed] [Google Scholar]
- Marg A, Shan Y, Meyer T, Meissner T, Brandenburg M, Vinkemeier U. Nucleocytoplasmic shuttling by nucleoporins Nup153 and Nup214 and CRM1-dependent nuclear export control the subcellular distribution of latent Stat1. J. Cell Biol. 2004;165:823–833. doi: 10.1083/jcb.200403057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur M, Tucker PW, Samuels HH. PSF is a novel corepressor that mediates its effect through Sin3A and the DNA binding domain of nuclear hormone receptors. Mol. Cell. Biol. 2001;21:2298–2311. doi: 10.1128/MCB.21.7.2298-2311.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JI, Curran T. Role of ion flux in the control of c-fos expression. Nature. 1986;322:552–555. doi: 10.1038/322552a0. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Worley PF, Baraban JM. L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron. 1991;7:625–635. doi: 10.1016/0896-6273(91)90375-a. [DOI] [PubMed] [Google Scholar]
- Sheng M, McFadden G, Greenberg ME. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron. 1990;4:571–582. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Shibasaki F, Price ER, Milan D, McKeon F. Role of kinases and the phosphatase calcineurin in the nuclear shuttling of transcription factor NF-AT4. Nature. 1996;382:370–373. doi: 10.1038/382370a0. [DOI] [PubMed] [Google Scholar]
- Venance L, Glowinski J, Giaume C. Electrical and chemical transmission between striatal GABAergic output neurones in rat brain slices. J. Physiol. 2004;559:215–230. doi: 10.1113/jphysiol.2004.065672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron. 1992;9:1099–1115. doi: 10.1016/0896-6273(92)90069-p. [DOI] [PubMed] [Google Scholar]
- Wu X, Yoo Y, Okuhama NN, Tucker PW, Liu G, Guan JL. Regulation of RNA-polymerase-II-dependent transcription by N-WASP and its nuclear-binding partners. Nat. Cell Biol. 2006;8:756–763. doi: 10.1038/ncb1433. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J. Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra F, Hengerer B, Leibrock J, Thoenen H, Lindholm D. Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J. 1990;9:3545–3550. doi: 10.1002/j.1460-2075.1990.tb07564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.