

Abstract
Two-electrode voltage-clamp electrophysiology has been used to study the actions of two amyloid peptides (Aβ1–42, Aβ1–40) on α7, α4β2 and α3β4 recombinant human neuronal nicotinic acetylcholine receptors (nicotinic AChRs), heterologously expressed in Xenopus laevis oocytes.
The application of Aβ1–42 or Aβ1–40 (1 pM–100 nM) for 5 s does not directly activate expressed human α7, α4β2 or α3β4 nicotinic AChRs.
Aβ1–42 and Aβ1–40 are antagonists of α7 nicotinic AChRs. For example, 10 nM Aβ1–42 and Aβ1–40 both reduced the peak amplitude of currents recorded (3 mM ACh) to 48±5 and 45±10% (respectively) of control currents recorded in the absence of peptide. In both the cases the effect is sustained throughout a 30 min peptide application and is poorly reversible.
Aβ1–42 and Aβ1–40 (10 nM) enhance currents recorded in response to ACh (3 mM) from oocytes expressing α4β2 nicotinic AChRs by 195±40 and 195±41% respectively. This effect is transient, reaching a peak after 3 min and returning to control values after a 24 min application of 10 nM Aβ1–42. We observe an enhancement of 157±22% of control ACh-evoked current amplitude in response to 100 nM Aβ1–42 recorded from oocytes expressing α4β2 nicotinic AChRs.
Aβ1–42 and Aβ1–40 (10 nM) were without antagonist actions on the responses of α3β4 nicotinic AChRs to ACh (1 nM–3 mM).
Keywords: Alzheimer's disease, β-amyloid peptides, nicotinic acetylcholine receptors, Xenopus laevis oocytes, two-electrode voltage-clamp
Introduction
Alzheimer's disease (AD) is the most common form of senile dementia, involving profound memory loss and disturbances in personality (Rossor et al., 1996). Patients show a selective degeneration of cholinergic neurons (Bowen et al., 1976) and brain lesions in the hippocampus, limbic and association cortices, which appear in the form of neuritic plaques and are composed mainly of the β-amyloid peptide (Aβ1–42) (see Selkoe, 2001 for review). The most common form of amyloid peptide in the nondiseased brain (Aβ1–40) has also been shown to form fibrils and to be neurotoxic in both in vitro (100 μg ml−1 peptide) and in vivo (13 mg ml−1 peptide) studies (Giordano et al., 1994), although the concentrations of Aβ1–40 used in these experiments are much higher than those found in the non-diseased human brain (0.3 pmol g−1, Wang et al., 1999).
The role of Aβ peptides is central to amyloid cascade hypothesis of AD (Hardy & Higgins, 1992). There is growing evidence that these peptides interact directly with nicotinic acetylcholine receptors (nicotinic AChRs) and that this interaction may contribute to the pathology of AD (Lena & Changeux, 1998; Romanelli & Gualtieri, 2003; Kar et al., 2004). The mechanism of interaction of Aβ peptides with nicotinic AChR merits further investigation, as some earlier work describes direct activation of nicotinic AChRs while others observe antagonistic effects. For example, Aβ1–42 has been reported to partially block native, postsynaptic α7 nicotinic AChRs in rat hippocampal brain slice preparations (Pettit et al., 2001) and rat hippocampal primary cell cultures (Liu et al., 2001). Similar antagonist actions of Aβ1–42 are also observed in studies on recombinant human α7 nicotinic AChRs expressed in either Xenopus laevis oocytes (Grassi et al., 2003) or SH-EP1 human epithelial cells (Wu et al., 2004), whereas other studies using the oocyte expression system have reported little, if any, suppression of amplitude of responses of rat α7 nicotinic AChRs to ACh (Tozaki et al., 2002). Others report a direct activation by Aβ1–42 of recombinant rat α7 receptors when expressed in oocytes (Dineley et al., 2002), native rat α7 receptors in synaptosomal preparations (Dougherty et al., 2003) and native α4-containing nicotinic AChRs in rat hippocampal slices (Fu & Jhamandas, 2003).
The apparently conflicting results emerging from studies on native neuronal nicotinic AChRs might be explained by species differences, variation in receptor subunit composition and the complications arising when several different nicotinic AChR subtypes are present in a single cell. Indeed, subunit composition is known to account for many differences in the physiology and pharmacology of native and recombinant nicotinic AChRs (Karlin, 2002). Neuronal nicotinic AChRs are assembled from some combinations of α (α2–10) and β (β2–4) subunits. Both homomeric and heteromeric nicotinic AChRs have been reported, with the majority of human central nervous system (CNS) nicotinic AChRs being of the α4β2 subtype, and the remainder largely made up of α7 subunit homomers and α3β4 heteromers, although many other combinations are also known (Lindstrom et al., 1995). While the actions of amyloid peptides on the α7 nicotinic AChR have been studied in detail, less attention has been paid to their actions on other nicotinic AChRs, including α4β2 and α3β4 subtypes. Studies on α4β2 nicotinic AChRs reported to date have shown that application of Aβ1–42 partially blocks the human α4β2 receptors when they are heterologously expressed in SH-EP1 cells (1 and 100 nM Aβ1–42, Wu et al., 2004); recombinant rat α4β2 receptors expressed in Xenopus oocytes are also bloked by 100 nM Aβ1–42 (Tozaki et al., 2002). To date there have been no studies of the actions of Aβ peptides on α3β4 nicotinic AChRs. In addition to the uncertainties of receptor subunit composition, interspecies differences and/or contributions from the different expression systems deployed, the aggregation state of the peptides (Chromy et al., 2003) may also account for some of the disparities between previous reports of amyloid peptide actions on nicotinic AChRs.
In this study, we have used the Xenopus oocyte expression system to compare directly the actions of Aβ1–42 and Aβ1–40 on three subtypes (α7, α4β2 and α3β4) of heterologously expressed human neuronal nicotinic AChRs.
Methods
cDNA preparation
cDNAs encoding the human α3, α4, β2 and β4 cDNAs were a generous gift from Prof. J. Lindstrom and human α7 was kindly donated by Prof. M. Ballivet. The α3 and β4 cDNAs were supplied in the pcDNA1 vector. The α4, α7 and β2 cDNAs were originally supplied in the pSP64 vector and were subsequently cloned into the pcDNA 3.1 vector under the control of the T7 promoter by the laboratory of Dr Isabel Bermudez to facilitate expression in Xenopus oocytes. All cDNAs were purified using an EndoFree Plasmid Maxi Kit (Qiagen Ltd, Crawley, U.K.).
Oocyte preparation
Mature X. laevis frogs (∼100 g body weight) were purchased from Blades Biological (U.K.). Ovaries were removed under general anaesthetic (0.2% 3-aminobenzoic ethyl ester) and every care was taken to (a) reduce suffering and (b) minimise the number of animals used in accordance with the U.K. Animals (Scientific Procedures) Act, 1986. Standard oocyte saline (see below) was used for superfusion of the oocytes in all physiological experiments. Stage V–VI oocytes were isolated by removal of the follicle cell layer using fine forceps (World Precision Instruments, forceps #5), following a 5 min incubation in a collagenase solution (Sigma, type IA, 0.2 mg ml−1 in SOS omitting Ca2+). Oocyte nuclei were injected with one of the following cDNAs or cDNA-combinations: (a) 23 nl of human α7 cDNA (1 ng nl−1); (b) 23 nl of α4 and β2 mixture (1 : 1 subunit ratio, 0.05 ng nl−1 final concentration); (c) 50 nl of α4 and β2 mixture (1 : 9 subunit ratio, 0.05 ng nl−1 final concentration); (d) 50 nl of α4 and β2 mixture (9 : 1 subunit ratio, 0.05 ng nl−1 final concentration); (e) 23 nl of α3 and β4 mixture (1 : 1 subunit ratio, 0.05 ng nl−1 final concentration); (f) 23 nl distilled water (dH2O). All cDNAs were diluted in dH2O to the final concentration for injection. Following injection, oocytes were incubated at 18°C in 0.2 μm-filtered SOS supplemented with sodium pyruvate (2.5 mM), penicillin (100 U ml−1), streptomycin (100 μg ml−1) and gentamycin (50 μg ml−1). The medium was changed daily, and electrophysiological experiments were performed 2–5 days after nuclear injection.
Saline, drugs and amyloid peptides
A fresh solution of 1.0 M ACh or 10 mM (−)-nicotine was prepared in dH2O for appropriate experiments. Stock solutions of 200 μM Aβ1–40 and Aβ1–42 (Sigma-Aldrich, U.K. and U.S. Peptide Inc., U.S.A.) were dissolved in 5% glacial acetic acid or dH2O and stored at −20°C. SOS containing (in mM) NaCl, 100; KCl, 2; CaCl2, 1.8; MgCl2, 1; HEPES, 5 adjusted to pH 7.6 with 5 M NaOH was prepared immediately before each experiment. All solutions were then diluted with SOS to final concentrations immediately before use. Standard extracellular solution (SES) had the composition (in mM) NaCl, 120; KCl, 3; CaCl2, 2; MgCl2, 2; HEPES, 10; D-Glucose, 25; sodium pyruvate, 2.5; adjusted to pH 7.4 with Tris-base. All chemicals, unless otherwise specified, were obtained from Sigma-Aldrich, U.K.
Voltage-clamp electrophysiology
Oocytes were secured in a Perspex chamber (100 μl volume) which was perfused continuously with SOS at a constant flow rate (5–7 ml min−1) via a gravity-fed system (Buckingham et al., 1994). Atropine (0.5 μM) was included in the saline to suppress any responses resulting from activation of endogenous muscarinic AChRs (Lupu-Meiri et al., 1990; Blake et al., 1993). Membrane currents were measured by the two-electrode voltage-clamp (TEVC) method, using 3 M KCl-filled electrodes (resistance=1–3 MΩ in SOS). Signals were amplified using either a Warner OC-725C Oocyte Clamp (Warner Instrument Corp., U.S.A.) or a Geneclamp 500 amplifier (Axon Instruments Inc., U.S.A.), with the oocyte membrane potential clamped at −60 mV. Signals were filtered at 1 kHz and digitised using a Digidata 1320A interface (Axon Instruments Inc., U.S.A.) with an acquisition rate of 10 kHz, stored on a PC using Axoscope v.8.1 and subsequently analysed off-line using Clampfit v.8.1 (Axon Instruments Inc., U.S.A.) software.
Only oocytes with resting membrane potentials more negative than −25 mV, that exhibited inward currents exceeding 100 nA in response to two successive, 5 s exposures to ACh (of concentrations close to the EC50 of ACh for the nicotinic AChR subtype expressed) and with <10% variation in amplitude between the two responses were used. At the beginning of each experiment oocytes were challenged with two (3 min interval), 5 s applications of an appropriate EC50 concentration of ACh to confirm the selection criteria of the experiment. At least two batches of oocytes from separate Xenopus were used in all electrophysiological experiments.
Experimental protocols
ACh and peptides were bath applied in SOS. ACh was applied for 5 s with 3 min intervals between successive applications to minimise receptor desensitization. ACh dose–response curves were constructed in the presence or absence of 10 nM Aβ peptide. To enable the collection of paired data for statistical analysis, the same oocyte was used in the construction of both dose–response curves. The oocyte was allowed to recover for at least 15 min after the maximal dose of ACh before the first application of ACh in the presence of either Aβ1–40 or Aβ1–42, whereupon each application was preceded by a 3 min preincubation with a 10 nM peptide (Figure 1).
Figure 1.

Actions of Aβ1–42 and Aβ1–40 on ACh-induced currents in Xenopus oocytes expressing recombinant human nicotinic AChRs. Each point represents the mean of six separate experiments using oocytes from at least two different frogs. ACh dose–response curves (10 nM–3 mM) for each receptor subtype were constructed in the absence or presence of 10 nM peptide. Inset traces show representative traces of inward current responses to 300 μM ACh in the absence (black traces) or presence (grey traces) of peptide.
As shown in Figure 2, 10 successive applications of 300 μM ACh were applied to oocytes expressing either α7 or α4β2 nicotinic AChRs in the continuous presence (30 min) of Aβ1–42 and the ACh-evoked responses measured. Five further applications of ACh were then carried out in the absence of peptide (Figure 2).
Figure 2.

The effect of a 30 min exposure of 10 nM Aβ1–42 on recombinant α7 (a) and α4β2 (b) nicotinic AChRs. In all, 10 successive applications of 300 μM ACh were applied in the continuous presence of 10 nM Aβ1–42. The perfusion buffer was then changed to SOS and the recovery of ACh-evoked responses recorded for five further applications. Each data point is shown as the mean±s.e.m. and expressed as a percentage of the control inward current. The continued decrease in agonist evoked current amplitude after peptide washout observed for α4β2 receptors is unlikely to be due to receptor rundown since control experiments do not mimic this effect over the same time period.
Oocytes expressing α4β2 nicotinic AChRs were challenged with two applications of ACh (10 μM) in the presence of 100 nM Aβ1–42 which had been initially dissolved in either dH2O or 5% glacial acetic acid. Two further applications of 10 μM ACh were applied in the absence of peptide (Figure 3). A similar protocol was employed to investigate the actions of 10 nM Aβ1–42 (initially dissolved in 5% acetic acid) on nicotine-evoked currents (data not shown). Stock solutions of Aβ1–42 or Aβ1–40 (200 μM) in each solvent were frozen at −20°C for at least 24 h before dilution to 100 nM in SOS immediately before use. All solutions of peptide were discarded 4 h after dilution.
Figure 3.

The effect of the aggregation state of 100 nM Aβ1–42 on ACh-evoked currents mediated by α4β2 nicotinic AChRs. Each application of ACh was separated by a 3 min interval to minimise receptor desensitization. Two 5 s applications of 10 μM ACh were applied to oocytes expressing α4β2 nicotinic AChRs and the perfusion media then changed to SOS containing 100 nM Aβ1–42 for 6 min. After 3 min preincubation with peptide the oocyte was challenged with two applications of 10 μM ACh containing 100 nM Aβ1–42 initially dissolved in either (a) 5% glacial acetic acid or (b) ddH2O. The perfusion media was then changed back to SOS and two further challenges of 10 μM ACh applied.
Transmission electron microscopy was then used to determine the aggregation state of water or acid dissolved Aβ1–40 and Aβ1–42 stock solutions (200 μM, centrifuged at 7000 × g for 1 min) and stock solutions (without centrifugation) diluted in SOS or SES to a concentration 100 nM. Two grids of each stock solution and 100 nM peptide were prepared on two separate days and 10 squares of each grid were scanned for fibril formation using negative staining with 1% uranyl acetate and viewed at 30,000 × magnification.
Electrophysiology data analysis
Peak current amplitudes were measured following agonist application and were determined as the maximum negative deflection of the current trace from the base line. To compensate for variations in receptor expression by individual oocytes, responses to applications of ACh in the presence and absence of Aβ peptide were expressed as a percentage of the maximum current response evoked in that oocyte by ACh alone. Each curve was fitted using GraphPad Prism version 4.0 (GraphPad Software Inc., U.S.A.) software to the expression:
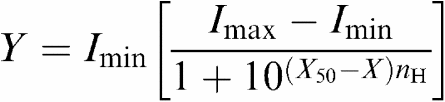 |
where Y is the normalised response amplitude, X is log10 ACh concentration (M), X50 is log10 of the EC50, Imax is the maximum value of Y, Imin is the minimum value of Y and nH is the Hill coefficient. Concentration–response plots for the action of ACh on α4β2 receptors expressed in oocytes injected with a 1 : 1 ratio of subunits was best fit with the sum of two sigmoidal curves with EC50 values of 7±2 and 357±37 μM, suggesting that two receptor populations with differing affinities were expressed. The EC50 of ACh in the presence or absence of 10 nM Aβ peptide was calculated for each nicotinic AChR using this equation. Plotted data points are the mean±s.e.m. and EC50 values are reported as the mean±s.d. Differences between the evoked current amplitudes to ACh in the presence and absence of 10 nM Aβ peptide were evaluated for statistical significance using a two-way analysis of variance (ANOVA) with Bonferroni post hoc test.
Comparison of the effects of 100 nM Aβ1–42 on α4β2 nicotinic AChRs when stock solutions were dissolved in ddH2O or 5% acetic acid and the actions of 10 nM Aβ1–42 on 10 μM nicotine-evoked currents mediated by the α4β2 nicotinic receptor were assessed for statistical significance using a one sample t-test. Results were considered significant if P<0.05.
Results
Bath application of ACh resulted in inwardly-directed currents in 90% of all oocytes injected with α4β2 or α3β4 nicotinic AChR subunit cDNA and 70% of oocytes injected with α7 cDNA (Figure 1, inset traces). Nicke et al. (2004) ascribes such differences in nicotinic AChR subtype expression to the oocyte's reduced efficiency to assemble α7 subunits compared to heteromeric receptor subunits. The absence of response to ACh in the remaining oocytes was attributed to a presumed failure to inject the nucleus with cDNA. No current responses were detected in response to ACh (300 μM) in uninjected or water-injected oocytes (n=6, data not shown). As previously reported (Chavez-Noriega et al., 1997), marked differences in desensitization rates were observed between the three receptor types (Figure 1).
Aβ peptides failed to activate recombinant human nicotinic AChRs (α7, α4β2 and α3β4)
The ability of Aβ1–42 or Aβ1–40 to directly activate human α7, α4β2 and α3β4 nicotinic AChR subtypes was tested. Bath application of either peptide (1 pM–100 nM) in all cases failed to evoke discernable currents in oocytes expressing any of the three receptor subtypes when cells were voltage clamped at −60 mV (n=4–10 for each receptor subtype, data not shown).
Differential actions of Aβ peptides on recombinant nicotinic AChRs (α7, α4β2 and α3β4)
The effect of Aβ1–42 and Aβ1–40 on ACh-induced currents mediated by the three recombinant receptor types was determined. A 3 min preincubation with either peptide (10 nM) resulted in receptor-specific effects on 10 nM–3 mM ACh dose–response relationships (Figure 1). Neither peptide had any significant effect on the Hill coefficient nor the EC50 for ACh for any of the receptor subtypes tested (Table 1).
Table 1.
EC50 and % change in maximal ACh-evoked current in the presence of Aβ peptides in oocytes expressing recombinant nicotinic AChRs
| EC50 (μM) | %ΔImax (Ach) | |||||
|---|---|---|---|---|---|---|
| Control | Aβ1–42 | Control | Aβ1–40 | Aβ1–42 | Aβ1–40 | |
| α7 | 390±87 | 413±97 | 261±66 | 206±22 | ||
| nH=1.3±0.1 | nH=1.8±0.4 | nH=1.9±0.2 | nH=1.5±0.02 | 52.3±5*** | 54.8±10*** | |
| α4β2 | 5.0±1.8 | 12.1±4 | 3±0.4 | 8.1±3 | ||
| 1:1a | nH=1.6±0.2 | nH=1.1±0.3 | nH=2.1±0.4 | nH=1.5±0.3 | 148.6±27 | 116.8±10 |
| α4β2 | 360±75 | 229±67 | 477±74 | 363±107 | ||
| 1:1b | nH=1.0±0.14 | nH=4.7±2.3 | nH=1.1±0.1 | nH=0.8±0.2 | 194.6±40*** | 194.8±41*** |
| α4β2 | 47±22 | 21±11 | ||||
| 1:9 | nH=0.92±0.4 | nH=0.94±0.1 | 128.8±11 | |||
| α4β2 | 165±41 | 281±120 | ||||
| 9:1 | nH=0.94±0.1 | nH=0.6±0.05 | 162.3±39*** | |||
| α3β4 | 509±77 | 404±61 | 387±88 | 412±81 | ||
| nH=1.4±0.1 | nH=1.4±0.1 | nH=1.4±0.1 | nH=1.3±0.1 | 96.0±1.2 | 99.1±7 | |
*P<0.05;
**P<0.01;
P<0.001.
Using a two-way repeated measures ANOVA with Bonferroni post-test. n⩾6.
Abbreviations: %ΔImax(ACh): Peptide induced percentage changes in maximal evoked ACh currents expressed relative to a control application of 3 mM ACh.
nH: Hill coefficient.
High affinity subpopulation.
Low affinity subpopulation.
α7
A 3 min preincubation with either peptide (10 nM) resulted in a reduction of the peak amplitude of evoked responses to ACh in α7 injected oocytes (Aβ1–42: 52.3±5%, P=0.001; Aβ1–40: 54.8±10%, P=0.001 of control responses to 3 mM ACh, Figure 1) suggesting that both peptides act as noncompetitive antagonists of α7 nicotinic AChRs. A 3 min exposure to 10 nM Aβ1–42 produced a reduction of 300 μM ACh-evoked current amplitude to 66±12% (n=6) of the control values (Figure 2). Further successive applications of ACh in the continuing presence of peptide reduced the current amplitude to 40±3% (n=6) of control values after 30 min. Little recovery was observed, with an average ACh-evoked response of 55±3% (n=6) of control after 15 min of peptide withdrawal.
α4β2
Preincubation with 10 nM Aβ1–42 or Aβ1–40 (3 min) resulted in an enhanced amplitude of ACh-evoked currents in α4β2 injected oocytes (Aβ1–42: 195±40%, P=0.001; Aβ1–40: 195±41%, P=0.008 of control responses to 3 mM ACh, Figure 1). We also tested the effects of 10 nM Aβ1–42 on 10 μM nicotine-evoked currents for comparison with the experiments of Wu et al. (2004). We found a significant enhancement of 151±15% of control nicotine-evoked current amplitude in the presence of peptide, mediated by α4β2 nicotinic receptors (P=0.03, data not shown). The presence of two receptor subpopulations, with differing affinities, in oocytes injected with a 1 : 1 ratio of α4 and β2 subunits is in accord with previous studies (Papke et al., 1989; Zwart & Vijverberg, 1998).
In the present study, these two receptor subpopulations of α4β2 nicotinic AChRs have been separated by injecting different ratios of the α4 and β2 subunits. By injecting different volume ratios (v v−1) of 1 : 9 and 9 : 1 of α4 : β2 subunit cDNA, respectively, it was possible to resolve the high- and low-ACh affinity subpopulations of α4β2 nicotinic receptors. Aβ1–42 (10 nM) enhanced the ACh (3 mM) evoked currents mediated by both the high- and low-ACh affinity receptors to 129±11 and 162±39% of control values, respectively, which agrees with the initial data obtained from the 1 : 1 injected ratio of receptor subunits. Thus, Aβ1–42 and Aβ1–40 enhanced both low- and high-affinity α4β2 nicotinic AChRs, with a significant action on the low-affinity receptor sub-population. The EC50 estimated for ACh for the high- and low-affinity α4β2 receptors, when preferentially expressed by injection of 9 : 1 and 1 : 9 ratios of α4 and β2 subunits, did not differ significantly from the estimated EC50 for ACh determined for the high- and low-affinity receptors expressed after an injection of 1 : 1 ratio of α4 and β2 subunits (P=0.36 and P=0.13 respectively, using a two-tailed one sample t-test). The respective estimated EC50s for ACh were not significantly affected by the presence of 10 nM Aβ1–42 (P=0.21 and P=0.53, respectively, using a two-tailed one sample t-test).
A 30 min exposure to 10 nM Aβ1–42 during repeated applications of 300 μM ACh to α4β2 nicotinic AChRs caused a rapid 156±7% increase in the amplitude of evoked current on the first application of ACh. This enhancement then steadily declined with repeated ACh applications until responses reached control values upon the eighth agonist challenge. ACh-evoked response amplitudes were not significantly different from control values during a 15 min wash-out with SOS (Figure 2).
A higher concentration (100 nM) of Aβ1–42 enhanced responses to 10 μM ACh in α4β2 injected oocytes (157±22% control responses, P=0.02, n=7, Figure 3). The observation of enhancement of human α4β2 ACh-evoked responses in the present study contrasts with a reported reduction in currents mediated by the same receptor subtype expressed in SH-EP1 cells (Wu et al., 2004). These differences might be attributable to differences in the aggregation state of the peptide, since Wu et al. (2004) used dH2O as the stock solute whereas the present study dissolved the peptides in 5% acetic acid to reduce fibril formation. To determine whether this enhancement was caused by Aβ1–42 fibril formation, the previous experiment was repeated with Aβ1–42 dissolved initially in dH2O rather than 5% acetic acid. An enhancement of ACh-evoked current was still observed (170±28% of control response, P=0.03, n=7, Figure 3).
Using transmission electron microscopy, the aggregation state of Aβ1–42 and Aβ1–40 stock solutions (200 μM) and experimental concentrations of 100 nM were assessed. When dissolved in dH2O, stock solutions of Aβ1–42 were found to produce large fibril aggregates in 40/40 of grid squares observed (average fibril length=435±121 nm). Similar sized aggregates were observed in 40/40 grid squares analysed, when Aβ1–42 was diluted to 100 nM in SES (mimicking the experimental conditions of Wu et al., 2004). In contrast, when 5% acetic acid was used as the solute, no aggregates were observed in 35/40 grid squares examined, although five of the grid squares contained a low number of small, uniform fibrils (average length=72±7.4 nm). When Aβ1–42 (200 μM in 5% acetic acid) was diluted in SOS to 100 nM, no fibril formation was seen in 40/40 grid squares analysed.
Whether Aβ1–40 was dissolved in dH2O or in 5% acetic acid (200 μM) fibrils were observed in 6/40 grid squares examined (dH2O dissolved Aβ1–40 average fibril length=435±93 nm; 5% acetic acid dissolved Aβ1–40 average fibril length=248±113 nm), but there were no large fibril aggregations. No fibrils were observed in 40/40 grid squares when Aβ1–40, dissolved in either solute, was diluted in SOS or SES to 100 nM. All fibrils were 10 nm in diameter. These findings are in accordance with a comprehensive study on these peptides (Stine et al., 2003).
α3β4
Both Aβ1–42 and Aβ1–40 failed to elicit a change in the current amplitude of ACh-evoked currents mediated by the ganglionic α3β4 nicotinic AChRs when bath applied at a concentration of 10 nM (Aβ1–42: 96±1%, Aβ1–40: 99±7% of control responses to 3 mM ACh, n=6). No significant change in the EC50 of ACh was observed in the presence of either peptide (ACh control=509±77 μM, ACh+Aβ1–42=404±61 μM; ACh control=387±88 μM, ACh+Aβ1–40=412±81 μM).
Discussion
We present new results to show that neither Aβ1–42 nor Aβ1–40, at concentrations ranging from 1 pM to 100 nM, directly activates α7, α4β2 or α3β4 recombinant receptors, thus confirming a recent study (Grassi et al., 2003) that showed that Aβ1–42 does not directly activate human α7 nicotinic AChRs over a similar concentration range (10 pM–1 μM). The absence of Aβ1–42 induced currents in human α7 nicotinic AChRs contrasts with the findings of Dineley et al. (2002), who reported that Aβ1–42 activated the recombinant rat α7 receptor on the first peptide application. Similarly, Dougherty et al. (2003) showed that low concentrations (1 pM) of Aβ1–42 activated native α7 containing synaptosomal preparations of the hippocampus, striata and cortex regions of the rat brain. Thus the activation of α7 nicotinic AChRs by Aβ1–42 appears to be species dependent, but Dougherty et al. (2003) suggested that, in rat synaptosomal preparations, Aβ1–42 could act as an agonist or antagonist depending on the concentration of peptide used. However, we found that concentrations of Aβ1–42 and Aβ1–40 ranging from 1 pM to 100 nM did not directly activate heterologously expressed human α7, α4β2 or α3β4 nicotinic AChRs. This range covers the concentrations reported for amyloid peptide in the plasma and cerebro-spinal fluid of AD patients (1–10 nM) (Mehta et al., 2001), although the concentration of amyloid peptide at cholinergic synapses is unknown. In this study, we have used 10 nM Aβ peptide, which is significantly higher than the reported affinity (4 pM) for Aβ1–42 binding to α7 nicotinic AChRs but is similar to its binding affinity for α4β2 (30 nM) (Wang et al., 2000), and therefore 10 nM Aβ1–42 would be expected to exert an effect upon both α7 and α4β2 receptors.
Our finding that Aβ peptides exert subtype-specific effects on α7, α4β2 or α3β4 nicotinic AChRs suggests that receptor subunit composition-dependent effects might account for some of the different actions reported for Aβ on neurons in vivo. In the case of the α7 nicotinic AChR, we found that 10 nM Aβ1–40 acted as a antagonist on this receptor subtype and confirmed previous reports that Aβ1–42 reduces α7-mediated ACh-evoked currents in a noncompetitive manner (Wang et al., 2000; Pettit et al., 2001; Grassi et al., 2003).
We showed that both Aβ peptides (10 nM) resulted in a significant enhancement of ACh-induced inward currents mediated by α4β2 nicotinic AChRs. Our detection of two α4β2 receptor sub-populations and their estimated EC50s for ACh agrees with the findings of Nelson et al. (2003) and Zwart & Vijverberg (1998) when the receptors (1 : 1 subunit ratio) are expressed in HEK cells and Xenopus oocytes, respectively. Zwart & Vijverberg (1998) suggest that the 1 : 1 injection ratio of α : β subunits results in two distinct receptor stoichiometries, both with differing sensitivities to agonists and antagonists, whereas an injection of 9 : 1 and 1 : 9 ratios of α : β subunits favour the production of a single population of low- and high-affinity receptors, respectively. When we preferentially expressed each of the receptor subpopulations by injecting 9 : 1 or 1 : 9 volume ratio of the respective subunit cDNAs, we found that 10 nM Aβ1–42 enhanced the 3 mM ACh-evoked currents mediated by both high- and low-affinity α4β2 nicotinic AChRs (129±12 and 162±44% of control values, respectively). The low affinity (9 : 1 ratio of α4 : β2 subunits) nicotinic AChR subtype is more sensitive to the actions of Aβ peptides as shown by the marked enhancement of 3 mM ACh-evoked currents (P<0.001) only observed in this subpopulation. The presence in vivo of functional receptors containing different α4 and β2 subunit compositions has not been confirmed, but we have demonstrated that the positive modulatory effects of Aβ1–42 and Aβ1–40 on ACh-evoked current amplitudes are still apparent in recombinant, heteromeric nicotinic AChRs generated from widely differing ratios (9 : 1 and 1 : 9) of human α4 and β2 subunits.
Our finding that the presence of 10 nM Aβ peptides enhance α4β2 current amplitude contrasts with previous studies, both of which (Tozaki et al., 2002; Wu et al., 2004) report that 100 nM Aβ1–42 cause an inhibition of 10 μM ACh-induced current amplitude mediated by human and rat (respectively) α4β2 nicotinic AChRs expressed in SH-EP1 cells and oocytes (32 and 31%, respectively). The disparity between our results and these previous studies cannot be explained by differences in Aβ1–42 concentration, as we also observe an ACh-evoked current enhancement in the presence of 100 nM Aβ1–42 when oocytes expressing α4β2 nicotinic AChRs were challenged with 10 μM ACh (157±23%, n=7 when stock was dissolved in 5% acetic acid and 170±29%, n=7 when stock was dissolved in dH2O). Our transmission electron microscopy studies confirm dilution of Aβ peptides in 5% glacial acetic acid (present study) slows the fibril formation of amyloid peptides, whereas the suspension of Aβ peptides in dH2O (Wu et al., 2004) leads to the immediate formation of a wide range of large peptide aggregates and extended fibrils (Stine et al., 2003). However, differences between the results of Wu et al. (2004) and this study cannot be explained by the aggregation state of the peptide as the enhancement of control responses were still observed in the presence of fibrils or aggregates of Aβ1–42. Our finding of an enhancement of nicotine-evoked current amplitudes contrasts with the findings of Wu et al. who reported a decrease to 75.5±2% of control nicotine-evoked current amplitude. Since both studies expressed human nicotinic AChRs the differences between the findings are most likely attributable to differences in the host cell environments (Xenopus oocytes and human epithelial SH-EP1 cells).
In direct contrast to the present study, Tozaki et al. (2002) expressed rat α4β2 nicotinic AChRs in Xenopus oocytes and observed a 31% inhibition of control ACh-evoked current amplitude. Our study expressed human α4β2 nicotinic AChRs in the same expression system but we observed a 95% increase in the amplitude of control ACh-evoked currents. As previous studies have shown direct Aβ1–42 activation of rat but not human α7 nicotinic AChRs (Dineley et al., 2002; Dougherty et al., 2003; Fu & Jhamandas, 2003; Grassi et al., 2003), it is possible that the discrepancy between the results of Tozaki et al. (2002) and the present study is due to species differences.
Transgenic rodent models of AD are widely used to investigate the effects of amyloid peptides in vivo and the results used to suggest explanations for the pathology of AD. The possibility of species dependent differences in the actions of amyloid peptides on human and rat nicotinic AChRs requires further investigation, as this would have profound implications on interpreting the results gained from animal models used in the drug discovery process.
In conclusion, we show that the two Aβ peptides, Aβ1–42 and Aβ1–40, have opposing effects on the α7 and α4β2 nicotinic AChRs, suggesting that these receptor subtypes have differing Aβ peptide interaction sites and that both α7 and α4β2 nicotinic AChRs are likely to have different roles in the neuronal responses of Aβ peptides in AD neuropathology. This study also finds that neither peptide, applied at a concentration of 10 nM, had any effect on the ACh-evoked responses of the α3β4 receptor subtype suggesting that the site of interaction for Aβ peptides present on the α7 and α4β2 nicotinic AChRs is absent from the α3β4 nicotinic AChR.
Acknowledgments
Aliquots of individual stock cDNAs (at concentrations ranging from 0.1 to 2 μg μl−1) were kindly supplied by the laboratories of Professors J. Lindstrom and M. Ballivet. We thank Dr Andrew Jones and Dr Laurence Brown for helpful discussions during this study. We express grateful thanks to Dr Anne Clarke for assistance with electron microscopy and Dr Isabel Bermudez for gifts of clones. This work was funded by the Medical Research Council and by a graduate student bursary from Merck, Sharp and Dohme, Harlow, U.K.
Abbreviations
- Aβ
β-amyloid
- ACh
acetylcholine
- AD
Alzheimer's disease
- nicotinic AChR
nicotinic acetylcholine receptor
- ANOVA
analysis of variance
- dH2O
distilled water
- s.d.
standard deviation
- s.e.m.
standard error of the mean
- TEVC
two-electrode voltage-clamp
References
- BLAKE A.D., ANTHONY N.M., CHEN H.H., HARRISON J.B., NATHANSON N.M., SATTELLE D.B. Drosophila nervous system muscarinic acetylcholine receptor: transient functional expression and localization by immunocytochemistry. Mol. Pharmacol. 1993;44:716–724. [PubMed] [Google Scholar]
- BOWEN D.M., SMITH C.B., WHITE P., DAVISON A.N. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain. 1976;99:459–496. doi: 10.1093/brain/99.3.459. [DOI] [PubMed] [Google Scholar]
- BUCKINGHAM S.D., HOSIE A.M., ROUSH R.L., SATTELLE D.B. Actions of agonists and convulsant antagonists on a Drosophila melanogaster GABA receptor (Rdl) homo-oligomer expressed in Xenopus oocytes. Neurosci. Lett. 1994;181:137–140. doi: 10.1016/0304-3940(94)90578-9. [DOI] [PubMed] [Google Scholar]
- CHAVEZ-NORIEGA L.E., CRONA J.H., WASHBURN M.S., URRUTIA A., ELLIOTT K.J., JOHNSON E.C. Pharmacological characterization of recombinant human neuronal nicotinic acetylcholine receptors hα2β2, hα2β4, hα3β2, hα3β4, hα4β2, hα4β4 and hα7 expressed in Xenopus oocytes. J. Pharmacol. Exp. Ther. 1997;280:346–356. [PubMed] [Google Scholar]
- CHROMY B.A., NOWAK R.J., LAMBERT M.P., VIOLA K.L., CHANG L., VELASCO P.T., JONES B.W., FERNANDEZ S.J., LACOR P.N., HOROWITZ P., FINCH C.E., KRAFFT G.A., KLEIN W.L. Self-assembly of Abeta(1–42) into globular neurotoxins. Biochemistry. 2003;42:12749–12760. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- DINELEY K.T., BELL K.A., BUI D., SWEATT J.D. β-Amyloid peptide activates α7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J. Biol. Chem. 2002;277:25056–25061. doi: 10.1074/jbc.M200066200. [DOI] [PubMed] [Google Scholar]
- DOUGHERTY J.J., WU J., NICHOLS R.A. β-Amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J. Neurosci. 2003;23:6740–6747. doi: 10.1523/JNEUROSCI.23-17-06740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FU W., JHAMANDAS J.H. β-Amyloid peptide activates non-α7 nicotinic acetylcholine receptors in rat basal forebrain neurons. J. Neurophysiol. 2003;90:3130–3136. doi: 10.1152/jn.00616.2003. [DOI] [PubMed] [Google Scholar]
- GIORDANO T, PAN J.B, MONTEGGIA L.M, HOLZMAN T.F., SNYDER S.W., KRAFFT G., GHANBARI H, KOWALL N.W. Similarities between β amyloid peptides 1–40 and 40–1: effects on aggregation, toxicity in vitro, and injection in young and aged rats. Exp. Neurol. 1994;125:175–182. doi: 10.1006/exnr.1994.1022. [DOI] [PubMed] [Google Scholar]
- GRASSI F., PALMA E., TONINI R., AMICI M., BALLIVET M., EUSEBI F. Amyloid β1–42 peptide alters the gating of human and mouse α-bungarotoxin-sensitive nicotinic receptors. J. Physiol. 2003;547:147–157. doi: 10.1113/jphysiol.2002.035436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARDY J.A., HIGGINS G.A. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- KAR S., SLOWIKOWSKI S.P., WESTAWAY D., MOUNT H.T. Interactions between β-amyloid and central cholinergic neurons: implications for Alzheimer's disease. J. Psychiatry Neurosci. 2004;29:427–441. [PMC free article] [PubMed] [Google Scholar]
- KARLIN A. Emerging structure of the nicotinic acetylcholine receptors. Nat. Rev. Neurosci. 2002;3:102–114. doi: 10.1038/nrn731. [DOI] [PubMed] [Google Scholar]
- LENA C., CHANGEUX J.P.Allosteric nicotinic receptors, human pathologies Torpedo electric organ to human brain: fundamental and applied aspects 1998Amsterdam: Elsevier Life Sciences; 63–74.Massoulie J. ed, pp [DOI] [PubMed] [Google Scholar]
- LINDSTROM J., ANAND R., PENG X., GERZANICH V., WANG F., LI Y. Neuronal nicotinic receptor subtypes. Ann. NY. Acad. Sci. 1995;757:100–116. doi: 10.1111/j.1749-6632.1995.tb17467.x. [DOI] [PubMed] [Google Scholar]
- LIU Q., KAWAI H., BERG D.K. β-Amyloid peptide blocks the response of α7-containing nicotinic receptors on hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 2001;98:4734–4739. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUPU-MEIRI M., SHAPIRA H., ORON Y. Extracellular calcium participates in responses to acetylcholine in Xenopus oocytes. FEBS Lett. 1990;262:165–169. doi: 10.1016/0014-5793(90)80180-q. [DOI] [PubMed] [Google Scholar]
- MEHTA P.D., PIRTTILA T., PATRICK B.A., BARSHATZKY M., MEHTA S.P. Amyloid β protein 1–40 and 1–42 levels in matched cerebrospinal fluid and plasma from patients with Alzheimer disease. Neurosci. Lett. 2001;304:102–106. doi: 10.1016/s0304-3940(01)01754-2. [DOI] [PubMed] [Google Scholar]
- NELSON M.E., KURYATOV A., CHOI C.H., ZHOU Y., LINDSTROM J. Alternate stoichiometries of α4β2 nicotinic acetylcholine receptors. Mol. Pharmacol. 2003;63:332–341. doi: 10.1124/mol.63.2.332. [DOI] [PubMed] [Google Scholar]
- NICKE A., THURAU H., SADTLER S., RETTINGER J., SCHMALZING G. Assembly of nicotinic α7 subunits in Xenopus oocytes is partially blocked at the tetramer level. FEBS Lett. 2004;575:52–58. doi: 10.1016/j.febslet.2004.08.035. [DOI] [PubMed] [Google Scholar]
- PAPKE R.L., BOULTER J., PATRICK J., HEINEMANN S. Single-channel currents of rat neuronal nicotinic acetylcholine receptors expressed in xenopus oocytes. Neuron. 1989;3:589–596. doi: 10.1016/0896-6273(89)90269-9. [DOI] [PubMed] [Google Scholar]
- PETTIT D.L., SHAO Z., YAKEL J.L. β-Amyloid1–42 peptide directly modulates nicotinic receptors in the rat hippocampal slice. J. Neurosci. 2001;21:RC120. doi: 10.1523/JNEUROSCI.21-01-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROMANELLI M.N., GUALTIERI F. Cholinergic nicotinic receptors: competitive ligands, allosteric modulators, and their potential applications. Med. Res. Rev. 2003;23:393–426. doi: 10.1002/med.10037. [DOI] [PubMed] [Google Scholar]
- ROSSOR M.N., FOX N.C., FREEBOROUGH P.A., HARVEY R.J. Clinical features of sporadic and familial Alzheimer's disease. Neurodegeneration. 1996;5:393–397. doi: 10.1006/neur.1996.0052. [DOI] [PubMed] [Google Scholar]
- SELKOE D.J. Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- STINE W.B., JR, DAHLGREN K.N., KRAFFT G.A., LADU M.J. In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J. Biol. Chem. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- TOZAKI H., MATSUMOTO A., KANNO T., NAGAI K., NAGATA T., YAMAMOTO S., NISHIZAKI T. The inhibitory and facilitatory actions of amyloid-β peptides on nicotinic ACh receptors and AMPA receptors. Biochem. Biophys. Res. Commun. 2002;294:42–45. doi: 10.1016/S0006-291X(02)00429-1. [DOI] [PubMed] [Google Scholar]
- WANG H.Y., LEE D.H., DAVIS C.B., SHANK R.P. Amyloid peptide Aβ1–42 binds selectively and with picomolar affinity to α7 nicotinic acetylcholine receptors. J. Neurochem. 2000;75:1155–1161. doi: 10.1046/j.1471-4159.2000.0751155.x. [DOI] [PubMed] [Google Scholar]
- WANG J., DICKSON D.W., TROJANOWSKI J.Q., LEE V.M. The levels of soluble versus insoluble brain Aβ distinguish Alzheimer's disease from normal and pathologic aging. Exp. Neurol. 1999;158:328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- WU J., KUO Y.P., GEORGE A.A., XU L., HU J., LUKAS R.J. β-Amyloid directly inhibits human α4β2-nicotinic acetylcholine receptors heterologously expressed in human SH-EP1 cells. J. Biol. Chem. 2004;279:37842–37851. doi: 10.1074/jbc.M400335200. [DOI] [PubMed] [Google Scholar]
- ZWART R., VIJVERBERG H.P. Four pharmacologically distinct subtypes of α4β2 nicotinic acetylcholine receptor expressed in Xenopus laevis oocytes. Mol. Pharmacol. 1998;54:1124–1131. [PubMed] [Google Scholar]