

Abstract
Recently proposed models that have gained wide acceptance posit that HIV-1 virion morphogenesis is initiated by targeting the major structural protein (Gag) to late endosomal membranes. Thereafter, late endosome-based secretory pathways are thought to deliver Gag or assembled virions to the plasma membrane (PM) and extracellular milieu. We present several findings that are inconsistent with this model. Specifically, we demonstrate that HIV-1 Gag is delivered to the PM, and virions are efficiently released into the extracellular medium, when late endosome motility is abolished. Furthermore, we show that HIV-1 virions are efficiently released when assembly is rationally targeted to the PM, but not when targeted to late endosomes. Recently synthesized Gag first accumulates and assembles at the PM, but a proportion is subsequently internalized via endocytosis or phagocytosis, thus accounting for observations of endosomal localization. We conclude that HIV-1 assembly is initiated and completed at the PM, and not at endosomal membranes.
Recently, it has been debated whether assembly of retroviral particles takes place at the plasma membrane or at endosomal membranes. The authors demonstrate that HIV assembly takes place at the plasma membrane.
Introduction
Human immunodeficiency virus type I (HIV-1) Gag is the major virion structural protein, and Gag expression alone in a variety of human cell types is sufficient to generate extracellular virus-like particles (VLPs). Gag exhibits several activities necessary for the formation of a complete particle [1], including an amino terminal myristoylated matrix (MA) domain that directs membrane binding, central capsid (CA) and nucleocapsid domains that drive multimerization and packaging of viral genomes, and a C-terminal domain (p6) that recruits class E vacuolar protein sorting (VPS) factors that are required for scission of the nascent virion bud [2,3].
Much confusion and controversy currently exist as to where within the cell HIV-1 assembly is initiated and completed. Historically, it was thought that almost all retrovirus Gag proteins were targeted selectively to the plasma membrane (PM) to induce efficient particle release. This view was based on electron microscopic or immunofluorescent detection of Gag proteins or budding structures at the PM of infected cells. However, numerous recent studies have challenged this notion, and it has become evident that considerable amounts of HIV-1 Gag protein and/or mature virion particles can be found in late endosomes. Apparent HIV-1 assembly in endosomes appears especially prominent in macrophages, in which very large numbers of virion particles are found within compartments containing the late endosomal markers CD63 and lysosome-associated membrane protein-1 (Lamp1) [4–7]. Moreover, extracellular macrophage-derived virions can be immunoprecipitated with antibodies specific for late endosomal membrane markers such as CD63 [4,6]. These data suggested that Gag is initially targeted to late endosomal membranes and that Gag protein or assembled virions are subsequently delivered to the extracellular milieu or PM via an endosome-based secretory pathway. Although intracellular particle assembly was initially thought to be a unique feature of macrophages, similar studies reporting localization of HIV-1 Gag and virions in intracellular CD63+ and/or Lamp1+ compartments in fibroblast, epithelial, and T-lymphocyte cell lines, as well as immunoprecipitation of extracellular virions with CD63 antibodies, have extended this notion, leading to the proposal that HIV-1 assembly is initiated on endosomal membranes in all cell types [8–13]. Moreover, several other works have reported the observation of murine leukemia virus and human T-cell leukemia virus type-1 particles in late endosomes [12,14–17], suggesting that assembly in late endosomes is a general feature of retroviruses.
Elaborations of the endosome assembly model have suggested that HIV-1 assembly intermediates that are seen at the cell surface are only observed there because the PM contains late endosome-derived microdomains that arise following the fusion of late endosomes with the PM [10,18,19]. This idea is conceptually attractive because it removes the need to invoke two distinct membrane-targeting capabilities for Gag proteins, and would reconcile the paradoxical divergence in assembly pathways into a single endosomal membrane-targeted pathway [20]. A further attraction to the notion that HIV-1 particle assembly occurs at endosomes, or endosome-like membrane domains, arises from the fact that virion budding requires factors that normally mediate the budding of vesicles into the late endosomal lumen [2,3]. Indeed, the Hrs protein, which targets endosomal membranes via a PI(3)P-binding Fab1/YOTB/Vac1/EEA1 (FYVE) domain to nucleate the recruitment of class E VPS factors, is functionally mimicked by HIV-1 Gag, in that both encode PTAP motifs that recruit endosomal sorting complex required for transport-I (ESCRT-I) to initiate budding events [21,22]. Thus, HIV-1 virions may be generated within endosomes or at endosome-like membrane domains in much the same way as the lumenal vesicles that are thought to be precursors of cellular exosomes.
Despite widespread acceptance of the notion that HIV-1 particles partially or completely assemble at endosomal membranes, this issue remains controversial because some studies have shown that HIV-1 virions can be endocytosed post-assembly if their release from the PM is inhibited in certain cell types [23,24] Moreover, other studies have suggested that PM localization may be achieved by Gag soon after its synthesis [25]. Thus, although endosomal Gag/virions have frequently been assumed to represent the precursors of extracellular particles, no previous study has conclusively established whether or not an endosomal intermediate is necessary for, or occurs at all, during HIV-1 virion assembly and release. Therefore, we adopted several complimentary approaches, including manipulation of endosome motility and inhibition of endocytic and phagocytic uptake, as well as rational targeting of the HIV-1 Gag protein to determine whether HIV-1 particle assembly indeed follows a pathway that is initiated on endosome membranes. Overall, these studies uniformly indicate that HIV-1 Gag specifically targets PM and not endosomal membranes and that targeting to the PM, but not to the endosomes, is essential for viral release in both an immortalized cell line and in primary macrophages. The endosomal localization of HIV-1 Gag and virions occurs as a result of internalization from the PM and is not a necessary intermediate for constitutive HIV-1 particle synthesis and release.
Results
Microtubules and Late Endosome Motility Are Dispensable for HIV-1 Particle Egress in 293T Cells
Most studies of HIV-1 Gag localization and assembly have examined infected or transfected cells at many hours or days after the onset of Gag expression, when a pseudo–steady state has been reached. We took a different approach, based on transient transfection and examination of the localization and release of very recently synthesized Gag-Pol or Gag–green fluorescent protein (GFP) molecules. This strategy allowed manipulation of cellular functions over a short time course (hours), and avoided the confounding effects of cellular toxicity. An example of an assay that follows the fate of HIV-1 Gag, expressed in the context of Gag-Pol in 293T cells, is shown in Figure 1A. The 55-kD HIV-1 Gag precursor became detectable within approximately 6 h of transfection. Assembly, evidenced by Gag-Pol processing and extracellular particle release, became detectable beginning at around 10–11-h post-transfection, or about 4 h after the onset of detectable Gag precursor synthesis (Figure 1A and 1B).
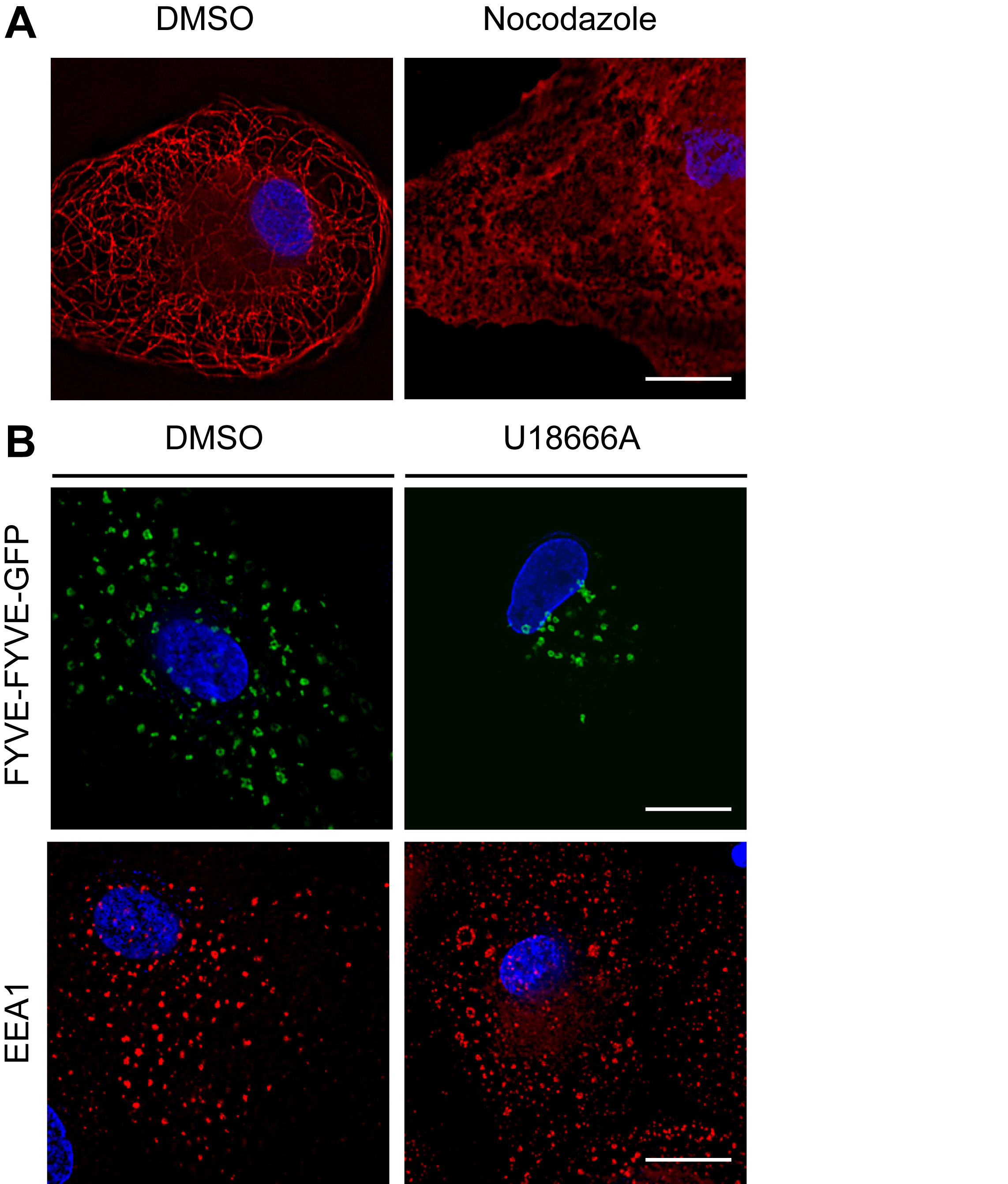
Figure 1. Efficient HIV-1 Particle Formation in the Absence of Microtubules.

(A and B) Western blot analysis of 293T cells expressing HIV-1 Gag-Pol in the absence (A) or presence (B) of nocodazole, added immediately after transfection. Samples were collected every hour, as indicated, and cell and virion lysates were probed with anti–HIV-1 CA antibodies. Numerical values below the blots indicate VLP p24CA signal intensities, derived by densitometry; absence of a value indicates undetectable signal.
(C) 293T cells expressing Gag-GFP (green) in the absence or presence of nocodazole were fixed 14-h post-transfection. Samples were stained with anti–α-tubulin antibodies (red) and with Hoechst 33258 (blue). Scale bars indicate 8 μm.
(D) 293T cells expressing VSV-G in the absence or presence of nocodazole were fixed 14-h post-transfection. Samples were stained with anti–VSV-G antibodies (green) and with Hoechst 33258 (blue). Scale bar indicates 8 μm.
Endosomes move between pericentriolar regions and the cell periphery by recruiting plus- and minus-end motors that direct their movement along microtubules [26,27]. If HIV-1 Gag is initially targeted to late endosomes to initiate assembly or transport to the PM, we reasoned that particle assembly and release should be dependent on endosome motility. Thus, nocodazole-induced microtubule depolymerization, which blocks endosome movement [26–29], should perturb HIV-1 Gag movement to the PM and particle release if endosomal membranes are the initial sites to which Gag is targeted. Within 30 min of nocodazole application, the microtubule network became undetectable by immunofluorescence, and this state was maintained for more than 14 h (Figure 1C), without cytotoxicity. Notably, when microtubules were disrupted throughout the duration of the 14-h assembly assay, the kinetics and extent of HIV-1 assembly, assessed by Gag processing and particle release, were completely unaffected (Figure 1B).
Microscopic examination of Gag-GFP in 293T cells at 14-h post-transfection revealed a diffuse cytoplasmic signal and intense accumulations at the PM (Figure 1C). This predominantly PM localization was unchanged in cells treated with nocodazole during the entire period of Gag-GFP synthesis (Figure 1C). In contrast, when nocodazole was applied during vesicular stomatitis virus glycoprotein (VSV-G) synthesis, the protein failed to be transported to the PM (Figure 1D). As described previously [30], VSV-G localized in a perinuclear area and at the PM in control cells, whereas in nocodazole-treated cells, the protein was found in vesicles that were dispersed throughout the cell (Figure 1D), indicating that the microtubule-dependent secretory pathway was effectively blocked in these experiments. Thus, the microtubule network, and by inference the active transport of endosomes within 293T cells, appeared not to be required for HIV-1 Gag transport to the PM or particle assembly.
In complementary experiments, we took a different approach to arrest late endosome movement. The lipid composition of endocytic compartments can determine their motility [29], and late endosome immobilization can be induced in HeLa cells by application of the hydrophobic amine U18666A. This causes cholesterol accumulation in late endosomal membranes [29], leading to defects in kinesin recruitment. Immunofluorescence examination of the endogenous late endosome marker Lamp1 in 293T cells revealed a characteristic collapse of dispersed Lamp1+ compartments to a perinuclear region of the cells within 2 to 4 h of U18666A treatment (Figure 2A). In contrast, the distribution of endogenous early endosomal antigen (EEA1) was unaffected (Figure 2A), demonstrating that U1866A interferes specifically with the localization of later endosomes. Additionally, dynamic observation of endosomes marked with a 2xHrsFYVE-GFP protein in live cells revealed that their movement was abolished following application of U18666A (Videos S1 and S2). Importantly, when U18666A was added to HIV-1 Gag-GFP–transfected cells during the entire 14-h time course of its synthesis, the ability of Gag-GFP to target the PM was unaffected (Figure 2B). This finding was obtained in numerous repetitions of the experiment, and contrasts with a previous report [31]. Moreover, when U18666A was applied to HIV-1 Gag-Pol–expressing cells, Gag processing and particle release were unaffected (Figure 2C). Thus, both microtubules and the movement of late endosomes within 293T cells appear entirely dispensable for HIV-1 Gag targeting to the PM and particle egress.
Figure 2. Efficient HIV-1 Particle Formation Does Not Require Late Endosome Motility.

(A) U18666A induces the collapse of late, but not early, endosomes toward a perinuclear region of 293T cells. Cells treated with U18666A or DMSO for 14 h were fixed and stained with anti-Lamp1 or anti-EEA1 antibodies (red) and Hoechst 33258 (blue). Scale bars indicate 8 μm.
(B) 293T cells expressing Gag-GFP (green) in the presence or absence of U18666A, which was added immediately after transfection. Samples were fixed at 14-h post-transfection and stained with Hoechst 33258 (blue). Scale bar indicates 4 μm.
(C) Western blot analysis of 293T cells expressing Gag-Pol in the absence or presence of U18666A, which was added immediately after transfection. Samples were collected and analyzed as in Figure 1A. Numerical values below the blots indicate VLP p24CA signal intensities, derived by densitometry; absence of a value indicates undetectable signal.
Endosomal Accumulation of HIV-1 Gag in 293T Cells Is Prevented by Inhibiting Endocytosis
Numerous studies have reported that HIV-1 Gag localizes to endosomes in transfected 293T and several other immortalized cell lines, and suggested that these concentrations are intermediates in particle egress [9–12,32]. We found three general patterns of Gag-GFP localization in transiently transfected 293T cells: in some cells, a diffuse cytoplasmic signal alone, that likely corresponds to non-membrane bound Gag-GFP [33], was observed, whereas in other cells, it was accompanied by PM accumulation or by accumulation at both internal compartments and at the PM (Figure 3A). Importantly, the apparent distribution of Gag-GFP in 293T cells was strongly dependent on the time after transfection at which they were fixed for imaging. Indeed, a temporal analysis revealed that cells exhibiting PM Gag-GFP accumulation were evident very early after transfection (4 h) and began to predominate between 8 and 10 h after transfection (Figure 3B). Conversely, cells exhibiting Gag-GFP accumulation at both the PM and internal sites became evident only later, and were always fewer than those in which only PM accumulation could be seen, maximally representing 30% at 24-h and 48-h post-transfection (Figure 3B).
Figure 3. Accumulation of Gag at the PM Precedes Endosomal Accumulation, Which Can Be Prevented by Blocking Endocytosis.

(A) Distribution of Gag-GFP in 293T cells at 24-h post-transfection. Examples of cells exhibiting diffuse Gag only (left), PM accumulations (center), or PM + internal (Int; right) accumulations are shown. Samples were stained with Hoechst 33258 (blue). Scale bars indicate 4 μm.
(B) Quantification of Gag-GFP distribution in 293T cells. The number of cells in which Gag-GFP accumulation was observed at the PM, as internal and PM accumulations, or as a diffuse cytoplasmic signal only was counted. Approximately 100 cells were evaluated at each time point.
(C) Gag-GFP accumulates at both early and late endosomes. 293T cells expressing Gag-GFP (green) were fixed at 24-h post-transfection and stained with anti-CD63, anti-Lamp1, or anti-EEA1 antibodies (red). Alternatively, cells were co-transfected with CherryFP-Rab5a (red). Cells were stained with Hoechst 33258 (blue). Insets show expanded views of individual Gag puncta. Scale bars, from top to bottom, indicate 4 μm, 2 μm, 8 μm, and 2 μm.
(D) Quantification of Gag-GFP distribution in 293T in the presence of endocytosis inhibitors. 293T cells expressing Gag-GFP together with the indicated proteins were fixed at 24-h post-transfection and the proportion of cells (out of 100 counted) in which Gag was found at the PM, or both at internal accumulations and at the PM was determined.
(E) Western blot analysis of 293T cells expressing HIV-1 Gag-Pol in the presence of endocytosis inhibitors. Samples were collected at 24-h post-transfection as indicated, and cell and virion lysates were probed with anti-HIV-1 CA antibodies. Numerical values below the blots indicate VLP p24CA signal intensities, derived by densitometry.
As expected from previous studies [4,10–12,32], a significant portion of intracellular Gag-GFP that was observed at late time points was associated with compartments that contained endogenous CD63 or Lamp1 (Figure 3C), both of which mark late endosomes [34,35]. However, some intracellular Gag-GFP concentrations were associated with endogenous EEA1+ compartments, or with co-expressed Rab5a-CherryFP (Figure 3C), both of which mark early endosomes [36,37]. In principle, accumulations of HIV-1 Gag in both early and late endosomes could arise through endocytosis [23]. Indeed this appeared to be the case, because endosomal accumulation of Gag-GFP at late time points was inhibited when Gag-GFP was co-expressed with dominant negative forms of EPS-15, which inhibits clathrin-mediated endocytosis [38], or with a mutant version of Rab5a (S34N), which blocks endocytic vesicle fusion (Figure 3D) [39]. Importantly, inhibiting endosomal localization of Gag using dominant negative version of EPS15 or Rab5a (S34N) had no effect on the efficiency with which particles were assembled and released (Figure 3E). This indicates that, in 293T cells, endosomal Gag represents a small fraction of the population and that this endosomal Gag is not required for HIV-1 assembly and release.
Rational Targeting of HIV-1 Gag to the PM, but Not Late Endosomal Membranes, Generates Extracellular Virions
The globular head of HIV-1 MA is responsible for membrane binding and contains the targeting information that governs the location of HIV-1 particle assembly [40,41]. Nevertheless, large deletions or insertions, or its removal is compatible with particle assembly, provided that a minimal myristoylated membrane binding domain is retained [33,42–44]. Although such manipulations can, in some cases, relax the membrane-targeting specificity of HIV-1 Gag, some MA-deleted HIV-1 Gag proteins can generate infectious extracellular particles. If HIV-1 Gag is initially targeted to endosomal membranes, we reasoned that replacement of the MA globular head with alternative membrane binding domains that target endosomes should result in Gag proteins whose localization mimics the wild-type protein. Moreover, such manipulations should be compatible with particle assembly and release.
This strategy exploited the fact that membranes of various cellular compartments are enriched in different phosphoinositides that are recognized by specific membrane-targeting domains [45]. For instance, early and late endosomal membranes are enriched in PI(3)P. FYVE domains are compact 70– to 80–amino acid domains and are found in a number of proteins such as Hrs and SARA that specifically recognize PI(3)P and are, consequently, targeted to endosomes [46,47]. Hrs is particularly noteworthy in this context since, like HIV-1 Gag, it binds Tsg101 (ESCRT-I) and initiates class E VPS factor recruitment, leading to intra-lumenal vesicle generation [21,22]. Thus, a Gag protein that has the targeting properties of Hrs-FYVE should accurately target the membranes to which ESCRT-I is normally recruited and therefore mimic authentic HIV-1 assembly and release into the lumen of endosomes, if that is what normally occurs. Additionally, the Phox homology (PX) domain of the p40phox subunit of NADPH oxidase also binds specifically and with high affinity to PI(3)P and localizes to endosomes [48,49]. Thus, both the Hrs FYVE domain and the p40phox PX domain were appended to HIV-1 Gag in place of the MA globular head, generating FYVE-Gag and PX-Gag, respectively (Figure 4A). Alternatively, protein kinase C conserved domain 2 (C2) domains are approximately 120-residue modules, found in a variety of proteins, that constitutively associate with a variety of membranes [50]. Although its precise lipid binding specificity is not known, we have previously shown that the C2 domain of the HECT ubiquitin ligase, WWP1, can direct a heterologous protein to the PM [51].
Figure 4. Retargeting HIV-1 Gag to Endosomes.

(A) Schematic representation of retargeted Gag proteins in which the globular head of matrix is replaced by specific membrane-binding domains.
(B) Distribution of retargeted Gag proteins in 293T cells. Cells expressing FYVE-Gag-GFP, PX-Gag-GFP, or C2-Gag-GFP (green) were fixed at 24-h post-transfection. Scale bars indicate 8 μm (left panel) or 4 μm (center and right panels).
(C) FYVE-Gag-GFP targets late endosomes. 293T cells expressing FYVE-Gag-GFP (green) were fixed at 24-h post-transfection and stained with anti–Lamp-1 or anti-CD63 antibodies (red). Alternatively, cells were co-transfected with mRed-Hrs or CherryFP-Rab5aQ79L (red). Insets show expanded views of individual FYVE-Gag-GFP accumulations. Scale bars indicate 4 μm.
Thus, when the MA globular head was replaced by the WWP1 C2 domain (Figure 4A), C2-Gag-GFP accumulated almost exclusively at the PM of 293T cells (Figure 4B). Conversely, both FYVE-Gag-GFP and PX-Gag-GFP were exclusively targeted to intracellular vesicular structures in 293T cells, and no PM localization was observed (Figure 4B). Analysis of FYVE-Gag-GFP distribution revealed extensive co-localization with endogenous CD63 and Lamp1 (Figure 4C). When FYVE-Gag-GFP was co-expressed with mRed-Hrs, both proteins targeted the same vesicles (Figure 4C), confirming that FYVE-Gag authentically targeted endosomal membranes. Live cell microscopy revealed that FYVE-Gag-GFP–containing endosomes were motile and moved rapidly in curvilinear paths within the cell interior . Moreover, when the formation of swollen endosomes was induced by over-expression of a “dominant active” Rab5a (Q79L) mutant [39], numerous fluorescent FYVE-Gag particles were observed inside the lumen of the Rab5a-bound structures (Figure 4C), suggesting that particles were able to bud into the lumen of endosomes. Importantly, these same endosomal markers (CD63, Lamp1, and Rab5a) have previously been reported to co-localize with wild-type HIV-1 Gag [4,10–12,23,32]. Similar endosomal localization patterns were observed with PX-Gag-GFP (Figure S1). Authentic intracellular assembly by FYVE-Gag-Pol was confirmed by electron microscopic examination of 293T cells co-expressing the Rab5a (Q79L) and FYVE-Gag-Pol. Indeed, as can be seen in Figure 5A, endosomes containing numerous, apparently mature, virion particles were present, as were nascent assembly intermediates at the limiting membranes of endosomes. Notably, however, unlike wild-type HIV-1 Gag-Pol, FYVE-Gag-Pol budding structures were not found at the PM.
Figure 5. HIV-1 Particles Are Not Released When Assembly Is Targeted to Endosomes.

(A) Electron microscopy analysis of 293T cells expressing CherryFP-Rab5aQ79L together with WT-Gag-Pol (left), in which a portion of the PM is shown, or FYVE-Gag-Pol (right), in which mature virions and assembly intermediates are found in intracellular vesicles. Scale bars indicate 200 nm.
(B) Western blot analysis of 293T cells expressing WT (wild type) or retargeted Gag-Pol or Gag-GFP proteins. Samples were collected at 24-h post-transfection. Cell and virion lysates were probed with anti–HIV-1 CA antibodies. Numerical values below the blots indicate VLP Gag-GFP or p24CA signal intensities, derived by densitometry.
(C) Infectivity measurements using supernatant collected from 293T cells expressing WT, G2A mutant, or retargeted Gag-Pol proteins along with a packageable HIV-1 GFP expression vector and VSV-G. The percentage of infected (GFP+) TE671 target cells is plotted as a function of inoculum volume.
Assembly assays in 293T cells revealed that Gag proteins that were rationally targeted to the PM, namely C2-Gag-GFP and C2-Gag-Pol, generated extracellular particles as efficiently as wild-type Gag-GFP and Gag-Pol (Figure 5B). Moreover, C2-Gag-Pol VLPs were accurately processed (Figure 5B) and contained predominantly mature, fully processed CA protein. Remarkably, when a packageable GFP-expressing HIV-1 vector genome and a VSV-G envelope protein were provided in trans, C2-Gag-Pol proved capable of generating virion particles that were almost as infectious as those generated by wild-type HIV-1 Gag-Pol (Figure 5C). Therefore, direct targeting of the PM by HIV-1 Gag is fully compatible with the generation of mature, infectious HIV-1 particles, and the MA globular head can be functionally replaced by a heterologous non-myristoylated membrane binding domain, provided that it targets an appropriate cell membrane.
In marked contrast, little or no extracellular virion release was observed from cells expressing FYVE-Gag or PX-Gag, in the context of Gag-GFP or Gag-Pol proteins (Figure 5B), even though the expected pattern of Gag processing was observed (Figure 5B). Occasionally, CA was barely detectable in the extracellular medium of FYVE-Gag-Pol expressing cells, and low levels of infectious particles could be detected, but this was less than 1% of that observed with wild type or C2-Gag-Pol (Figure 5C). Overall, therefore, when fully formed HIV-1 particles were generated in motile CD63+ and Lamp1+ compartments in 293T cells, they remained largely trapped therein and were not destined for extracellular release.
HIV-1 Gag Accumulates at the PM prior to Late Endosomes in Primary Macrophages
Although apparent assembly and accumulation of HIV-1 particles in CD63+ endosomes has been observed in 293T cells and many other immortalized cell lines [10–13,32], late endosomal localization of HIV-1 virions is especially evident in primary macrophages [4–7]. However, because macrophages are not easily transfected, studies of Gag localization have heretofore relied on immunofluorescent or electron microscopic detection of Gag protein and particles in cells that were infected several days or weeks previously. As is demonstrated above, when Gag is expressed for extended periods, its localization changes and may not accurately reflect sites of particle assembly. Therefore, we applied recently developed methods for efficient transfection of primary human macrophages to determine HIV-1 Gag-GFP localization at time points very soon after its synthesis. With this approach, Gag-GFP expression became easily detectable by Western blotting or fluorescent microscopy at around 4-h post-transfection (Figure 6A, 6B, and 6C). Particle release into the extracellular medium became detectable approximately 2 h later (Figure 6B).
Figure 6. HIV-1 Gag Accumulation at the PM Precedes Accumulation in Primary Human Macrophage Endosomes.

(A) Distribution of Gag-GFP in macrophages. Cells expressing Gag-GFP (green) were fixed at 24-h post-transfection and stained with Hoechst 33258 (blue). Examples of cells exhibiting diffuse Gag only (left), PM accumulations (center), or PM + internal (Int; right) accumulations are shown. Insets show individual Gag-GFP puncta at the PM coverslip interface. Scale bar indicates 10 μm.
(B) Western blot analysis of macrophages expressing Gag-GFP. Samples were collected every 2 h, from 4- to 12-h post-transfection. Cell and extracellular particle lysates were probed with anti–HIV-1 CA antibodies. Numerical values below the blots indicate VLP signal intensities, derived by densitometry.
(C) Quantification of Gag-GFP distribution in macrophages. The proportion of cells (of 100 counted at each time point) in which Gag-GFP was observed in accumulations only at the PM, as internal and PM accumulations, or as a diffuse cytoplasmic signal only were counted.
(D) Intracellular Gag-GFP is observed on early and late endosomes in macrophages. Cells expressing Gag-GFP (green) were fixed at 24-h post-transfection and stained with anti-EEA1 (red). Alternatively, cells were co-transfected with mRed-Hrs (red). Inset in the bottom right panel shows an individual EEA1+ endosome and associated Gag-GFP. Scale bars indicate 10 μm (top panel) and 4 μm (bottom panel).
As was the case in 293T cells, three readily distinguishable patterns of Gag-GFP localization (diffuse only, diffuse accompanied by PM accumulations, and diffuse accompanied by both PM and intracellular accumulations) were evident in macrophages (Figure 6A). Strikingly, at 4- to 6-h post-transfection, Gag-GFP localization was either entirely diffuse or accompanied by accumulations only at the PM (Figure 6A and 6C). A clear PM accumulation of Gag-GFP was observed when its distribution was analyzed with deconvolved optical sections acquired approximately at center of the cell's vertical dimension, and at the cell–coverslip interface (Figure 6A). Temporal analysis revealed that the population of cells containing only diffuse Gag-GFP was progressively replaced by cells in which Gag-GFP was also located at the PM (Figure 6C). These events preceded the detection of Gag accumulation at internal sites, which was maximal at 24-h post-transfection and reached 10% to 30% of all transfected cells, depending on the donor (Figure 6C and unpublished data). However, Gag was also detected at the PM in these cells. Co-localization studies, done at 24-h post-transfection, revealed that internal Gag-GFP particles associated with large vesicular structures positive for mRed-Hrs (Figure 6D), which is found on both late endosomes and phagosomes in macrophages [52]. As was the case in 293T cells, some Gag-GFP was also found in association with early endosomes containing endogenous EEA1 (Figure 6D).
Microtubules and Late Endosomes Do Not Mediate HIV-1 Assembly in Macrophages
To determine whether late endosomes are functional sites for HIV-1 assembly in macrophages, we adopted essentially the same approaches as in 293T cells. We first verified that nocodazole caused dissolution of the microtubule network in macrophages and that U18666A caused endosomes marked by 2XFYVE-Hrs-GFP, but not EEA1+ early endosomes, to collapse into a perinuclear cluster (Figure S2). In macrophages expressing Gag-GFP that were incubated in medium containing nocodazole or U18666A for the entire duration of Gag-GFP synthesis (up to 24 h), Gag-GFP localized primarily to the PM, both in untreated cells or in cells treated with nocodazole or U18666A (Figure 7A). Moreover, a temporal analysis of Gag-GFP particle generation revealed that these treatments had little or no effect on the kinetics or efficiency of particle release (Figure 7B). Thus, just as was the case in 293T cells, microtubules and late endosome motility appeared not to be required for HIV-1 particle release from macrophages.
Figure 7. HIV-1 Assembly Does Not Required Microtubules or Endosome Motility in Primary Human Macrophages.

(A) Macrophages expressing Gag-GFP were treated with nocodazole or U18666A for the entire duration of Gag-GFP synthesis. Cells were fixed at 12-h post-transfection and stained with Hoechst 33258 (blue). Scale bars indicate 10 μm.
(B) Western blot analysis of macrophages expressing Gag-GFP in the presence of DMSO, nocodazole, or U18666A. Samples were collected every 2 h, from 4- to 12-h post-transfection. Cell and virion lysates were probed with anti–HIV-1 CA antibodies. Numerical values below the blots indicate VLP signal intensities, derived by densitometry.
We next determined whether targeting HIV-1 assembly to macrophage endosomal compartments would result in the release of extracellular virions. Using precisely the same Gag-GFP constructs as previously (Figure 4A), C2-Gag-GFP, like wild-type Gag-GFP, targeted the macrophage PM, whereas FYVE-Gag-GFP and PX-Gag-GFP localized in intracellular compartments (Figure 8A). Co-localization studies showed that FYVE- and PX-Gag-GFP targeted CD63+ and Lamp1+ endosomes (Figure 8B). Importantly, Gag-GFP proteins that were targeted to macrophage CD63+ endosomes failed to generate extracellular particles, whereas C2-Gag-GFP that targeted the PM efficiently generated extracellular particles (Figure 8C). Overall, these experiments indicate that the macrophage PM supports efficient HIV-1 particle release, whereas CD63+/Lamp1+ endosomes appear to be non-viable compartments in which to initiate extracellular HIV-1 particle assembly.
Figure 8. Late Endosomes Are Not Productive Sites of HIV-1 Particle Formation in Primary Human Macrophages.

(A) Distribution of retargeted Gag-GFP proteins in macrophages. Cells expressing C2-Gag-GFP, FYVE-Gag-GFP, or PX-Gag-GFP (green) were fixed at 24-h post-transfection. Scale bars indicate 10 μm.
(B) FYVE-Gag-CherryFP targets late endosomes. Macrophages expressing FYVE-Gag-CherryFP (red) and CD63-GFP or Lamp1-GFP (green) were fixed at 24-h post-transfection and stained with Hoechst 33258 (blue). Insets are expanded views of individual accumulations of FYVE-Gag-CherryFP. Scale bar indicates 10 μm.
(C) Western blot analysis of macrophages expressing Gag-GFP WT or mutants. Samples were collected at 24-h post-transfection. Cell and virion lysates were probed with anti–HIV-1 CA antibodies. Numerical values below the blots indicate VLP signal intensities, derived by densitometry.
HIV-1 Gag Localization, but Not Particle Release, Is Dramatically Affected by Actin Depolymerization in Macrophages
As their name suggests, macrophages are professional phagocytes, and we reasoned that this property might be responsible for the unusually prominent intracellular accumulations of HIV-1 particles that have been observed therein. Phagocytosis is an actin-dependent process [53], and drugs that disrupt actin filaments, such as cytochalasin D, are frequently used to arrest phagocytosis in macrophages. Notably, application of cytochalasin D to macrophages during wild-type HIV-1 Gag-GFP expression effectively removed actin filaments (Figure 9A), but did not inhibit particle release (Figure 9B). Importantly, however, the accumulation of Gag-GFP in intracellular compartments at late time points was almost completely abolished when macrophages were treated with cytochalasin D (Figure 9C).
Figure 9. Actin Influences HIV-1 Gag Localization but Not Particle Release in Primary Human Macrophages.

(A) Disruption of microfilaments in macrophages. Macrophages treated with DMSO or cytochalasin-D were fixed 12 h after treatment and stained with phalloidin (red) and Hoechst 33258 (blue). Scale bars indicate 20 μm.
(B) Western blot analysis of macrophages expressing Gag-GFP in the presence of DMSO or cytochalasin-D. Samples were collected every 2 h, from 4- to 12-h post-transfection. Cell and virion lysates were probed with anti–HIV-1 CA antibodies. Numerical values below the blots indicate VLP signal intensities, derived by densitometry.
(C) Quantification of Gag-GFP distribution in macrophages in the absence of microfilaments. Macrophages expressing Gag-GFP were treated with DMSO or cytochalasin D for the entire duration of Gag-GFP synthesis. Cells were fixed at 12-h post-transfection, and the proportion of cells (of 100 counted) in which Gag was found at the PM, or at both internal sites and the PM was counted.
We performed a similar analysis of Gag localization macrophages that were infected with a full-length, single-cycle infectious HIV-1. Although the kinetics of Gag expression are slower in infected as compared to transfected macrophages, we designed relatively short time-course experiments for this analysis, based on the approximate time taken to complete a single cycle of HIV-1 infection. This approach contrasts with most previous studies of HIV-1–infected macrophages in which Gag and virions accumulated over several days [4,5]. Specifically, macrophages were inoculated with a VSV-G–pseudotyped, Env-defective HIV-1 for 12–16 h. Thereafter, carrier or cytochalasin D was applied to induce actin depolymerization for the ensuing 24 h, at which point cells were fixed. Immunofluorescent detection revealed that the distribution of Gag was either diffuse only, or accompanied by prominent internal accumulations of Gag in 40% of the cells (Figure 10A and 10B). Some Gag puncta were also detected at the PM in about 60% of cells and were usually accompanied by intracellular accumulation (Figure 10B). However, these cell-surface puncta were generally few in number (e.g., see Figure 10A). In marked contrast, intracellular accumulation of Gag was dramatically inhibited in cytochalasin D–treated macrophages (Figure 10A and 10B). Although some intracellular accumulations were found in some cells (about 40%) (Figure 10B), these were much less prominent than in carrier-treated cells (Figure 10A). Moreover, massive accumulation of Gag puncta, presumably representing viral particles, was seen at the PM of virtually all cytochalasin D–treated infected macrophages. (Figure 10A).
Figure 10. Actin Influences HIV-1 Gag Localization but Not Particle Release in Infected Macrophages.

(A) Immunofluorescent detection of HIV-1 CA in macrophages, 38 h after infection by pseudotyped NL4–3 (Env−). Deconvolved optical sections acquired at the center of the vertical dimension of the cell, and at the cell–coverslip interface are shown for examples of infected cells cultured in the presence of DMSO or cytochalasin D. Cells were stained with anti–HIV-1 CA antibodies (red) and Hoechst 33258 (blue). Inset shows expanded view at the PM–coverslip interface and individual Gag puncta. Scale bar indicates 10 μm.
(B) Quantitative analysis of Gag localization in NL4–3 (Env−) infected macrophages. The proportion of cells exhibiting each Gag distribution among 44 to 49 cells counted is plotted.
(C) Distribution of Gag in NL4–3 (MA/YFP) infected macrophages, as in (A) except that no immunostaining was done. Inset shows expanded view at the PM–coverslip interface and individual Gag (MA/YFP) puncta. Scale bar indicates 10 μm.
(D) Western blot analysis of HIV-1 release from NL4–3 (Env−) infected cells in the presence or absence of cytochalasin D, as indicated. Numerical values below the blots indicate p24CA signal intensities in cells and VLPs, derived by densitometry.
Like most HIV-1 Gag-specific antibodies, the anti-p24CA antibody used in these experiments does not efficiently stain accumulations of unprocessed HIV-1 Gag, i.e., assembling particles, presumably because of epitope occlusion (unpublished data) [54]. The rareness with which we observed Gag puncta at the PM of HIV-1–infected but otherwise untreated macrophages prompted us to examine Gag localization using an antibody-independent technique that enables visualization of both unprocessed and processed Gag proteins with equal efficiency. To this end, we used an HIV-1 clone, termed NL4–3 (MA/YFP), in which YFP was inserted into the HIV Gag protein in the stalk region of MA. When pseudotyped with VSV-G, NL4–3 (MA/YFP) produced only approximately 2-fold fewer infectious particles as compared to unmodified HIV-1 (unpublished data). Infection of primary macrophages by VSV-G–pseudotyped NL4–3(MA/YFP) and inspection 36 h later revealed a distribution of Gag(MA/YFP) that was similar to that detected by immunofluorescence (compare Figure 10A and Figure 10C). However, in addition to diffuse fluorescence and intracellular Gag(MA/YFP) accumulations, we also observed significant accumulation of Gag(MA/YFP) at the macrophage PM, and evident Gag puncta at the PM and cell–coverslip interface in many cells (Figure 10C). As previously, there was massive accumulation of particles at the PM of infected cells upon treatment with cytochalasin D during the final 24 h of the assay (Figure 10C). Thus, the prominent intracellular accumulation of HIV-1 Gag that is observed in macrophages clearly occurs via an actin-dependent process, most likely internalization from the PM.
Importantly, actin filament disruption with cytochalasin D had no effect on the levels or processing of cell-associated Gag, nor on the yield of extracellular virions in HIV-1–infected macrophages (Figure 10D). Thus, accumulation of particles at the PM in actin-disrupted cells was not a result of retention of virions that otherwise would be released. Rather, the loss of intracellular Gag accumulation, accompanied by prominent accumulation at the cell surface upon a manipulation that inhibits phagocytosis, suggests that many nascent HIV-1 particles are normally internalized from the surface of macrophages, resulting in accumulation at intracellular sites.
Discussion
Given the considerable uncertainty over where HIV-1 assembles within cells, we adopted a variety of experimental strategies to test the hypothesis that endosomal compartments are functional sites for HIV-1 assembly. Several lines of evidence suggest that they are not. First, Gag localization early after expression is observed only at the PM, suggesting it is the initial membrane targeted by Gag. Second, arresting late endosome movement within cells using two pharmacological inhibitors with distinct mechanisms of action had no effect on accumulation of Gag-GFP at the PM, or on extracellular particle release. Third, rational targeting of HIV-1 Gag using a membrane binding domain that targets endosomes failed to deliver Gag or particles to the PM or extracellular milieu. Although it is formally possible that these retargeted Gag proteins might assemble aberrantly, or target a particular endosomal compartment that is different from that hypothesized to be targeted by wild-type HIV-1 Gag, we believe that this is unlikely. Indeed, we used a membrane-targeting domain that is ordinarily responsible for nucleating ESCRT protein recruitment to endosomes. Moreover, our manipulations resulted in the assembly of complete assembled virions within the lumen of motile CD63+ late endosomal compartments that could clearly be observed by fluorescent and electron microscopy. Conversely a Gag protein that was rationally targeted to the PM closely mimicked the localization of wild-type Gag and allowed efficient release of infectious particles. Importantly, the results presented herein were generated using a commonly used immortalized cell line (293T cells), and using primary macrophages, which are physiological targets of HIV-1 infection. Although it is possible that other cell types might behave differently, the idea that HIV-1 assembly is initiated in endosomes arose from studies done in these, or similar, cell types.
The findings documented herein are difficult to reconcile with the hypothesis that HIV-1 Gag ordinarily targets the endosomal or endosome-like membrane domains normally targeted by ESCRT proteins, and thereby follows an active exosome-like release pathway. Rather, these data suggest that the intrinsic PM-selective binding of the MA Gag domain determines the site at which HIV-1 assembly is initiated. Indeed, recent evidence suggests that the MA domain of HIV-1 Gag binds specifically to PM-resident phospholipids, in particular PI(4,5)P2 [55,56]. Moreover, ESCRT proteins can be recruited from a cytoplasmic pool to membranes where they are not ordinarily located [57], and it is therefore not necessary to invoke endosomal membranes as intermediates to explain HIV-1 release.
Although these studies provide several lines of evidence that HIV-1 assembly occurs primarily at the PM, it is nevertheless true that numerous previous studies, as well as our own observations, have demonstrated that accumulated HIV-1 particles can be observed within endosomes of many cells at steady state (see Introduction). Indeed, this localization predominates in long-term infected, but otherwise unperturbed, macrophages. How can this be explained in terms of a model in which particle assembly occurs at the PM? In fact, these previous studies can readily be reconciled with the findings described herein if one invokes internalization of nascent or completely assembled virions from the PM. Internalization of PM occurs in essentially all cell types via endocytic or phagocytic mechanisms, but the predominant mechanism and the rate of PM turnover likely varies with cell type. Thus, the observation of virion particles in endosomal compartments does not necessarily mean that they were assembled therein. Indeed, this and previous work clearly demonstrates that endosomal virions can arise via endocytosis [23].
Inevitably, the overall steady-state cellular localization of HIV-1 Gag and virion particles is governed by several kinetic factors, including the rate at which Gag moves to the PM following its synthesis. HIV-1 Gag:membrane interaction is highly cooperative and rather inefficient at low Gag concentration due to the presence of the “myristoyl switch” [33,56,58,59]. Once Gag and/or virions are at the PM, the rates at which they are either released as extracellular virions or internalized are also crucial kinetic factors determining steady-state cellular distribution. In principle, therefore, a slow rate of synthesis—which would result in a slow rate of PM binding and assembly—coupled with a high rate of PM internalization would, over time, result in a steady-state distribution in which most HIV-1 Gag or virions accumulate in endosomal compartments. We surmise that this is precisely what occurs in HIV-1–infected professional phagocytes, i.e., macrophages. Consistent with this idea, inhibition of actin polymerization, which reduces the rate of phagocytosis, resulted in very dramatic accumulation of HIV-1 particles at the PM instead of in endosomes in infected primary macrophages. Importantly, this apparent redistribution occurred in the complete absence of any effects on Gag expression or particle release.
Similar alterations in the distribution of HIV-1 Gag in other cell types can be effected by manipulating alternative internalization pathways. For example, in HeLa cells, in which a large proportion of cells contain CD63+ endosome-associated Gag, release of virions is inefficient due to the action of an unknown dominant inhibitor that is counteracted by the HIV-1 Vpu protein [60]. In that instance, endosomal virion accumulation could be prevented either by ablating the effect of the release inhibitor, via expression of Vpu, or alternatively, by inhibiting endocytosis [23]. In a similar way, this study shows that blockade of endocytosis in 293T cells clearly reduced the already quite small fraction of cells in which intracellular accumulation of HIV-1 Gag-GFP is evident.
Overall, it seems likely that the intracellular HIV-1 particles that are observed in a variety of cell types can be accounted for, perhaps in all cases, by internalization from the PM by endocytic or phagocytic mechanisms. Notably, no study that has reported intracellular concentrations of retroviral Gag proteins or virions has excluded this possibility.
It is possible that HIV-1 particles whose assembly was initiated at the PM but were then internalized could retain infectivity for some time and eventually find their way to the extracellular milieu. This would require some physiological stimulus, e.g., antigen presentation or calcium influx, which induces endosome fusion with the cell surface. Indeed, an antigen presentation-related phenomenon may occur in dendritic cells that capture virions from the extracellular environment for dissemination to other cells [61]. Infected macrophages could also retain HIV-1 particles within endosomes for extended periods [62]. Nonetheless, our findings strongly suggest that endosome-based HIV-1 virion release is not a constitutive process by which most extracellular HIV-1 particles are generated. Rather, several lines of evidence presented herein lead to the conclusion that the initiating site for constitutive HIV-1 assembly and release is the PM. The subsequent fate of the nascent particle appears to be governed by whether that PM domain remains at the surface, enabling particle release, or internalized, resulting in particle retention in endosomes.
Materials and Methods
Plasmid derivation.
Plasmids expressing HIV-1 Gag-Pol and Gag-GFP proteins, namely pCR3.1/SYNGP and pCR3.1/Gag-GFP, were previously described [33] and were used to generate plasmids expressing retargeted Gag proteins. First, a truncated version of HIV-1 Gag lacking the MA globular head (encoding amino acids 116–501) was generated using PCR and inserted between the XhoI–NotI sites of pCR3.1/GFP. Hrs-FYVE (amino acids 147–224), WWP1-C2 (amino acids 21–187), and p40phox PX (amino acids 19–140) domains were amplified from previously described expression plasmids [51,63] or from human placental cDNA. These fragments were inserted as EcoR1–Xho1 fragments into the Gag(116–501)-GFP plasmid. The corresponding Gag-Pol derivatives of these fusion proteins were made by replacing the original EcoR1–AflIII fragment of pCR3.1/SYNGP by fragments encoding the Gag fusion proteins. 2xFYVE-GFP was generated by inserting a second FYVE domain between the XhoI–NotI sites of pCR3.1/FYVE-GFP.
A full-length infectious NL4–3–derived proviral plasmid, NL4–3(MA/YFP), that incorporated YFP into the stalk region of MA was derived essentially as described [64], except that YFP was used in place of GFP.
Plasmids expressing red-fluorescent fusion proteins were derived from pCR3.1/GFP by replacing GFP by CherryFP (provided by R. Tsien) or mRed (BD Biosciences, San Jose, California, United States). Human CD63 was amplified from human placental cDNA and inserted as an EcoR1–XhoI fragment into pCR3.1/GFP to express CD63 with GFP fused to its C-terminus. Plasmids expressing CherryFP-Rab5a fusion proteins and mutant derivatives have been described previously [23], as has the dominant negative EPS-15 expression plasmid, a gift from S. Polo [65]. Hrs was transferred from previously described pCR3.1/YFP-Hrs [63] into pCR3.1/mRed to express an mRed-Hrs fusion protein. The plasmid expressing Lamp1-GFP has been described previously [66].
Cells, transfection, and drug treatment.
293T and TE671 cells were maintained in DMEM supplemented with 10% fetal calf serum. Monocyte-derived macrophages were isolated from the adherent fraction of human peripheral blood mononuclear cells (PBMCs). PBMCs were plated at a density of 5 × 107 per T-75 poly-D-lysine–coated flask (BD BioCoat, San Jose, California, United States) and were cultured for 2 h in RPMI supplemented with 10% human serum (Sigma, St. Louis, Missouri, United States). The non-adherent fraction was removed, and fresh medium containing 600 U/ml of macrophage colony-stimulating factor (Peprotech, Rocky Hill, New Jersey, United States) was added. Cell differentiation was completed after 4 to 5 d, and transfection or infection was performed at day 7.
293T cells were transfected using polyethylenimine (PolySciences, Warrington, Pennsylvania, United States) as described previously [67]. Macrophages were transfected by nucleofection using the AMAXA device (Amaxa, Gaithersburg, Maryland, United States). About 5–7 × 105 cells were transfected with 5 μg of DNA using the Y-010 program. In experiments in which fluorescent microscopy was performed, cells were plated on poly-D-lysine–coated glass-bottomed dishes (Mattek, Ashland, Massachusetts, United States).
Nocodazole (Sigma) and U18666A (Biomol, Plymouth Meeting, Pennsylvania, United States) were used at a final concentration of 10 μg/ml, irrespective of cell type. Cytochalasin D (Sigma) was used at 5 μM (macrophages). All the aforementioned drugs were dissolved in DMSO and diluted in medium immediately prior to use. Control cultures were treated with an equal amount of DMSO.
Virus release assays and infectivity assays.
Transfection-based virus release assays were performed by transfecting 293T or macrophages as described above, and then harvesting cells and extracellular virus particles at various time points thereafter, as described previously [23]. Drugs were added to culture media either immediately after transfection (293T) or 1 h later (macrophages). Alternatively, infectious virions were generated by transfecting 293T cells with NL4–3(Env−) or NL4–3(MA/YFP) along with a VSV-G expression plasmid. These virus stocks were used to inoculate primary macrophages. Virus was removed and cells washed thoroughly 12 h later. At 16 h after inoculation, cytochalasin D or DMSO was added, and 24 h later, cultures were processed for analysis of virus release or microscopy. Virions and cell lysates were separated on 10% acrylamide gels, and proteins were probed with anti–HIV-1-p24 (183-H12-5C) antibodies. Subsequently, blots were probed with species-specific HRP-conjugated goat secondary antibodies. Western blot signal intensities were quantitated using densitometry (AlphaImager 2200; Alpha Innotech, San Leandro, California, United States).
To measure infectious virion generation by C2-, FYVE-, and PX-appended Gag proteins, each Gag-Pol expression plasmid was cotransfected in 293T cells with a V1/GFP, a packageable HIV-1 vector [68] and a VSV-G expression plasmid. Virions were harvested after 48 h. Infectivity was measured using TE671 target cells and (FACS) analysis 36 h later as previously described [69].
Microscopy and antibodies.
Samples for microscopy analysis were fixed, processed, and stained as described previously [23]. Antibodies were as follows: mouse anti-CD63 (Chemicon, Temecula, California, United States), mouse anti-EEA1 (BD Biosciences), mouse anti-Lamp1 clone H4A3 (Developmental Studies Hybridoma Bank, Iowa University, Iowa City, Iowa, United States), mouse anti–VSV-G clone P5D4 (Roche Applied Science, Basel, Switzerland), mouse anti–α-tubulin (Sigma), anti–HIV-1-p24 (183-H12-5C, National Institutes of Health AIDS reagent program [http://www.aidsreagent.org/)], and anti-mouse Alexafluor 594 (Molecular Probes, Eugene, Oregon, United States). Phalloidin-Alexafluor 594 (Molecular Probes) was used to stain actin. Nuclei were stained with Hoechst 33258. Fluorescent imaging of fixed cells was done using an Olympus IX70–based Deltavision microscopy suite (Applied Precision, Issaquah, Washington, United States), as previously described [51].
Electron microscopy.
Cells were fixed 44-h post-transfection in buffered 2.5% glutaraldehyde, post-fixed with 1% osmium tetroxide, and dehydrated through a graded series of acetone washes. Dehydrated samples were transitioned into EPON:SPURR (1:1) and baked overnight at 60 °C. Embedded samples were thin sectioned, post-stained with uranyl acetate and Sato's triple lead stain, and viewed on a JEOL 1200 EX transmission electron microscope (JEOL, Peabody, Massachusetts, United States).
Live cell imaging.
Total internal reflection fluorescence microscopy (TIR-FM) images were acquired as previously described [70,71]. Briefly, TIR illumination was accomplished using illumination through the objective (Apo 60X NA 1.45; Olympus America, Center Valley, Pennsylvania, United States) on an inverted epifluorescence microscope (IX-70; Olympus). The evanescent field decayed with a space constant of less than 100 nm. Cells were maintained at 37 °C in a home-built temperature-controlled chamber. All cells were placed in Cell Imaging Media (CIM; HBSS + 10 mM Hepes + 5% FBS [pH 7.4]) immediately before image acquisition.
GFP constructs were excited with the 514-nm line of an Argon laser (Omnichrome, model 543-AP A01; Melles Griot, Carlsbad, California, United States) reflected off a dichroic mirror (442/514PC). Emitted light was collected through an emission band-pass filter (HQ550/50M). The filters were obtained from Chroma Technologies Corp (Rockingham, Vermont, United States). Images were acquired with a 12-bit cooled CCD ORCA-ER (Hamamatsu Photonics, Hamamatsu City, Japan) with a resolution of 1,280 × 1,024 pixels with a pixel size of 6.45 μm × 6.45 μm. The camera and mechanical shutters (Uniblitz; Vincent Associates, Rochester, New York, United States) were controlled using MetaMorph (Molecular Devices, Sunnyvale, California, United States). Images were acquired with 20- or 50-ms exposures per frame either streaming for 200 frames or acquired in time lapse mode every 4 s for 10 min. Videos are displayed at 10 frames/s.
Supporting Information
293T cells expressing PX-Gag-GFP (green) were fixed at 24-h post-transfection and stained with anti–Lamp-1 or anti-CD63 antibodies (red). Alternatively, cells were co-transfected with mRed-Hrs or CherryFP-Rab5aQ79L (red). Nuclei were stained with Hoechst 33258 (blue). Scale bar indicates 4 μm.
(1.5 MB JPG)
{kind=link}
(A) Disruption of microtubules in macrophages. Macrophages treated with nocodazole or DMSO were fixed 12 h after treatment and stained with anti–α-tubulin antibodies (red) and with Hoechst 33258 (blue). Scale bar indicates 10 μm.
(B) U18666A induces the collapse of late, but not early, endosomes in the perinuclear region of macrophages. Cells expressing 2xFYVE-GFP (green) were treated with U18666A or DMSO for 12 h prior to fixation. Alternatively, cells treated with U18666A or DMSO were stained with anti-EEA1 antibodies (red). Nuclei were stained with Hoechst 33258 (blue). Scale bars indicate 10 μm.
(975 KB JPG)
{kind=link}
293T cells expressing 2xFYVE-GFP in the presence of DMSO, which was added immediately after transfection, were imaged at 14-h post-transfection. Images were acquired in the TIR field as described in Materials and Methods, with 50-ms exposure per frame every 4 s for 10 min. The video is displayed at 10 frames/s.
(48 MB AVI)
293T cells expressing with 2xFYVE-GFP in the presence of U18666A, which was added immediately after transfection, were imaged at 14-h post-transfection. Images were acquired in the TIR field as described in Materials and Methods, with 50-ms exposure per frame every 4 s for 10 min. The video is displayed at 10 frames/s.
(83 MB AVI)
293T cells expressing FYVE-Gag-GFP were imaged at 24-h post-transfection. Images were acquired in the TIR field as described in Materials and Methods, with 20-ms exposure, streaming for 200 frames. The video is displayed at 10 frames/s.
(155 MB AVI)
Acknowledgments
We thank members of the Bieniasz and the Simon groups for various reagents and helpful discussions.
Abbreviations
- C2
protein kinase C conserved domain-2
- CA
capsid
- EEA1
early endosomal antigen-1
- ESCRT
endosomal sorting complex required for transport
- FYVE
Fab1/YOTB/Vac1/EEA1
- GFP
green fluorescent protein
- Lamp1
lysosome-associated membrane protein-1
- MA
matrix
- PM
plasma membrane
- PX
Phox homology
- VLP
virus-like particle
- VPS
vacuolar protein sorting
- VSV-G
vesicular stomatitis virus glycoprotein
Funding Statement
The work was supported by grants from the National Institutes of Health (NIH) (R01AI50111) to PDB and grants from the NIH (GM072015) and from National Science Foundation (NSF) BES 00520813 to SMS. NJ is supported by the Rockefeller University Women & Science Fellowship program. PDB is an Elizabeth Glaser Scientist of the Elizabeth Glaser Pediatric AIDS foundation.
References
- 1.Gottlinger HG. The HIV-1 assembly machine. AIDS. 2001;15(Suppl 5):S13–20. doi: 10.1097/00002030-200100005-00003. [DOI] [PubMed] [Google Scholar]
- 2.Bieniasz PD. Late budding domains and host proteins in enveloped virus release. Virology. 2006;344:55–63. doi: 10.1016/j.virol.2005.09.044. [DOI] [PubMed] [Google Scholar]
- 3.Morita E, Sundquist WI. Retrovirus budding. Annu Rev Cell Dev Biol. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- 4.Pelchen-Matthews A, Kramer B, Marsh M. Infectious HIV-1 assembles in late endosomes in primary macrophages. J Cell Biol. 2003;162:443–455. doi: 10.1083/jcb.200304008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raposo G, Moore M, Innes D, Leijendekker R, Leigh-Brown A, et al. Human macrophages accumulate HIV-1 particles in MHC II compartments. Traffic. 2002;3:718–729. doi: 10.1034/j.1600-0854.2002.31004.x. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen DG, Booth A, Gould SJ, Hildreth JE. Evidence that HIV budding in primary macrophages occurs through the exosome release pathway. J Biol Chem. 2003;278:52347–52354. doi: 10.1074/jbc.M309009200. [DOI] [PubMed] [Google Scholar]
- 7.Ono A, Freed EO. Cell-type-dependent targeting of human immunodeficiency virus type 1 assembly to the plasma membrane and the multivesicular body. J Virol. 2004;78:1552–1563. doi: 10.1128/JVI.78.3.1552-1563.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Resh MD. Intracellular trafficking of HIV-1 Gag: How Gag interacts with cell membranes and makes viral particles. AIDS Rev. 2005;7:84–91. [PubMed] [Google Scholar]
- 9.Goff A, Ehrlich LS, Cohen SN, Carter CA. Tsg101 control of human immunodeficiency virus type 1 Gag trafficking and release. J Virol. 2003;77:9173–9182. doi: 10.1128/JVI.77.17.9173-9182.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nydegger S, Foti M, Derdowski A, Spearman P, Thali M. HIV-1 egress is gated through late endosomal membranes. Traffic. 2003;4:902–910. doi: 10.1046/j.1600-0854.2003.00145.x. [DOI] [PubMed] [Google Scholar]
- 11.Perlman M, Resh MD. Identification of an intracellular trafficking and assembly pathway for HIV-1 Gag. Traffic. 2006;7:731–745. doi: 10.1111/j.1398-9219.2006.00428.x. [DOI] [PubMed] [Google Scholar]
- 12.Sherer NM, Lehmann MJ, Jimenez-Soto LF, Ingmundson A, Horner SM, et al. Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic. 2003;4:785–801. doi: 10.1034/j.1600-0854.2003.00135.x. [DOI] [PubMed] [Google Scholar]
- 13.Dong X, Li H, Derdowski A, Ding L, Burnett A, et al. AP-3 directs the intracellular trafficking of HIV-1 Gag and plays a key role in particle assembly. Cell. 2005;120:663–674. doi: 10.1016/j.cell.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 14.Houzet L, Gay B, Morichaud Z, Briant L, Mougel M. Intracellular assembly and budding of the Murine Leukemia Virus in infected cells. Retrovirology. 2006;3:12. doi: 10.1186/1742-4690-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blot V, Perugi F, Gay B, Prevost MC, Briant L, et al. Nedd4.1-mediated ubiquitination and subsequent recruitment of Tsg101 ensure HTLV-1 Gag trafficking towards the multivesicular body pathway prior to virus budding. J Cell Sci. 2004;117:2357–2367. doi: 10.1242/jcs.01095. [DOI] [PubMed] [Google Scholar]
- 16.Dorweiler IJ, Ruone SJ, Wang H, Burry RW, Mansky LM. Role of the human T-cell leukemia virus type 1 PTAP motif in Gag targeting and particle release. J Virol. 2006;80:3634–3643. doi: 10.1128/JVI.80.7.3634-3643.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basyuk E, Galli T, Mougel M, Blanchard JM, Sitbon M, et al. Retroviral genomic RNAs are transported to the plasma membrane by endosomal vesicles. Dev Cell. 2003;5:161–174. doi: 10.1016/s1534-5807(03)00188-6. [DOI] [PubMed] [Google Scholar]
- 18.Nydegger S, Khurana S, Krementsov DN, Foti M, Thali M. Mapping of tetraspanin-enriched microdomains that can function as gateways for HIV-1. J Cell Biol. 2006;173:795–807. doi: 10.1083/jcb.200508165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Booth AM, Fang Y, Fallon JK, Yang JM, Hildreth JE, et al. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J Cell Biol. 2006;172:923–935. doi: 10.1083/jcb.200508014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gould SJ, Booth AM, Hildreth JE. The Trojan exosome hypothesis. Proc Natl Acad Sci U S A. 2003;100:10592–10597. doi: 10.1073/pnas.1831413100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bache KG, Brech A, Mehlum A, Stenmark H. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol. 2003;162:435–442. doi: 10.1083/jcb.200302131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pornillos O, Higginson DS, Stray KM, Fisher RD, Garrus JE, et al. HIV Gag mimics the Tsg101-recruiting activity of the human Hrs protein. J Cell Biol. 2003;162:425–434. doi: 10.1083/jcb.200302138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neil SJ, Eastman SW, Jouvenet N, Bieniasz PD. HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2006;2:e39. doi: 10.1371/journal.ppat.0020039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harila K, Prior I, Sjoberg M, Salminen A, Hinkula J, et al. Vpu and Tsg101 regulate intracellular targeting of the human immunodeficiency virus type 1 core protein precursor Pr55gag. J Virol. 2006;80:3765–3772. doi: 10.1128/JVI.80.8.3765-3772.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudner L, Nydegger S, Coren LV, Nagashima K, Thali M, et al. Dynamic fluorescent imaging of human immunodeficiency virus type 1 gag in live cells by biarsenical labeling. J Virol. 2005;79:4055–4065. doi: 10.1128/JVI.79.7.4055-4065.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aniento F, Emans N, Griffiths G, Gruenberg J. Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J Cell Biol. 1993;123:1373–1387. doi: 10.1083/jcb.123.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bomsel M, Parton R, Kuznetsov SA, Schroer TA, Gruenberg J. Microtubule- and motor-dependent fusion in vitro between apical and basolateral endocytic vesicles from MDCK cells. Cell. 1990;62:719–731. doi: 10.1016/0092-8674(90)90117-w. [DOI] [PubMed] [Google Scholar]
- 28.Wubbolts R, Fernandez-Borja M, Jordens I, Reits E, Dusseljee S, et al. Opposing motor activities of dynein and kinesin determine retention and transport of MHC class II-containing compartments. J Cell Sci 112 ( Pt 6) 1999. pp. 785–795. [DOI] [PubMed]
- 29.Lebrand C, Corti M, Goodson H, Cosson P, Cavalli V, et al. Late endosome motility depends on lipids via the small GTPase Rab7. EMBO J. 2002;21:1289–1300. doi: 10.1093/emboj/21.6.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, et al. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- 31.Lindwasser OW, Resh MD. Human immunodeficiency virus type 1 Gag contains a dileucine-like motif that regulates association with multivesicular bodies. J Virol. 2004;78:6013–6023. doi: 10.1128/JVI.78.11.6013-6023.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grigorov B, Arcanger F, Roingeard P, Darlix JL, Muriaux D. Assembly of infectious HIV-1 in human epithelial and T-lymphoblastic cell lines. J Mol Biol. 2006;359:848–862. doi: 10.1016/j.jmb.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 33.Perez-Caballero D, Hatziioannou T, Martin-Serrano J, Bieniasz PD. Human immunodeficiency virus type 1 matrix inhibits and confers cooperativity on Gag precursor-membrane interactions. J Virol. 2004;78:9560–9563. doi: 10.1128/JVI.78.17.9560-9563.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobayashi T, Vischer UM, Rosnoblet C, Lebrand C, Lindsay M, et al. The tetraspanin CD63/lamp3 cycles between endocytic and secretory compartments in human endothelial cells. Mol Biol Cell. 2000;11:1829–1843. doi: 10.1091/mbc.11.5.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Escola JM, Kleijmeer MJ, Stoorvogel W, Griffith JM, Yoshie O, et al. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J Biol Chem. 1998;273:20121–20127. doi: 10.1074/jbc.273.32.20121. [DOI] [PubMed] [Google Scholar]
- 36.Stenmark H, Aasland R, Toh BH, D'Arrigo A. Endosomal localization of the autoantigen EEA1 is mediated by a zinc-binding FYVE finger. J Biol Chem. 1996;271:24048–24054. doi: 10.1074/jbc.271.39.24048. [DOI] [PubMed] [Google Scholar]
- 37.Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2:107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- 38.Benmerah A, Bayrou M, Cerf-Bensussan N, Dautry-Varsat A. Inhibition of clathrin-coated pit assembly by an Eps15 mutant. J Cell Sci 112 ( Pt 9) 1999. pp. 1303–1311. [DOI] [PubMed]
- 39.Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, et al. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ono A, Freed EO. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J Virol. 1999;73:4136–4144. doi: 10.1128/jvi.73.5.4136-4144.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ono A, Orenstein JM, Freed EO. Role of the Gag matrix domain in targeting human immunodeficiency virus type 1 assembly. J Virol. 2000;74:2855–2866. doi: 10.1128/jvi.74.6.2855-2866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang CT, Zhang Y, McDermott J, Barklis E. Conditional infectivity of a human immunodeficiency virus matrix domain deletion mutant. J Virol. 1993;67:7067–7076. doi: 10.1128/jvi.67.12.7067-7076.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Facke M, Janetzko A, Shoeman RL, Krausslich HG. A large deletion in the matrix domain of the human immunodeficiency virus gag gene redirects virus particle assembly from the plasma membrane to the endoplasmic reticulum. J Virol. 1993;67:4972–4980. doi: 10.1128/jvi.67.8.4972-4980.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee PP, Linial ML. Efficient particle formation can occur if the matrix domain of human immunodeficiency virus type 1 Gag is substituted by a myristylation signal. J Virol. 1994;68:6644–6654. doi: 10.1128/jvi.68.10.6644-6654.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hurley JH, Misra S. Signaling and subcellular targeting by membrane-binding domains. Annu Rev Biophys Biomol Struct. 2000;29:49–79. doi: 10.1146/annurev.biophys.29.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hayakawa A, Hayes SJ, Lawe DC, Sudharshan E, Tuft R, et al. Structural basis for endosomal targeting by FYVE domains. J Biol Chem. 2004;279:5958–5966. doi: 10.1074/jbc.M310503200. [DOI] [PubMed] [Google Scholar]
- 47.Blatner NR, Stahelin RV, Diraviyam K, Hawkins PT, Hong W, et al. The molecular basis of the differential subcellular localization of FYVE domains. J Biol Chem. 2004;279:53818–53827. doi: 10.1074/jbc.M408408200. [DOI] [PubMed] [Google Scholar]
- 48.Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, et al. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol. 2001;3:675–678. doi: 10.1038/35083070. [DOI] [PubMed] [Google Scholar]
- 49.Zhan Y, Virbasius JV, Song X, Pomerleau DP, Zhou GW. The p40phox and p47phox PX domains of NADPH oxidase target cell membranes via direct and indirect recruitment by phosphoinositides. J Biol Chem. 2002;277:4512–4518. doi: 10.1074/jbc.M109520200. [DOI] [PubMed] [Google Scholar]
- 50.Cho W. Membrane targeting by C1 and C2 domains. J Biol Chem. 2001;276:32407–32410. doi: 10.1074/jbc.R100007200. [DOI] [PubMed] [Google Scholar]
- 51.Martin-Serrano J, Eastman SW, Chung W, Bieniasz PD. HECT ubiquitin ligases link viral and cellular PPXY motifs to the vacuolar protein-sorting pathway. J Cell Biol. 2005;168:89–101. doi: 10.1083/jcb.200408155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vieira OV, Harrison RE, Scott CC, Stenmark H, Alexander D, et al. Acquisition of Hrs, an essential component of phagosomal maturation, is impaired by mycobacteria. Mol Cell Biol. 2004;24:4593–4604. doi: 10.1128/MCB.24.10.4593-4604.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Niedergang F, Chavrier P. Signaling and membrane dynamics during phagocytosis: many roads lead to the phagos(R)ome. Curr Opin Cell Biol. 2004;16:422–428. doi: 10.1016/j.ceb.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 54.Ono A, Waheed AA, Joshi A, Freed EO. Association of human immunodeficiency virus type 1 gag with membrane does not require highly basic sequences in the nucleocapsid: use of a novel Gag multimerization assay. J Virol. 2005;79:14131–14140. doi: 10.1128/JVI.79.22.14131-14140.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A. 2004;101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saad JS, Miller J, Tai J, Kim A, Ghanam RH, et al. From the cover: Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci U S A. 2006;103:11364–11369. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martin-Serrano J, Bieniasz PD. A bipartite late-budding domain in human immunodeficiency virus type 1. J Virol. 2003;77:12373–12377. doi: 10.1128/JVI.77.22.12373-12377.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Resh MD. A myristoyl switch regulates membrane binding of HIV-1 Gag. Proc Natl Acad Sci U S A. 2004;101:417–418. doi: 10.1073/pnas.0308043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, et al. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci U S A. 2004;101:517–522. doi: 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Varthakavi V, Smith RM, Bour SP, Strebel K, Spearman P. Viral protein U counteracts a human host cell restriction that inhibits HIV-1 particle production. Proc Natl Acad Sci U S A. 2003;100:15154–15159. doi: 10.1073/pnas.2433165100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GC, et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- 62.Sharova N, Swingler C, Sharkey M, Stevenson M. Macrophages archive HIV-1 virions for dissemination in trans. Embo J. 2005;24:2481–2489. doi: 10.1038/sj.emboj.7600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martin-Serrano J, Yarovoy A, Perez-Caballero D, Bieniasz PD. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc Natl Acad Sci U S A. 2003;100:12414–12419. doi: 10.1073/pnas.2133846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muller B, Daecke J, Fackler OT, Dittmar MT, Zentgraf H, et al. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. J Virol. 2004;78:10803–10813. doi: 10.1128/JVI.78.19.10803-10813.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Poupon V, Polo S, Vecchi M, Martin G, Dautry-Varsat A, et al. Differential nucleocytoplasmic trafficking between the related endocytic proteins Eps15 and Eps15R. J Biol Chem. 2002;277:8941–8948. doi: 10.1074/jbc.M108385200. [DOI] [PubMed] [Google Scholar]
- 66.Jaiswal JK, Andrews NW, Simon SM. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J Cell Biol. 2002;159:625–635. doi: 10.1083/jcb.200208154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Durocher Y, Perret S, Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002;30:E9. doi: 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Simon V, Zennou V, Murray D, Huang Y, Ho DD, et al. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog. 2005;1:e6. doi: 10.1371/journal.ppat.0010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hatziioannou T, Cowan S, Goff SP, Bieniasz PD, Towers GJ. Restriction of multiple divergent retroviruses by Lv1 and Ref1. EMBO J. 2003;22:385–394. doi: 10.1093/emboj/cdg042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lampson MA, Schmoranzer J, Zeigerer A, Simon SM, McGraw TE. Insulin-regulated release from the endosomal recycling compartment is regulated by budding of specialized vesicles. Mol Biol Cell. 2001;12:3489–3501. doi: 10.1091/mbc.12.11.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schmoranzer J, Goulian M, Axelrod D, Simon SM. Imaging constitutive exocytosis with total internal reflection fluorescence microscopy. J Cell Biol. 2000;149:23–32. doi: 10.1083/jcb.149.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
293T cells expressing PX-Gag-GFP (green) were fixed at 24-h post-transfection and stained with anti–Lamp-1 or anti-CD63 antibodies (red). Alternatively, cells were co-transfected with mRed-Hrs or CherryFP-Rab5aQ79L (red). Nuclei were stained with Hoechst 33258 (blue). Scale bar indicates 4 μm.
(1.5 MB JPG)
(A) Disruption of microtubules in macrophages. Macrophages treated with nocodazole or DMSO were fixed 12 h after treatment and stained with anti–α-tubulin antibodies (red) and with Hoechst 33258 (blue). Scale bar indicates 10 μm.
(B) U18666A induces the collapse of late, but not early, endosomes in the perinuclear region of macrophages. Cells expressing 2xFYVE-GFP (green) were treated with U18666A or DMSO for 12 h prior to fixation. Alternatively, cells treated with U18666A or DMSO were stained with anti-EEA1 antibodies (red). Nuclei were stained with Hoechst 33258 (blue). Scale bars indicate 10 μm.
(975 KB JPG)
293T cells expressing 2xFYVE-GFP in the presence of DMSO, which was added immediately after transfection, were imaged at 14-h post-transfection. Images were acquired in the TIR field as described in Materials and Methods, with 50-ms exposure per frame every 4 s for 10 min. The video is displayed at 10 frames/s.
(48 MB AVI)
293T cells expressing with 2xFYVE-GFP in the presence of U18666A, which was added immediately after transfection, were imaged at 14-h post-transfection. Images were acquired in the TIR field as described in Materials and Methods, with 50-ms exposure per frame every 4 s for 10 min. The video is displayed at 10 frames/s.
(83 MB AVI)
293T cells expressing FYVE-Gag-GFP were imaged at 24-h post-transfection. Images were acquired in the TIR field as described in Materials and Methods, with 20-ms exposure, streaming for 200 frames. The video is displayed at 10 frames/s.
(155 MB AVI)