

Abstract
The US is experiencing an epidemic of cocaine use entangled with HIV-1 infection. Normal human astrocytes (NHA) are susceptible to HIV-1 infection. We utilized LTR-R/U5 amplification, p24 antigen assay and the proteomic method of difference gel electrophoresis (DIGE) combined with protein identification through HPLC-MS/MS to investigate the effect of cocaine on HIV-1 infectivity and the proteomic profile of NHA, respectively. Data demonstrate that cocaine significantly upregulates HIV-1 infection in NHA as measured by LTR-R/U5 amplification and p24 antigen assay. Further, our results show for the first time that cocaine differentially regulates the expression of a number of proteins by NHA that may play a role in the neuropathogenesis of HIV-1 disease.
Keywords: Cocaine, Normal human astrocytes (NHA), Difference gel electrophoresis (DIGE), High performance liquid, chromatography-tandem mass, spectrometry (HPLC-MS/MS)
1. Introduction
The CNS is a major target for HIV-1 infection. Within days of infection, HIV-1 can enter the CNS where various resident cell populations serve as reservoirs for HIV-1 (Brack-Werner, 1999; Canki et al., 2001; Clarke et al., 2006; Davis et al., 1992; Speth et al., 2005). Macrophage and microglial cells are the primary sources of HIV-1 replication in CNS (Gonzalez-Scarano and Martin-Garcia, 2005; Kaul et al., 2001; Kramer-Hammerle et al., 2005; Minagar et al., 2002). Astrocytes are also reported to be susceptible to HIV-1 infection albeit at lower levels (Brack-Werner, 1999; Canki et al., 2001; Conant et al., 1994). Astrocytes are integral components of the CNS; they maintain a homeostatic environment and actively participate in a bidirectional communication with neurons (Brack-Werner, 1999; Dong and Benveniste, 2001; Hansson and Ronnback, 2003). Astrocytes share many receptors and functions with immune cells (Dong and Benveniste, 2001). Astrocytes express functional receptors for chemokines, neurotransmitters and voltage and ligand gated channels. They synthesize cytokines, chemokines, proteases and adhesion molecules (Dong and Benveniste, 2001; Hansson and Ronnback, 2003; Minagar et al., 2002; Sabri et al., 2003).
After initial infection with HIV-1, astrocytes exhibit a transient burst of viral replication that diminishes to low levels and remains at a persistent state of infection (Brack-Werner, 1999; Canki et al., 2001; Conant et al., 1994). It was reported that the number of astrocytes in the CNS ranges between 1×1011 to 2×1012 cells and in HIV-1 infected patients, up to 20% of these cells may be infected with HIV-1 and remain as reservoirs for latent HIV-1 (Canki et al., 2001). Upon activation, HIV-1 infected astrocytes may spread the infection or induce cellular damage to neighboring cells of the CNS through the release of viral and cellular products. Consequently, disruption of astrocyte function could lead to severe neuroimmunopathogenesis.
In 2004, according to the National Household Survey on Drug Abuse, 34.9 million Americans (aged 12 or older) had tried cocaine at least once in their lifetimes (SAMSHA, 2004). During the last decade, an intertwined epidemic of cocaine abuse and HIV-1 infections has emerged. Previous studies suggest that drug abuse, particularly with crack cocaine, is a risk factor for contracting HIV-1 infection and has been shown to be independently associated with progression to clinical AIDS (Centers for Disease Controls Task Force on Acquired Immune Deficiency Syndrome, 1983; Des Jarlais et al., 1987; Des Jarlais and Friedman, 1988; Li et al., 2005; Nath et al., 2001; Nath et al., 2002). Some AIDS patients develop severe neurological symptoms referred to as HIV-1 encephalopathy (HIVE). HIVE is characterized by multinucleated giant cells, microglial nodules and astrogliosis (Gendelman et al., 1994; Li et al., 2005; Nath et al., 2001). However, it is unclear whether ongoing drug abuse is a co-factor in HIV-1 disease progression to HIVE. Previous studies have shown that cocaine enhanced the replication of HIV-1 in peripheral blood leukocytes and microglial cell cultures (Bagasra and Pomerantz, 1993; Gekker et al., 2004; Peterson et al., 1991; Peterson et al., 1992; Peterson et al., 1993) supporting the hypothesis that cocaine is a co-factor in the neuropathogenesis of HIV-1 disease and associated HIVE. Because a significant number of astrocytes can be infected with HIV-1 in the CNS, and cocaine may act as a cofactor in HIVE, we hypothesize that cocaine-induced increases in HIV-1 susceptibility and progression to HIVE are mediated via dysregulation of specific proteins that foster the neuroimmunopathogenesis of HIV-1 infection.
Cocaine-induced differences in the protein profile of normal human astrocyte (NHA) cultures were investigated using the proteomic method of difference gel electrophoresis (DIGE). The identification of specific, cocaine-responsive proteins by proteomic analysis may identify novel bio-markers for diagnostic, preventive and therapeutic intervention in cocaine-using, HIV-1 seropositive populations.
2. Results
2.1. Cocaine enhances HIV-1 replication in NHA
Data presented in Fig. 1A show the stimulatory effects of cocaine on HIV-1 replication in NHA. NHA were treated with and without cocaine (10−10 to 10−6 M) for 48 h, infected with HIV-1 strain Ba-L, washed and cultured again for 48 h at which RNA was subsequently extracted. The HIV-LTR-R/U5 region was amplified by real-time Q-PCR using primers specific for a 180 bp fragment of the region as described (Secchiero et al., 2000). This method is designed to detect early stages of reverse transcription of HIV-1. Data shown in Fig. 1A demonstrate that cocaine at 10−8 (TAI=2.59, p=0.001) and 10−6 M (TAI=3.89, p=0.002) significantly upregulated HIV-LTR-R/U5 region compared to the untreated HIV-1-infected control culture (TAI=1.0).
Fig. 1.
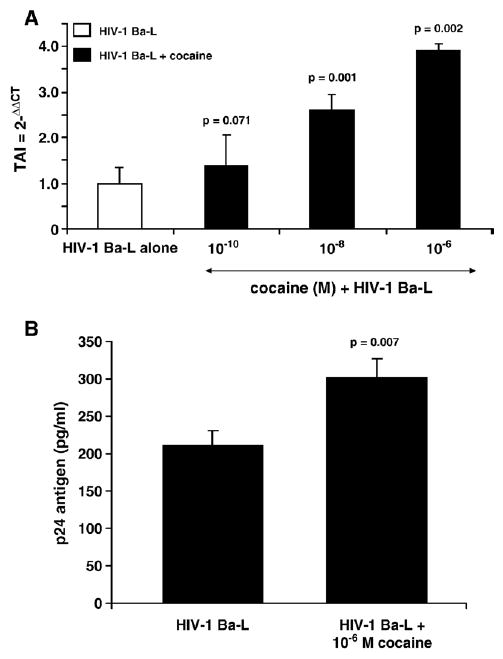
Effect of cocaine on HIV-1 replication in NHA. (A) NHA were treated with or without cocaine (48 h), infected with HIV-1 overnight, washed and cultured for 48 h. RNA was extracted and reverse transcribed followed by quantitative, real-time Q-PCR using primers specific for the LTR R/U5 region of the HIV-1 genome. Data are presented as the mean±SD of 3 independent experiments. Statistical significance was calculated by Student's 't'-test. (B) NHA were treated with or without cocaine (10−6 M, 48 h), infected overnight, washed and cultured for 15 days. The culture supernatants were quantitated for p24 antigen using Retrotek, HIV-1 p24 ELISA from ZeptoMetrix (Cat # 0801111, Buffalo, NY) with a minimum detection range of 1 pg p24 antigen/ml. Data are presented as the mean±SD of 3 independent experiments from 3 separate NHA cultures. Statistical significance was calculated by Student's t-test.
In subsequent experiments, NHA were treated with and without cocaine for 48 h, infected with HIV-1 strain Ba-L washed and cultured again for 15 days when levels of p24 antigen in culture supernatants were measured. Data presented in Fig. 1B show the levels of p24 Antigen in the culture supernatants of NHA infected with HIV-1 in the presence or absence of cocaine (10−6 M). NHA infected with HIV-1 Ba-L in the absence of cocaine produced 210 pg/ml of p24 antigen whereas NHA infected with HIV-1 Ba-L in the presence of cocaine significantly upregulated the production of p24 antigen (304 pg/ml, p=0.007). Although NHA showed only low level of infection with HIV-1 Ba-L which is consistent with other studies (Brack-Werner, 1999; Canki et al., 2001), the production of p24 antigen was significantly upregulated by cocaine and thus supports the LTR R/U5 amplification as presented in Fig. 1A. These results show direct evidence that cocaine upregulates HIV-1 infection in NHA.
2.2. Cocaine differentially induces the expression of several proteins in NHA
Data presented in Fig. 2 show differences in protein expression between cocaine treated and untreated NHA cultures. Several protein spots were identified that were differentially expressed. The protein spots were excised from the gel and identified by HPLC-MS/MS. Table 1 lists 22 proteins that were identified by HPLC-MS/MS. The protein spots that were identified by HPLC-MS/MS that had increased expression levels in NHA treated with cocaine compared to untreated control were: chaperonin containing TCP1 (37.51% upregulation, p=0.005); IMPDH/pyruvate kinase (23.72% upregulation, p=0.008); enolase/sorting nexin 5 (16.33% upregulation, p=0.002); phosphoglycerate kinase I (20% upregulation, p = 0.009); AA (25.71% upregulation, p=0.006); malate dehydrogenase/endothelial monocyte-activating protein II (18.26% upregulation, p = 0.004); and S100A13 (32.29% upregulation, p=0.002). Several protein spots that showed a significant decrease in expression in NHA treated with cocaine compared to untreated control NHA were: DnaJ (Hsp40)/galactokinase 1 (19.31% down-regulation, p=0.021); hRNPC/serine–threonine kinase receptor-associated protein (31.31% downregulation, p=0.001); B23/hepatoma-derived growth factor (40.53% downregulation, p=0.001); EF1/HSP60 (29.24% downregulation, p=0.013); annexin I/nuclear ribonucleoprotein homolog JKTBP (31.39% downregulation, p=0.001); and aldolase C (24.36% down-regulation, p=0.004).
Fig. 2.
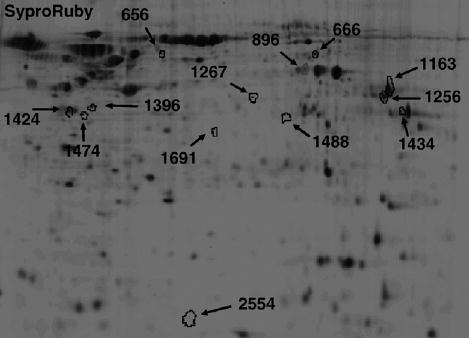
Two-dimensional analyses of cocaine-induced differentially expressed proteins in NHA cells. Cells were treated with 10−6 M cocaine for 48 h. Total protein was isolated and subjected to DIGE analyses as described in Experimental procedures (n=3 independent experiments). Data are shown as a representative 2D SYPRO-Ruby stained gel image of cocaine (10−6 M, 48 h) treated NHA cells. Arrows represent statistically significant differentially expressed proteins (Student's t-test.). The pH increases from left to right and the molecular mass decreases from the top to the bottom of the gels.
Table 1.
Differential proteins induced by cocaine in NHA
| Spot # | Protein name | % change | p value | Function |
|---|---|---|---|---|
| Upregulated proteins | ||||
| 656 | Chaperonin containing TCP1, subunit 5 | 37.51±0.03 | 0.005 | Molecular chaperone: protein folding |
| 666 | Inosine-5′-monophosphate dehydrogenase 2 (IMPDH) | 23.72±3.77 | 0.008 | GMP biosynthesis Regulation of cell growth. |
| 666 | Pyruvate kinase | 23.72±3.77 | 0.008 | Glycolysis |
| 896 | Enolase 1 | 16.33±1.09 | 0.002 | Glycolysis |
| 896 | Sorting nexin 5 | 16.33±1.09 | 0.002 | intracellular trafficking |
| 1163 | Phosphoglycerate kinase I | 20.0±0.38 | 0.009 | Glycolysis |
| 1256 | Aldolase A (AA) | 25.71±3.37 | 0.006 | Glycolysis |
| 1256 | Fructose-1,6-(bis)phosphate aldolase covalently bound to the substrate dihydroxyacetone phosphate | 25.71±3.37 | 0.006 | Glycolysis intermediate |
| 1434 | Malate dehydrogenase | 18.26±1.91 | 0.004 | Citric acid cycle |
| 1434 | Endothelial monocyte-activating protein II | 18.26±1.91 | 0.004 | Cytokine |
| 2554 | S100 calcium binding protein A13 (S100A13) | 32.29±2.79 | 0.002 | Calcium binding |
| Downregulated proteins | ||||
| 1267 | DnaJ (Hsp40) homolog | 19.31±0.92 | 0.021 | Molecular chaperone: protein folding |
| 1267 | Galactokinase 1 | 19.31±0.92 | 0.021 | Galactose metabolism |
| 1396 | Heterogeneous nuclear ribonucleoprotein C (hRNPC) | 31.31±1.21 | 0.001 | RNA binding protein |
| 1396 | Serine–threonine kinase receptor-associated protein | 31.31±1.21 | 0.001 | Cell cycle regulator |
| 1424 | Nucleolar phosphoprotein B23 (B23) | 40.53±2.65 | 0.001 | Molecular chaperone |
| 1424 | Hepatoma-derived growth factor (HDGF) | 40.53±2.65 | 0.001 | Growth factor |
| 1474 | Eukaryotic translation elongation factor 1 (EF1) | 29.24±5.92 | 0.013 | Guanine-nucleotide exchange factor |
| 1474 | 60 kDa heat shock protein (HSP60) | 29.24±5.92 | 0.013 | Molecular chaperone |
| 1488 | Annexin I | 31.39±1.59 | 0.001 | Ca2+-dependent phospholipid binding protein |
| 1488 | Nuclear ribonucleoprotein homolog JKTBP | 31.39±1.59 | 0.001 | mRNA biogenesis |
| 1691 | Aldolase C | 24.36±2.69 | 0.004 | Glycolysis |
To confirm that the changes in cocaine-induced protein expression by NHA as identified by DIGE were specific, the expression of select proteins was further examined by western blot and quantitated by densitometry (Fig. 3). Cocaine treatment had no effect on β-actin expression. Fig. 3A demonstrates a 29.28% upregulation in the protein expression of AA (129.28±2.12, p<0.01) induced by treatment of NHA with cocaine compared to the untreated control (100). Fig. 3B demonstrates that cocaine induces a 12.65% upregulation of protein expression for enolase by NHA (112.65±4.05, p=0.05) compared to the untreated control. Fig. 3C demonstrates a 19.6% downregulation of protein expression for B23 (81.40±61, p=0.05) induced by treatment of NHA with cocaine while Fig. 3D demonstrates a 23.23% downregulation of protein expression for HSP60 (76.77±7.7, p<0.01) induced by treatment of NHA with cocaine compared to the untreated control. These results are consistent with the effects of cocaine on protein expression by NHA as determined by DIGE (Table 1).
Fig. 3.
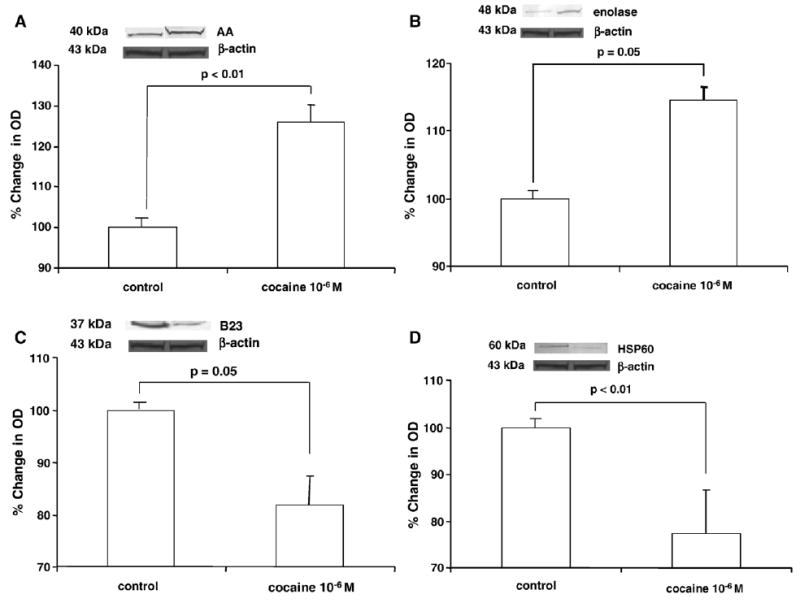
Western blot analyses of differentially expressed proteins. Cells were treated with 10−6 M cocaine for 48 h. Total protein was isolated and subjected to western blot analyses as described in Experimental procedures. Densitometry analyses were done using a Syngene Image Analyzer with Gene Tools Analysis Software version 3.02.00 (Syngene, Frederick, MD). Data were normalized to levels of β-actin (control OD=1000 units; treated=1015 units). The graphs show the mean±SD of % change in OD as measured by densitometry of bands from western blots from 5 independent experiments. Note: Graphs are truncated. Statistical significance was determined by Student's t-test. (A) Representative western blot for AA. AA migrates as a 40 kDa protein. (B) Representative western blot for enolase. Enolase migrates as a 48 kDa protein. (C) Representative western blot for B23. B23 migrates as a 37 kDa protein. (D) Representative western blot for HSP60. HSP60 migrates as a 60 kDa protein.
2.3. Cocaine modulates differential gene expression in NHA
To associate the differential expression of cocaine-induced protein modulation with respective mRNA expression, we investigated the effect of cocaine on mRNA levels of selected proteins by Q-PCR analyses. NHA were cultured for 48 h with 10−6 M cocaine, RNA was extracted, reverse transcribed and cDNA was amplified by Q-PCR using primers shown in Table 2. Data presented in Fig. 4 show that cocaine treatment had no effect on 18s RNA expression which is used as control RNA. However, cocaine upregulates the gene expression of IMPDH (TAI=2.41±0.57, 141% upregulation, p<0.01); enolase (TAI=1.55±0.07, 55% upregulation, p<0.001); AA (TAI=1.55± 0.34, 55% upregulation, p<0.01); and S100A13 (TAI=1.30±0.11, 30% upregulation, p<0.01) compared to the respective untreated control culture (TAI=1.00). Similarly, cocaine significantly downregulated the gene expression of DNAJ (Hsp40) homolog (TAI=0.51± 0.097, 49% downregulation, p<0.01); hRNPC (TAI=0.53±0.11, 47% downregulation, p<0.001); B23 (TAI=0.73±0.12, 27% downregulation, p<0.001); EF1 (TAI=0.40±0.07, 60% downregulation, p<0.001); HSP60 (TAI=0.42±0.07, 58% downregulation, p < 0.001); and annexin I (TAI = 0.54 ± 0.10, 46% downregulation, p<0.01) compared to the respective untreated control culture (TAI=1.0). These data suggest that changes in protein expression levels are reflective of changes at the mRNA level.
Table 2.
Primer sequences for real-time Q-PCR
| Primer | PCR product size (bp) | Primer sequences |
|---|---|---|
| β-actin | 548 | 5′, 5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′
3′, 5-AGTCATAGTCCGCCTAGAAGCATTTGCGGT-3′ |
| HIV-1 LTR RU/5 | 180 | 5′, 5′-TCT CTC TGG TTA GAC CAG ATC TG-3′
3′, 5′-ACT GCT AGA GAT TTT CCA CAC TG-3′ |
| IMPD2 | 118 | 5′, 5′-TCTGCATTACGCAGGAAGTG-3′
3′, 5′-TTTTGGATTCCTCCATCAGC-3′ |
| Enolase 1 | 597 | 5′, 5′-ATAAAGAAGGCCTGGAGCTGCTGA-3′
3′, 5′-TGCCCAGCTCCTCTTCAATTCTGA-3′ |
| AA | 341 | 5′, 5′-AACATGACCCACCTGTCCATGCTA-3′
3′, 5′-TGGATATTGGTAGGGCATGGTGCT-3′ |
| DnaJ (HSP40) | 295 | 5′, 5′-TGGGGATTTTGGTTTCATGT-3′
3′, 5′-CAGCGTTCGTTCTTCATTCA-3′ |
| hRNPC | 254 | 5′, 5′-AGAGGCTGAGGAAGGAGAGG-3′
3′, 5′-AGGAGCGTCAAAGGAAGTGA-3′ |
| B23 | 269 | 5′, 5′-GTTCAGGGCCAGTGCATATT-3′
3′, 5′-TTTCTTCACTGGCGCTTTTT-3′ |
| EF1 | 200 | 5′, 5′-ACAAGGTGGGGACAGACTTG -3′
3′, 5′-TTTTTGCCGGTCTCAGTCTT-3′ |
| HSP60 | 651 | 5′ 5′-TACTGGTGGTGCAGTGTTTGGAGA-3′
3′ 5′-GCCACACCAGCAGCATCCAATAAA-3′ |
| Annexin I | 203 | 5′, 5′-GCAGGCCTGGTTTATTGAAA-3′
3′, 5′-GCTGTGCATTGTTTCGCTTA-3′ |
| S100A13 | 128 | 5′, 5′-AAGGATAGCCTCAGCGTCAA-3′
3′, 5′-TTGAGCTCCGAGTCCTGATT-3′ |
| ERK2 | 186 | 5′, 5′-CCACCCATATCTGGAGCAGT-3′
3′, 5′-AGGACCAGGGGTCAAGAACT-3′ |
Fig. 4.
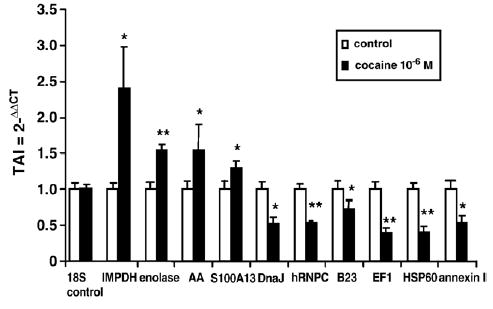
Cocaine-modulation of gene expression. NHA were treated with 10−6 M cocaine for 48 h. Total RNA was isolated and subjected to Q-PCR. Gene expression for 18S control RNA, Inosine-5′-monophosphate dehydrogenase 2 (IMPDH), enolase, aldolase A (AA), S100 calcium binding protein A13 (S100A13), DnaJ (Hsp40) homolog (DNAJ), heterogeneous nuclear ribonucleoprotein C (hRNPC), nucleolar phosphoprotein B23 (B23), eukaryotic translation elongation factor 1 (EF1), heat shock protein 60 (HSP60) and annexin I were determined using Q-PCR. Data are presented as the mean±SD of 3 independent experiments. Statistical significance was calculated by Student's t-test. *p<0.01; **p<0.001.
2.4. Cocaine modulates extracellular signal related kinase (ERK)
Signal transduction via mitogen-activated protein kinases (MAP kinases) plays a significant role in cellular immune responses. Therefore, we investigated the effect of cocaine on activation and gene expression of ERK2 as a possible mechanism of protein regulation. Data shown in Fig. 5A demonstrate that cocaine (10−6 M) increased the phosphorylation of ERK2 in a time-dependent manner, with the maximum level of phosphorylation peaking at 15 min and decreasing to levels similar to control by 30 and 60 min. Furthermore, data shown in Fig. 5B demonstrate that at 24 h, cocaine at 10−8 (TAI=1.51, p=0.008) and 10−6 M (TAI=2.38, p=0.003) significantly increased gene expression for ERK2 compared to the untreated control (TAI=1.0) as determined by Q-PCR. Similarly, cocaine at 10−8 (TAI=1.57, p =0.002) and 10−6 M (TAI=2.11, p =0.009) significantly increased gene expression for ERK2 compared to the untreated control (TAI=1.0) at 48 h. These results show that cocaine modulates the signal transduction molecule ERK2.
Fig. 5.
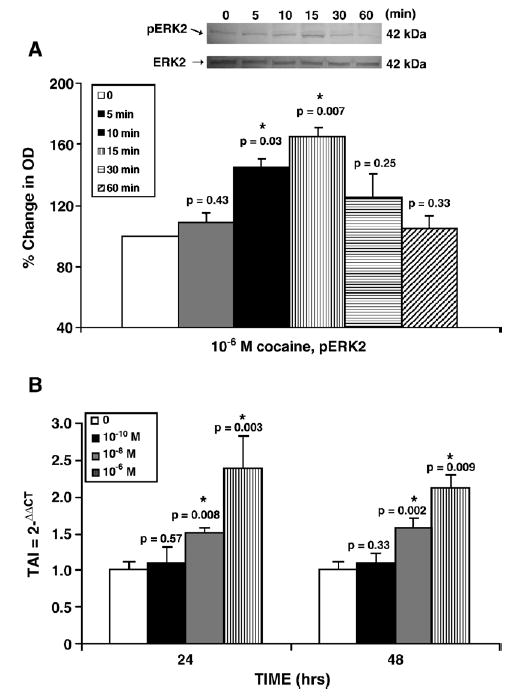
Cocaine modulation of ERK2. (A) Representative western blot of cocaine (10−6 M) on pERK2 at various times (min) compared to control; Graph represents % change in OD of pERK2, where control is 100%. Data are normalized to total ERK2 levels. (B) Total RNA was extracted and ERK2 gene expression was analyzed using Q-PCR. Data are represented as mean±standard deviation. Statistical significance was calculated by Student's t-test.
3. Discussion
It is well established that parenteral drug abuse is a significant risk factor for contracting HIV-1 infection and subsequently developing AIDS (Centers for Disease Controls Task Force on Acquired Immune Deficiency Syndrome, 1983; Des Jarlais et al., 1987; Des Jarlais and Friedman, 1988; Nath et al., 2001; Nath et al., 2002). With progression of HIV-1, infections patients subsequently can develop HIVE (Gendelman et al., 1994; Li et al., 2005). Previous studies have shown that drugs of abuse such as cocaine enhance the replication of HIV-1 in vitro (Bagasra and Pomerantz, 1993; Gekker et al., 2004; Nair et al., 2005; Peterson et al., 1991; Peterson et al., 1992; Peterson et al., 1993), supporting the hypothesis that drug abuse is a co-factor in the pathogenesis of HIV-1 disease. The current study demonstrates that pretreatment of NHA with cocaine prior to HIV-1 infection enhances viral replication as demonstrated by a significant increase in LTR–R/U5 gene expression as well as increased p24 antigen levels compared to the non-cocaine treated HIV-1 infection control. These studies demonstrate that cocaine potentiates HIV-1 replication in NHA and possibly acts as a co-factor in HIV-1 mediated neuropathogenesis.
The mechanisms and the role of cocaine abuse underlying the neuropathogenesis of HIV-1 infections remain undetermined. Since astrocytes are integral components of the CNS that can be infected with HIV-1 and cocaine acts as a cofactor in the pathogenesis of HIVE, it is reasonable to suggest that several proteins may be involved in the neuropathogenesis of HIVE. Our results show that the expression of several proteins was differentially regulated by cocaine. These proteins were identified by HPLC-MS/MS. Below we discuss the potential relevance of the differential expression of various cocaine regulated proteins to HIV-1 infection of NHA.
Our studies show that the expression of several intracellular signaling molecules in NHA was differentially regulated by cocaine. These include: sorting nexin 5, an intracellular regulator of endocytosis; serine–threonine kinase receptor-associated protein, a cell cycle regulator; endothelial monocyte-activating protein II, a cytokine; annexin I, a Ca2+-dependent phospholipid binding protein; hepatoma-derived growth factor; and S100A13, a calcium binding protein. Previous studies show that the anti-allergy drugs amlexanox, cromolyn and tranilast bind to S100A13 and S100A12 to inhibit degranulation of mast cells exerting an anti-inflammatory response (Shishibori et al., 1999). Our study demonstrates that cocaine upregulates S100A13 in NHA, suggesting that cocaine may inhibit the inflammatory response in NHA potentially decreasing the local immune response against HIV-1 infection.
Eukaryotic translation elongation factor 1 (EF1) is responsible for the enzymatic delivery of aminoacyl tRNAs to the ribosome. Cimarelli et al. demonstrated that EF1 interacts with HIV-1 matrix protein and binds to the entire HIV-1 Gag polyprotein suggesting that these interactions may play a role in releasing viral RNA from polysomes, thereby permitting the RNA to be packaged into virions (Cimarelli and Luban, 1999). Our findings demonstrate that EF1 protein expression in NHA is decreased by cocaine. While this observation is somewhat counterintuitive to our hypothesis that cocaine increases the pathogenesis of HIV-1 infections, it is well known that various proteins have dual functions. For example, the signaling molecule JNK can be either pro- or anti-apoptotic depending on cell type, and stimulus (Liu and Lin, 2005). Furthermore, MCP-1 enhances transmigration of HIV-1 infected leukocytes across the blood–brain barrier by increasing its permeability allowing for infiltration of the CNS with HIV-1 infected leukocytes (Eugenin et al., 2006). MCP-1 also has been shown to be neuroprotective, inhibiting NMDA and tat-induced apoptosis in astrocytes and neurons (Eugenin et al., 2003). Nevertheless, it further demonstrates that cocaine can modulate the expression of host proteins associated with HIV-1 infections. However, the role of EF1 protein in the context of HIVE remains to be delineated.
Our current study also demonstrates that several molecular chaperones including TCP1, DNAJ (HSP40) homolog, nucleolar phosphoprotein B23 and HSP60 are differentially regulated. Heat shock proteins (HSP) also known as stress proteins, belong to the family of molecular chaperones that can be induced upon cellular injury. Heat shock proteins are classified by their molecular weight, comprising six general families: HSP110, HSP90, HSP70, HSP60, small molecular weight HSPs and immunophilins (Gullo and Teoh, 2004; Prohaszka and Fust, 2004). Studies show that HSPs have diverse functions including chaperone activity, regulation of the redox state and modulation of protein turnover (Fust et al., 2005; Prohaszka and Fust, 2004). HSP60 is a mitochondrial chaperonin, highly preserved during evolution, and is responsible for protein folding and assembly of newly imported mitochondrial proteins (Prohaszka and Fust, 2004). HSP60 may function as a signaling molecule in the innate immune system. HSP60 induces IL-1 production and secretion in macrophage (Retzlaff et al., 1994). In primary human astrocytes, IL-1β, TNFα, IL-4, IL-6 and IL-10 increase the expression of HSP60 (Bajramovic et al., 2000). Furthermore, acute cocaine treatment (5 h) increased immunostaining for HSPs (HSP27, HSP60, HSP70, HSC70) in hippocampal neurons and decreased immunostaining for HSP60 at 48 h (Hayase 2003a,b). Moreover, previous studies show that HSP60 and HSP70 are incorporated into the membrane of HIV-1 virions through an interaction with HIV-1 Gag (Gurer et al., 2002). Findings from our current study support the previous observation that cocaine decreases HSP60 protein expression. The clinical significance of HSP incorporation into HIV-1 virions and the observation that cocaine decreases HSP60 expression has yet to be elucidated. Nucleolar phosphoprotein B23 functions similar to a molecular chaperone by importing ribosomal proteins to the nucleus (Fankhauser et al., 1991; Szebeni et al., 1997). B23 binds to Rev and acts as a chaperone for its import to the nucleus or nucleolus (Fankhauser et al., 1991). Furthermore, B23 is a factor in the nuclear localization of Tat (Li, 1997) and is spatially associated with Tat in the cytoplasm, nucleolus, and plasma membrane (Marasco et al., 1994). Our study finds that cocaine decreases the expression of B23 in NHA suggesting that cocaine can modulate the expression of host proteins associated with the transport of ribosomal proteins to the nucleus.
Replication of HIV-1 depends on host cell production of deoxynucleoside triphosphates. Inosine monophosphate dehydrogenase (IMPDH) is involved in the de novo synthesis of purines. IMPDH is the first enzyme in the conversion of IMP to GMP which is subsequently converted to GDP, GTP and dGTP (Borroto-Esoda et al., 2004). A reduction in the intracellular levels of GTP has been shown with inhibitors of IMPDH (Lowe et al., 1977; Yalowitz and Jayaram, 2000). An inhibitor of IMPDH, mycophenolic acid suppresses HIV-1 replication in lymphocytes and monocytes by reducing dGTP levels (Borroto-Esoda et al., 2004; Chapuis et al., 2000; Ichimura and Levy, 1995). The findings from our study that the levels of IMPDH are increased in NHA treated with cocaine, suggest that cocaine, by increasing the levels of IMPDH, may allow for increased production of dGTP and increased viral replication once NHA become infected with HIV-1.
MAP kinases are used to transduce extracellular signals into intracellular responses. MAP kinases regulate cellular activities from proliferation and differentiation, to gene expression, mitosis, movement and metabolism as well as programmed cell death (Johnson and Lapadat, 2002). HIV-1 replication and viral entry in host cells have been associated with MAP kinases. Activation of ERK1/2 was required for viral entry into brain microvascular endothelial cells (Liu et al., 2002). Viral assembly and release were regulated by ERK2 in 293T cells (Hemonnot et al., 2004). Moreover, recent evidence indicates that cocaine activates MAP kinases to induce cellular changes. Acute cocaine administration increased the phosphorylation of ERK in the mouse caudoputamen and striatum (Berhow et al., 1996; Valjent et al., 2000). The DAT (dopamine transporter) protein is the primary target for cocaine in the brain. Investigation of cellular and molecular mechanisms associated with DAT function and regulation indicate that several protein kinases including ERK, interact with DAT and that these interactions might be important in the regulation of transporter function (Beaulieu et al., 2006). ERKs are essential component of a signaling pathway involved in synaptic plasticity and in long-term effects of drugs of abuse. Our study shows a direct association between cocaine treatment and activation of ERK2 and we speculate that these ERK kinases interact with the DAT protein thereby regulating its function. This study suggests that through the activation of MAP kinases, cocaine may increase the susceptibility of NHA to HIV-1 infection. Further studies are necessary to demonstrate the role of MAP kinases in cocaine-induced increases in HIV-1 infection of NHA.
In summary, using proteomic analyses, we were able to identify 22 proteins in NHA that were differentially regulated by cocaine which directly or indirectly play a supportive role in the neuropathogenesis of HIV-1 infection. Unique, cocaine-responsive proteins may be potential bio-markers or targets for use in the diagnosis, prevention and/or therapy of HIVE in cocaine-using, HIV-1 infected patients.
4. Experimental procedures
4.1. Normal human astrocyte (NHA) cultures
Primary NHA cultures isolated from human fetal brain cortex tissue (independent cultures) were obtained from Cell Systems (Kirkland, WA). NHA cells were grown in complete Astrocyte Basal Medium (Cell Systems) as recommended by the manufacturer. Astrocytes were added to 6 well tissue culture plates at densities of 1×106 cells/ml in DMEM+10% FBS. By immunocytochemistry, astrocytes were >95% GFAP positive and were >98% viable by trypan blue exclusion criteria.
4.2. Drug treatment
NHA were treated with and without cocaine-hydrochloride (Sigma-Aldrich, St. Louis MO); time and concentration used were based on previous studies that produced a maximum biological response without causing toxicity to the target cells (Nair et al., 2000; Nair et al., 2001; Nair et al., 2004; Nair et al., 2005). Cocaine was dissolved in sterile distilled water and was subsequently diluted in media to the required concentrations. For all experiments, cells treated with vehicle alone (media alone) were used as untreated controls. Kinase kinetic studies: NHA (1×106 cells/ml) were plated overnight and then incubated with cocaine in serum free media for various times (0, 5, 10, 15, 30 and 60 min and 24 and 48 h) at different concentrations as indicated.
4.3. Treatment of human NHA with HIV-1
NHA (1×106 cells/ml) were treated with cocaine for 48 h and then infected with native HIV-1 Ba-L (NIH AIDS Research and Reference Reagent Program, Cat # 510) at a concentration of 103.5 TCID50/ml cells, overnight and washed 3 times with Hank's balanced salt solution (Invitrogen) before being returned to culture. A post infection period of 48 h was used in the study to amplify the LTR-R/U5 region which represents early stages of reverse transcription of HIV-1 (Secchiero et al., 2000). In separate experiments, NHA were treated with cocaine for 48 h, infected with HIV-1 Ba-L overnight and washed, cultured for 15 days. The culture supernatants were quantitated for p24 antigen using a p24 ELISA kit (ZeptoMetrix Corporation, Buffalo, NY) on day 15.
4.4. RNA extraction and real-time quantitative PCR (Q-PCR)
NHA treated with and without cocaine were washed with 1× PBS (Invitrogen) and cytoplasmic RNA was extracted using an acid guanidinium–thiocyanate–phenol–chloroform method (Chomczynski and Sacchi, 1987). The final RNA pellet was dried and resuspended in diethyl pyrocarbonate (DEPC) water and the concentration of RNA was determined using a spectrophotometer at 260 nm. DNA contamination in the RNA preparation was removed by treating the RNA preparation with DNAse (1 IU/μg of RNA, Promega, Madison, WI) for 30 min at 37 °C, followed by proteinase K digestion at 37 °C for 15 min and subsequent extraction with phenol/chloroform and NH4OAc/ETOH precipitation. DNA contamination of the RNA preparation was checked by including a control in which reverse transcriptase enzyme was not added in the PCR amplification procedure. The isolated RNA was stored at −70 °C until used. Gene expressions for LTR R/U5, inosine-5′-monophosphate dehydrogenase 2 (IMPDH), enolase, aldolase A (AA), DnaJ (Hsp40) homolog, heterogeneous nuclear ribonucleoprotein C (hRNPC), nucleolar phosphoprotein B23 (B23), eukaryotic translation elongation factor 1 (EF1), 60 kDa heat shock protein (HSP60), annexin I, S100 calcium binding protein A13 (S100A13), extracellular signal related kinase (ERK2) and β-actin were quantitated using Q-PCR. QuantumRNA Universal 18S Internal Standard (Ambion, Austin, TX) was used as a control. Relative abundance of each mRNA species was assessed using the SYBR green master mix from Stratagene (La Jolla, CA) to perform Q-PCR using the ABI Prism 5700 instrument that detects and plots the increase in fluorescence versus PCR cycle number to produce a continuous measure of PCR amplification. To provide precise quantification of initial target in each PCR reaction, the amplification plot is examined at a point during the early log phase of product accumulation. This is accomplished by assigning a fluorescence threshold above background and determining the time point at which each sample's amplification plot reaches the threshold (defined as the threshold cycle number or CT). Differences in threshold cycle number are used to quantify the relative amount of PCR target contained within each tube (Shively et al., 2003). Relative mRNA species expression was quantitated and expressed as transcript accumulation index (TAI=2−ΔΔCT), calculated using the comparative CT method (Mahajan et al., 2003). All data were controlled for quantity of RNA input by performing measurements on an endogenous reference gene, β-actin. In addition, results with RNA from treated samples were normalized to results obtained with RNA from the control, untreated sample.
4.5. DIGE
After stimulation, cells were washed 2 times with 1× PBS (Invitrogen, Grand Island, NY). Total protein was extracted using standard cell lysis buffer [30 mM Tris–Cl; 8 M Urea; 4% (w/v) CHAPS, adjusted to pH 8.5] for 10 min on ice. Cell lysate was centrifuged at 4 °C for 10 min at 12,000×g and lysate was further purified by precipitation with chloroform/methanol as described (Wessel and Flugge, 1984). Samples were resus-pended in standard cell lysis buffer. Final cell lysate protein concentrations were determined using Coomassie Protein Reagent (Bio-Rad, Hercules Ca) and used for protein determination by DIGE analysis.
Ettan DIGE technique developed by Amersham Pharmacia Biotech (http://www1.amershambiosciences.com/aptrix/upp00919.nsf/Content/Proteomics+DIGE+Protocols) was used to detect differences in protein abundance between normal and experimental samples. The Ettan DIGE system uses three CyDye DIGE fluors (Cy2, Cy3, Cy5), each with a unique fluorescent wavelength, matched for mass and charge. CyDyes form a covalent bond with the free epsilon amino group on lysine residues of the sample proteins. CyDyes label approximately 2% of the lysine residues. This system allows for two experimental samples and an internal standard to be simultaneously separated on the same gel. The internal standard is comprised of a pool of an equal amount of all the experimental samples. The use of an internal standard facilitates accurate inter-gel matching of spots, and allows for data normalization between gels to minimize gel to gel experimental variability (http://www1.amershambiosciences.com/aptrix/upp00919.nsf/Content/Proteomics+DIGE+Protocols; Tonge et al., 2001).
Cell lysates were labeled with CyDye per the manufacturer. All reagents used were from GE Healthcare (Amersham Biosciences, Piscataway, NJ). Briefly, 50 μg of cell lysate was labeled with 400 pmol of either Cy3 or Cy5 or Cy2 (Cy2 was used to label the internal standard) on ice for 30 min and then quenched with a 50-fold molar excess of free lysine. Cy3, Cy5 and Cy2 labeled samples and unlabelled protein (500–800 μg) were then pooled. An equal volume of 2× sample buffer was added [8 M Urea; 2% (v/v) Pharmalytes 3–10; 2% (w/v) dithiothreitol (DTT); 4% (w/v) CHAPS] incubated on ice for 10 min. The total volume of sample was adjusted to 450 μl with rehydration buffer [4% (w/v) CHAPS; 8 M Urea; 1% (v/v) Pharmalytes 3–10 nonlinear (NL); 13 mM DTT]. Samples were applied to immobilized pH gradient (IPG) strips (24 cm, pH 3–10 NL), and absorbed by active rehydration at 30 V for 13 h. Isoelectric focusing was carried out using an IPGphor IEF system with a three phase program; first phase at 500 V for 1 h, second phase at 1000 V for 1 h and third phase (linear gradient) 8000 V to 64000 V for 2 h (50 μA maximum per strip). Prior to separation in the second dimension, strips were equilibrated for 15 min in equilibration buffer I [50 mM Tris–HCl, 6 M Urea, 30% (v/v) glycerol, 2% (w/v) SDS, 0.5% (w/v) DTT]. The strips were again equilibrated for 15 min in the equilibration buffer II [50 mM Tris–HCl, 6 M Urea, 30% (v/v) glycerol, 2% (w/v) SDS, 4.5% (w/v) iodoacetamide] and the equilibrated IPG strips were then transferred onto 18×20 cm, 12.5% uniform polyacrylamide gels poured between low fluorescence glass plates. Gels were bonded to inner plates using Bind-Silane solution according to the manufacturer. Strips were overlaid with 0.9% agarose in 1× running buffer containing bromophenol blue and were run for 16 h (1.8 W per gel, overnight) at 15 °C, in Ettan DALT electrophoresis system. After the run was completed, the 2D gels were scanned three times with a Typhoon 9410 imager, each time at different excitation wavelengths. Images were cropped in ImageQuant v5.2, and then imported into DeCyder Differential In-gel Analysis (DIA) software v5.0 from GE Healthcare for spot identification, and normalization of spot intensities within each gel.
Gels were fixed in 30% (v/v) methanol, 7.5% (v/v) acetic acid for 3 h and then stained with SYPRO-Ruby dye (Molecular Probes, Eugene, OR) overnight at room temperature. Gels were destained in water and then scanned using the Typhoon 9410 scanner. Spots of interest were excised from the gel using the Ettan Spot Picker.
The DeCyder software (GE Healthcare) was specifically developed for use with Ettan DIGE. The DeCyder software allows for automatic detection of spots, background subtraction, quantitation, normalization, internal standardization and integral matching. DeCyder, DIA draws boundaries around spots in a composite gel image obtained from the intra-gel overlap of the Cy2, Cy3 and Cy5 scanned images, and normalizes the data from each CyDye to account for differences in dye fluorescence intensity, scanner sensitivity. The abundance difference between samples run on the same gel is then analyzed. The biological variation analysis (BVA) component of DeCyder is then used to match all image comparisons from in-gel analysis for a cross-gel statistical analysis. DeCyder BVA initially calculates normalized intensities ("standard abundance") for all spots by comparison to the internal standard, and from this an average volume ratio and a Student's paired t-test derived p value are calculated for each spot. A paired t-test derived p value of ≤ 0.05 was considered statistically significant (http://www1.amershambiosciences.com/aptrix/upp00919.nsf/Content/Proteomics+DIGE+Protocols; Tonge et al., 2001).
4.6. HPLC-MS/MS
Excised spots were sent to the Proteomic Analysis Laboratory at the University of Arizona for analysis. In-gel digestion and HPLC-MS/MS were performed as described by Breci et al., 2005 (Breci et al., 2005). Briefly, gel slices were destained (Breci et al., 2005; Cooper et al., 2003; Gharahdaghi et al., 1999) and digested with trypsin (Wilm et al., 1996). The tryptic peptides were extracted with 5% formic acid/50% CH3CN. HPLC was performed using a microbore system (Surveyor, ThermoFinnigan, San Jose, CA). The HPLC column eluate was directed into a ThermoFinnigan LCQ Deca XP Plus ion trap mass spectrometer. Automated peak recognition, dynamic exclusion and daughter ion scanning of the two most intense ions were performed using Xcalibur software (Andon et al., 2002; Haynes et al., 1998). Spectra were scanned over the range 400–1400 mass units. MS/MS data are analyzed using SEQUEST software and searched against the latest version of the National Center for Biotechnology's public non-redundant protein database (Breci et al., 2005).
4.7. Western blot
Briefly, 40 μg of protein was separated by electrophoresis using 4–20% Tris–glycine Express gels (ISC Bioexpress, Kaysville, UT) and transferred to polyvinylidene fluoride (PVDF) membranes (Sigma-Aldrich). Membranes were blocked for 2 h with 5% nonfat dry milk in Tris-buffered saline with Tween 20 [150 mM NaCl, 20 mM Tris, pH 7.5, 0.1% Tween 20], and then incubated with primary antibodies overnight at 4 °C, with gentle rocking. The primary antibodies used were anti-B23 mouse monoclonal, anti-enolase rabbit polyclonal, anti-aldolase A goat polyclonal, anti-HSP60 mouse monoclonal, anti-phosphorylated ERK2 mouse monoclonal, anti-ERK2 mouse monoclonal and anti-actin goat polyclonal (Santa Cruz Biotech, Santa Cruz CA). Antibody concentrations used were based on the manufacturer's specifications. After incubation with primary antibodies, membranes were washed and incubated with a biotin-conjugated secondary antibody (goat anti-mouse IgG or donkey anti-goat IgG or goat anti-rabbit IgG, Santa Cruz Biotech). After secondary antibody incubations, the membranes were washed 3 times, for 10 min each, in 1× TBS with 0.5% Tween 20 and then incubated for another 30 min with a streptavidin–alkaline phosphatase conjugate (Invitrogen) followed by colorimetric detection using NBT/BCIP reagent (Roche, Indianapolis, IN). Densitometry analyses were done using a Syngene Image Analyzer with Gene Tools Analysis Software version 3.02.00 (Syngene, Frederick, MD). Data were normalized to levels of β-actin.
4.8. Statistics
Statistical significance was determined using Student's t-test (Sigmastat, SPSS, Inc). p ≤ 0.05 was considered significant.
References
- Andon NL, Hollingworth S, Koller A, Greenland AJ, Yates JR, III, Haynes PA. Proteomic characterization of wheat amyloplasts using identification of proteins by tandem mass spectrometry. Proteomics. 2002;2:1156–1168. doi: 10.1002/1615-9861(200209)2:9<1156::AID-PROT1156>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Bagasra O, Pomerantz RJ. Human immunodeficiency virus type 1 replication in peripheral blood mononuclear cells in the presence of cocaine. J Infect Dis. 1993;168:1157–1164. doi: 10.1093/infdis/168.5.1157. [DOI] [PubMed] [Google Scholar]
- Bajramovic JJ, Bsibsi M, Geutskens SB, Hassankhan R, Verhulst KC, Stege GJ, de Groot CJ, van Noort JM. Differential expression of stress proteins in human adult astrocytes in response to cytokines. J Neuroimmunol. 2000;106:14–22. doi: 10.1016/s0165-5728(99)00260-x. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Gainetdinov RR, Caron MG. Paradoxical striatal cellular signaling responses to psychostimulants in hyperactive mice. J Biol Chem. 2006 doi: 10.1074/jbc.M606062200. (electronic publication). [DOI] [PubMed] [Google Scholar]
- Berhow MT, Hiroi N, Nestler EJ. Regulation of ERK (extracellular signal regulated kinase), part of the neurotrophin signal transduction cascade, in the rat mesolimbic dopamine system by chronic exposure to morphine or cocaine. J Neurosci. 1996;16:4707–4715. doi: 10.1523/JNEUROSCI.16-15-04707.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack-Werner R. Astrocytes: HIV cellular reservoirs and important participants in neuropathogenesis. AIDS. 1999;13:1–22. doi: 10.1097/00002030-199901140-00003. [DOI] [PubMed] [Google Scholar]
- Breci L, Hattrup E, Keeler M, Letarte J, Johnson R, Haynes PA. Comprehensive proteomics in yeast using chromatographic fractionation, gas phase fractionation, protein gel electrophoresis, and isoelectric focusing. Proteomics. 2005;5 (8):2018–2028. doi: 10.1002/pmic.200401103. [DOI] [PubMed] [Google Scholar]
- Borroto-Esoda K, Myrick F, Feng J, Jeffrey J, Furman P. In vitro combination of amdoxovir and the inosine monophosphate dehydrogenase inhibitors mycophenolic acid and ribavirin demonstrates potent activity against wild-type and drug-resistant variants of human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2004;48:4387–4394. doi: 10.1128/AAC.48.11.4387-4394.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canki M, Thai JN, Chao W, Ghorpade A, Potash MJ, Volsky DJ. Highly productive infection with pseudotyped human immunodeficiency virus type 1 (HIV-1) indicates no intracellular restrictions to HIV-1 replication in primary human astrocytes. J Virol. 2001;75:7925–7933. doi: 10.1128/JVI.75.17.7925-7933.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Controls Task Force on Acquired Immune Deficiency Syndrome. United States Morb Mortl Weekly Report. 1983:32. 30. [Google Scholar]
- Chapuis AG, Paolo Rizzardi G, D'Agostino C, Attinger A, Knabenhans C, Fleury S, Acha-Orbea H, Pantaleo G. Effects of mycophenolic acid on human immunodeficiency virus infection in vitro and in vivo. Nat Med. 2000;6:762–768. doi: 10.1038/77489. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Cimarelli A, Luban J. Translation elongation factor 1-alpha interacts specifically with the human immunodeficiency virus type 1 Gag polyprotein. J Virol. 1999;73:5388–53401. doi: 10.1128/jvi.73.7.5388-5401.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JN, Lake JA, Burrell CJ, Wesselingh SL, Gorry PR, Li P. Novel pathway of human immunodeficiency virus type 1 uptake and release in astrocytes. Virology. 2006;348 (1):141–155. doi: 10.1016/j.virol.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Conant K, Tornatore C, Atwood W, Meyers K, Traub R, Major EO. In vivo and in vitro infection of the astrocyte by HIV-1. Adv Neuroimmunol. 1994;4:287–289. doi: 10.1016/s0960-5428(06)80269-x. [DOI] [PubMed] [Google Scholar]
- Cooper B, Eckert D, Andon NL, Yates JR, Haynes PA. Investigative proteomics: identification of an unknown plant virus from infected plants using mass spectrometry. J Am Soc Mass Spectrom. 2003;14:736–741. doi: 10.1016/S1044-0305(03)00125-9. [DOI] [PubMed] [Google Scholar]
- Davis LE, Hjelle BL, Miller VE, Palmer DL, Llewellyn AL, Merlin TL, Young SA, Mills RG, Wachsman W, Wiley CA. Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology. 1992;42:1736–1739. doi: 10.1212/wnl.42.9.1736. [DOI] [PubMed] [Google Scholar]
- Des Jarlais DC, Friedman SR. HIV and intravenous drug use. AIDS. 1988;2S1:S65–S69. doi: 10.1097/00002030-198800001-00010. [DOI] [PubMed] [Google Scholar]
- Des Jarlais DC, Friedman SR, Marmor M. Development of AIDS, HIV seroconversion and potential cofactors for T4 cell loss in a cohort of intravenous drug users. AIDS. 1987;1:105–111. [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. GLIA. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, D'Aversa TG, Lopez L, Calderon TM, Berman JW. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J Neurochem. 2003;85:1299–1311. doi: 10.1046/j.1471-4159.2003.01775.x. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood–brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci. 2006;26:1098–1106. doi: 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fankhauser C, Izaurralde E, Adachi Y, Wingfield P, Laemmli UK. Specific complex of human immunodeficiency virus type 1 rev and nucleolar B23 proteins: dissociation by the Rev response element. Mol Cell Biol. 1991;11:2567–2575. doi: 10.1128/mcb.11.5.2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fust G, Beck Z, Banhegyi D, Kocsis J, Biro A, Prohaszka Z. Antibodies against heat shock proteins and cholesterol in HIV infection. Mol Immunol. 2005;42:79–85. doi: 10.1016/j.molimm.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Gekker G, Hu S, Wentland MP, Bidlack JM, Lokensgard JR, Peterson PK. Kappa-opioid receptor ligands inhibit cocaine-induced HIV-1 expression in microglial cells. J Pharmacol Exp Ther. 2004;309:600–606. doi: 10.1124/jpet.103.060160. [DOI] [PubMed] [Google Scholar]
- Gendelman HE, Lipton SA, Tardieu M, Bukrinsky MI, Nottet HS. The neuropathogenesis of HIV-1 infection. J Leukoc Biol. 1994;56:389–398. doi: 10.1002/jlb.56.3.389. [DOI] [PubMed] [Google Scholar]
- Gharahdaghi F, Weinberg CR, Meagher DA, Imai BS, Mische SM. Mass spectrometric identification of proteins from silver-stained polyacrylamide gel: a method for the removal of silver ions to enhance sensitivity. Electrophoresis. 1999;20:601–605. doi: 10.1002/(SICI)1522-2683(19990301)20:3<601::AID-ELPS601>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Gullo CA, Teoh G. Heat shock proteins: to present or not, that is the question. Immunol Lett. 2004;94:1–10. doi: 10.1016/j.imlet.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Gurer C, Cimarelli A, Luban J. Specific incorporation of heat shock protein 70 family members into primate lentiviral virions. J Virol. 2002;76:4666–4670. doi: 10.1128/JVI.76.9.4666-4670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson E, Ronnback L. Glial neuronal signaling in the central nervous system. FASEB J. 2003;17:348–351. doi: 10.1096/fj.02-0429rev. [DOI] [PubMed] [Google Scholar]
- Hayase T, Yamamoto Y, Yamamoto K, Muso E, Shiota K, Hayashi T. Similar effects of cocaine and immobilization stress on the levels of heat-shock proteins and stress-activated protein kinases in the rat hippocampus, and on swimming behaviors: the contribution of dopamine and benzodiazepine receptors. Behav Pharmacol. 2003a;14:551–562. doi: 10.1097/00008877-200311000-00008. [DOI] [PubMed] [Google Scholar]
- Hayase T, Yamamoto Y, Yamamoto K, Muso E, Shiota K. Stressor-like effects of cocaine on heat shock protein and stress-activated protein kinase expression in the rat hippocampus: interaction with ethanol and anti-toxicity drugs. Leg Med. 2003b;5S1:S87–S90. doi: 10.1016/s1344-6223(02)00093-7. [DOI] [PubMed] [Google Scholar]
- Haynes PA, Gygi SP, Figeys D, Aebersold R. Proteome analysis: biological assay or data archive? Electrophoresis. 1998;19:1862–1871. doi: 10.1002/elps.1150191104. [DOI] [PubMed] [Google Scholar]
- Hemonnot B, Cartier C, Gay B, Rebuffat S, Bardy M, Devaux C, Boyer V, Briant L. The host cell MAP kinase ERK-2 regulates viral assembly and release by phosphorylating the p6gag protein of HIV-1. J Biol Chem. 2004;279:32426–32434. doi: 10.1074/jbc.M313137200. [DOI] [PubMed] [Google Scholar]
- http://www1.amershambiosciences.com/aptrix/upp00919.nsf/Content/Proteomics+DIGE+Protocols.
- Ichimura H, Levy JA. Polymerase substrate depletion: a novel strategy for inhibiting the replication of the human immunodeficiency virus. Virology. 1995;211:554–560. doi: 10.1006/viro.1995.1437. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Kramer-Hammerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005;111:194–213. doi: 10.1016/j.virusres.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Li YP. Protein B23 is an important human factor for the nucleolar localization of the human immunodeficiency virus protein Tat. J Virol. 1997;71:4098–4102. doi: 10.1128/jvi.71.5.4098-4102.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Galey D, Mattson MP, Nath A. Molecular and cellular mechanisms of neuronal cell death in HIV dementia. Neurotox Res. 2005;8:119–134. doi: 10.1007/BF03033824. [DOI] [PubMed] [Google Scholar]
- Liu J, Lin A. Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 2005;15:36–42. doi: 10.1038/sj.cr.7290262. [DOI] [PubMed] [Google Scholar]
- Liu NQ, Lossinsky AS, Popik W, Li X, Gujuluva C, Kriederman B, Roberts J, Pushkarsky T, Bukrinsky M, Witte M, Weinand M, Fiala M. Human immunodeficiency virus type 1 enters brain microvascular endothelia by macropinocytosis dependent on lipid rafts and the mitogen-activated protein kinase signaling pathway. J Virol. 2002;76:6689–6700. doi: 10.1128/JVI.76.13.6689-6700.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe JK, Brox L, Henderson JF. Consequences of inhibition of guanine nucleotide synthesis by mycophenolic acid and virazole. Cancer Res. 1977;37:736–743. [PubMed] [Google Scholar]
- Mahajan S, Schwartz S, Nair MP. Immunological assays for chemokine detection in in vitro culture of CNS cells. Biol Proced Online. 2003;5:90–102. doi: 10.1251/bpo50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marasco WA, Szilvay AM, Kalland KH, Helland DG, Reyes HM, Walter RJ. Spatial association of HIV-1 tat protein and the nucleolar transport protein B23 in stably transfected Jurkat T-cells. Arch Virol. 1994;39:133–154. doi: 10.1007/BF01309460. [DOI] [PubMed] [Google Scholar]
- Minagar A, Shapshak P, Fujimura R, Ownby R, Heyes M, Eisdorfer C. The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J Neurol Sci. 2002;202:13–23. doi: 10.1016/s0022-510x(02)00207-1. [DOI] [PubMed] [Google Scholar]
- Nair MPN, Kailash C, Hewitt RG, Mahajan SD, Sweet A, Schwartz SA. Cocaine differentially modulates chemokine production by mononuclear cells from normal donors and human immunodeficiency virus type 1-infected patients. Clin Diag Lab Immunol. 2000;7:96–100. doi: 10.1128/cdli.7.1.96-100.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair MP, Mahajan S, Chadha KC, Nair NM, Hewitt RG, Pillai SK, Chadha P, Sukumaran PC, Schwartz SA. Effect of cocaine on chemokine and CCR-5 gene expression by mononuclear cells from normal donors and HIV-1 infected patients. Adv Exp Med Biol. 2001;493:235–240. doi: 10.1007/0-306-47611-8_28. [DOI] [PubMed] [Google Scholar]
- Nair MP, Schwartz SA, Mahajan SD, Tsiao C, Chawda RP, Whitney R, Sykes D, Hewitt R. Drug abuse and neuropathogenesis of HIV infection: role of DC-SIGN and IDO. J Neuroimmunol. 2004;157:56–60. doi: 10.1016/j.jneuroim.2004.08.040. [DOI] [PubMed] [Google Scholar]
- Nair MP, Mahajan SD, Schwartz SA, Reynolds J, Whitney R, Bernstein Z, Chawda RP, Sykes D, Hewitt R, Hsiao CB. Cocaine modulates dendritic cell-specific C type intercellular adhesion molecule-3-grabbing nonintegrin expression by dendritic cells in HIV-1 patients. J Immunol. 2005;174:6617–6626. doi: 10.4049/jimmunol.174.11.6617. [DOI] [PubMed] [Google Scholar]
- Nath A, Maragos WF, Avison MJ, Schmitt FA, Berger JR. Acceleration of HIV dementia with methamphetamine and cocaine. J Neurovirology. 2001;7:66–71. doi: 10.1080/135502801300069737. [DOI] [PubMed] [Google Scholar]
- Nath A, Hauser KF, Wojna V, Booze RM, Maragos W, Prendergast M, Cass W, Turchan JT. Molecular basis for interactions of HIV and drugs of abuse. J Acquired Immune Defic Syndr. 2002;31S2:S62–S69. doi: 10.1097/00126334-200210012-00006. [DOI] [PubMed] [Google Scholar]
- Peterson PK, Gekker G, Chao CC, Schut R, Molitor TW, Balfour HH., Jr Cocaine potentiates HIV-1 replication in human peripheral blood mononuclear cell cocultures. Involvement of transforming growth factor-beta. J Immunol. 1991;146:81–84. [PubMed] [Google Scholar]
- Peterson PK, Gekker G, Chao CC, Schut R, Verhoef J, Edelman CK, Erice A, Balfour HH., Jr Cocaine amplifies HIV-1 replication in cytomegalovirus-stimulated peripheral blood mononuclear cell cocultures. J Immunol. 1992;149:676–680. [PubMed] [Google Scholar]
- Peterson PK, Gekker G, Schut R, Hu S, Balfour HH, Jr, Chao CC. Enhancement of HIV-1 replication by opiates and cocaine: the cytokine connection. Adv Exp Med Biol. 1993;335:181–188. doi: 10.1007/978-1-4615-2980-4_26. [DOI] [PubMed] [Google Scholar]
- Prohaszka Z, Fust G. Immunological aspects of heat-shock proteins-the optimum stress of life. Mol Immunol. 2004;41:29–44. doi: 10.1016/j.molimm.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Retzlaff C, Yamamoto Y, Hoffman PS, Friedman H, Klein TW. Bacterial heat shock proteins directly induce cytokine mRNA and interleukin-1 secretion in macrophage cultures. Infect Immun. 1994;62:5689–5693. doi: 10.1128/iai.62.12.5689-5693.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabri F, Titanji K, De Milito A, Chiodi F. Astrocyte activation and apoptosis: their roles in the neuropathology of HIV infection. Brain Pathol. 2003;13:84–94. doi: 10.1111/j.1750-3639.2003.tb00009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAMSHA. 2004. Office of Applied Studies, National Household Survey on Drug Abuse. [Google Scholar]
- Secchiero P, Zella D, Curreli S, Mirandola P, Capitani S, Gallo RC, Zauli G. Engagement of CD28 modulates CXC chemokine receptor 4 surface expression in both resting and CD3-stimulated CD4+ T cells. J Immunol. 2000;164:4018–4024. doi: 10.4049/jimmunol.164.8.4018. [DOI] [PubMed] [Google Scholar]
- Shishibori T, Oyama Y, Matsushita O, Yamashita K, Furuichi H, Okabe A, Maeta H, Hata Y, Kobayashi R. Three distinct anti-allergic drugs, amlexanox, cromolyn and tranilast, bind to S100A12 and S100A13 of the S100 protein family. Biochem J. 1999;338:583–589. [PMC free article] [PubMed] [Google Scholar]
- Shively L, Chang L, LeBon JM, Liu Q, Riggs AD, Singer-Sam J. Real-time PCR assay for quantitative mismatch detection. BioTechniques. 2003;34:498–502. doi: 10.2144/03343st01. [DOI] [PubMed] [Google Scholar]
- Speth C, Dierich MP, Sopper S. HIV-infection of the central nervous system: the tightrope walk of innate immunity. Mol Immunol. 2005;42:213–228. doi: 10.1016/j.molimm.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Szebeni A, Mehrotra B, Baumann A, Adam SA, Wingfield PT, Olson MO. Nucleolar protein B23 stimulates nuclear import of the HIV-1 Rev protein and NLS-conjugated albumin. Biochemistry. 1997;36:3941–3949. doi: 10.1021/bi9627931. [DOI] [PubMed] [Google Scholar]
- Tonge R, Shaw J, Middleton B, Rowlinson R, Rayner S, Young J, Pognan F, Hawkins E, Currie I, Davison M. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics. 2001;1:377–396. doi: 10.1002/1615-9861(200103)1:3<377::AID-PROT377>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Pages C, Besson MJ, Maldonado R, Caboche J. Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J Neurosci. 2000;20:8701–8709. doi: 10.1523/JNEUROSCI.20-23-08701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel D, Flugge UI. Method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- Wilm M, Shevchenko A, Houthaeve T, Breit S, Schweigerer L, Fotsis T, Mann M. Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature. 1996;379:466–469. doi: 10.1038/379466a0. [DOI] [PubMed] [Google Scholar]
- Yalowitz JA, Jayaram HN. Molecular targets of guanine nucleotides in differentiation, proliferation and apoptosis. Anticancer Res. 2000;20:2329–2338. [PubMed] [Google Scholar]