

Abstract
The effect of denaturation and/or extraction of nonintegral membrane proteins by 7 M urea on the binding of the antagonist [3H]cyclopentyl-1,3-dipropylxanthine 8 dipropyl-2,3 ([3H]DPCPX), and the agonists adenosine, (−)-N6-(2-phenylisopropyl)-adenosine (R-PIA) and N6-cyclohexyladenosine (CHA), was investigated at human A1 adenosine receptors stably expressed in CHO cells.
Pretreatment with urea caused a 56% reduction in membrane proteins. Compared to controls, the use of adenosine deaminase (ADA), 100 μM 5′-guanylylimidodiphosphate (Gpp(NH)p) or urea each caused equivalent increases in specific [3H]DPCPX binding.
Neither the binding kinetics nor the affinity of [3H]DPCPX were significantly different in urea-pretreated compared to ADA-pretreated membranes.
At 25°C in ADA-pretreated membranes, the competition isotherms for R-PIA and CHA were characterized by two affinity states. Gpp(NH)p (100 μM) reduced, but did not abolish, the value of the high-affinity dissociation constant. Similar results were obtained after treatment with urea for R-PIA, whereas the high-affinity state for CHA was abolished.
At 37°C, urea pretreatment, but not 100 μM Gpp(NH)p, abolished high-affinity agonist competition binding. There was no significant effect of any of the treatments on the low-affinity agonist binding state.
In urea-pretreated membranes, exogenously added adenosine competed according to a simple mass-action model with a pKL of 5.66±0.05 (n=3).
Compared to the more common approaches of ADA treatment and/or use of guanine nucleotides, our findings suggest that urea pretreatment represents an inexpensive and useful approach for investigating the binding properties of adenosine A1 ligands (including adenosine) to the G protein-uncoupled form of the receptor.
Keywords: Adenosine, A1 receptor, drug discovery, G protein-coupled receptor, kinetics, radioligand binding, urea
Introduction
The purine nucleoside, adenosine, plays a vital role in regulating many physiological processes, particularly in the cardiovascular and central nervous systems. In pathophysiological situations, adenosine is generally regarded as being a tissue protective molecule. Many of the protective actions of adenosine result from it acting via the adenosine A1 receptor, one of four G protein-coupled receptor (GPCR) subtypes recognized by adenosine. The A1 receptor is widely expressed throughout the body, having its highest expression in the brain, spinal cord, atria and adipose tissue (Baraldi et al., 2000; Kourounakis et al., 2001). Therapies that promote or enhance the actions of adenosine at the A1 receptor are thought to improve tissue protection during a heart attack or stroke (Kollias-Baker et al., 1994; Mizumura et al., 1996; Tranberg et al., 2002), provide antinociception during chronic pain states (Pan et al., 2001; Li et al., 2002; 2003) and alleviate epileptic seizure activity (Haas & Selbach, 2000). For these reasons, compounds acting at this receptor subtype have generated much interest as potentially novel therapeutic agents.
Despite recent advances in functional cellular assays for GPCR ligands, radioligand-binding assays remain a common and useful method for screening novel ligands at established GPCR targets, such as the A1 receptor. However, and in contrast to most other GPCR-based binding assays, studies of adenosine receptor-binding properties are complicated by high levels of endogenous adenosine that are constantly produced by all cell types and can distort the results of binding assays at adenosine receptors. A common approach to removing the influence of endogenous adenosine is to promote its metabolism using adenosine deaminase (ADA). Unfortunately, this method precludes the subsequent use of exogenously added adenosine in the binding assay, and thus relies solely on the use of surrogate adenosine agonists that are chemically resistant to the effects of ADA. An alternative method for removing the impact of endogenous adenosine is to promote the uncoupling of the A1 receptor from its cognate G protein(s) using either (a) GTP or nonhydrolysable guanine nucleotide analogues such as 5′-guanylylimidodiphosphate (Gpp(NH)p), (b) pretreatment of cells with pertussis toxin, which uncouples Gi/o-family proteins from their cognate receptors (e.g. A1 receptor) via ADP ribosylation or (c) the use of the sulphydryl modifying agent, N-ethylmaleimide, which alkylates Gi/o proteins on the same cysteine residue that is recognized by pertussis toxin (Böhm et al., 1993). Although these treatments do not actually remove the adenosine, they promote a conformation of the receptor that generally has too low an affinity for the endogenous agonist to bind appreciably (e.g., see Cohen et al., 1996a, 1996b; Lorenzen et al., 2000).
Another, generally underused, approach for uncoupling G proteins from receptors involves pretreating cell membranes with a high concentration of the chaotropic agent, urea (Willardson et al., 1993). Chaotropic agents are thought to cause denaturation of all nonintegral proteins, such as G proteins, by disrupting the hydrophobic interactions that stabilize their native conformation (Lim & Neubig, 2001). It has previously been shown that 7 M urea is sufficient to functionally inactivate G protein subunits, while not affecting receptor function if G proteins are reconstituted back into the system (Lim & Neubig, 2001). An additional advantage of using a method that uncouples receptors from G proteins for ligand screening is that the resulting binding parameters should reflect the affinity of the test ligand for the free receptor, rather than a system-dependent measure influenced by the complement and stoichiometry of G proteins. Therefore, the aim of the current study was to investigate the impact of chaotropic extraction of nonintegral membrane proteins with urea on the ligand-binding properties of human A1 adenosine receptors, and to compare this method with more commonly used approaches for studying ligand binding at these receptors.
Methods
Materials
Dulbecco's modified Eagle medium (DMEM) and geneticin were purchased from Invitrogen (Carlsbad, CA, U.S.A.). Foetal bovine serum (FBS) was purchased from ThermoTrace (Melbourne, VIC, Australia). [3H]cyclopentyl-1,3-dipropylxanthine 8 dipropyl-2,3 ([3H]DPCPX) (108.3 Ci mmol−1) was purchased from Perkin-Elmer (Boston, MA, U.S.A.) and ADA, derived from calf intestine, was purchased from Roche (Basel, Switzerland). All other reagents were purchased from Sigma-Aldrich (St Louis, MO, U.S.A.).
Cell culture
Chinese hamster ovary (CHO-K1) cells, stably transfected with the human adenosine A1 receptor (A1 CHO cells), were kindly provided by Professor Peter Schofield from the Garvan Institute of Medical Research, Sydney, Australia. Cells were grown to 80% confluence and maintained in DMEM containing 20 mM HEPES, 10% FBS and 50 μg ml−1 geneticin at 37°C in a humidified incubator containing 5% CO2 : 95% O2. Cells were then harvested by trypsinization, followed by centrifugation (300 × g, 3 min), and resuspension of the pellet in the appropriate buffer (described below) prior to membrane preparation.
Cell membrane preparation
No pretreatment
The intact cell pellet was suspended in homogenization buffer (50 mM Tris(hydroxymethyl)aminomethane (Tris(Base)), 0.1 mg ml−1 saponin, pH 7.7 with HCl) and centrifuged (300 × g, 3 min). Cells were then resuspended in homogenization buffer and homogenized using a Polytron PT1200 homogenizer for two 10-s intervals at maximum setting (6), with 30-s cooling periods on ice between each burst. The homogenate was then centrifuged (30,000 × g, 30 min, 4°C), the pellet resuspended in Tris(HCl) buffer (50 mM Tris(Base), pH 7.7 with HCl) and centrifuged again (30,000 × g, 30 min, 4°C). The resulting pellet was resuspended in 5 ml of Tris(HCl) buffer, and the protein content determined using the method of Bradford (1976). The homogenate was then divided into 1-ml aliquots and either used immediately or stored frozen at −80°C until required for radioligand-binding assays.
ADA pretreatment
Intact A1 CHO cell pellets were treated as described in the preceding paragraph. However, after the second centrifugation at 30,000 × g, the pellet was resuspended in Tris(HCl) buffer containing 10 U ml−1 ADA (Bruns & Fergus, 1990), incubated at 37°C for 30 min and then re-centrifuged (30,000 × g, 30 min, 4°C). The resulting pellet was resuspended in Tris(HCl) buffer and centrifuged again (30,000 × g, 30 min, 4°C). This final pellet was then resuspended in 5 ml of Tris(HCl) buffer, and the protein content determined as described above.
Urea pretreatment
Intact A1 CHO cell pellets were suspended in solution A (10 mM 4-morpholinepropanesulphonic acid (MOPS) pH 7.5, 1 mM ethylene glycol-bis-aminoethylethertetraacetic acid (EGTA), 100 μM 4-(2-aminoethyl) benzenesulphonyl fluoride (AEBSF)) and left at 4°C for 30 min. Cells were then homogenized using a Polytron homogenizer for two 10-s intervals at a maximum setting with 30 s cooling periods on ice between each burst. The homogenate was then centrifuged (1600 × g, 10 min), the pellet discarded and the supernatant recentrifuged at 30,000 × g for 40 min at 4°C. The resulting pellet was then resuspended in solution A containing 7 M urea and left on ice for 30 min. Membrane suspension was then diluted two-fold with solution A and centrifuged at 142,000 × g for 30 min at 4°C. The resulting pellet was then resuspended in solution A and the procedure repeated. The final pellet was resuspended in 3 ml of Tris(HCl) buffer, and the protein content determined as described above. For some experiments, a mock extraction was also performed, which was identical to the preceding procedure except for the inclusion of urea.
Saturation-binding assays
Initial binding experiments utilized a single concentration of [3H]DPCPX (1 nM) to ascertain the influence of endogenous adenosine under three different conditions, namely, (i) no membrane pretreatment; (ii) membrane pretreatment with ADA; (iii) membrane pretreatment with urea (see above). For these experiments, 50 μg ml−1 (no pretreatment or ADA pretreated) or 20 μg ml−1 (urea pretreated) of A1 CHO cell membranes were incubated for 90 min at 25°C in 1 ml total volume of Tris(HCl) buffer containing 1 nM of [3H]DPCPX with or without 0.1 U ml−1 ADA and/or 100 μM Gpp(NH)p (as indicated in the Results). Nonspecific binding was defined using 10 μM (−)-N6-(2-phenylisopropyl)-adenosine (R-PIA). Incubation was terminated by rapid filtration through Whatman GF/C filters using a Brandell cell harvester (Gaithersburg, MD, U.S.A.). Filters were washed three times with 3-ml aliquots of ice-cold Tris(HCl) buffer and dried before the addition of 4 ml of scintillation cocktail (Ultima Gold; Packard Bioscience, Meriden, CT, U.S.A.). Vials were then left to stand until the filters became uniformly translucent, before radioactivity was determined using scintillation counting.
Subsequent experiments involved the determination of the saturation-binding properties of [3H]DPCPX. For these experiments, 50 μg ml−1 (no pretreatment or ADA pretreated) or 20 μg ml−1 (urea pretreated) of A1 CHO cell membranes was incubated for 90 min at either 25 or 37°C (as indicated in the Results) in a 1-ml total volume of Tris(HCl) buffer (also containing 0.1 U ml−1 ADA for ADA-pretreated membranes only), and concentrations of [3H]DPCPX ranging from 0.01 to 20 nM. Determination of nonspecific binding, termination of the reaction and determination of radioactivity were performed as described above.
Association kinetic assays
In all, 50 μg ml−1 (no pretreatment or ADA pretreated) or 20 μg ml−1 (urea pretreated) of A1 CHO cell membranes was incubated with 0.02 nM [3H]DPCPX in a 1-ml total volume of Tris(HCl) buffer (also containing 0.1 U ml−1 ADA for ADA-pretreated membranes only) for various time periods (see Results) at 25°C. The concentration of radioligand was based on the lowest concentration to be used in subsequent saturation-binding experiments. Determination of nonspecific binding, termination of the reaction and determination of radioactivity were performed as described above.
Dissociation kinetic assays
In all, 50 μg ml−1 (no pretreatment or ADA pretreated) or 20 μg ml−1 (urea pretreated) of A1 CHO cell membranes was equilibrated for 90 min at 25°C in a 1-ml total volume of Tris(HCl) buffer with 1 nM [3H]DPCPX (also containing 0.1 U ml−1 ADA for ADA-pretreated membranes only). Then, 10 μl of R-PIA (yielding a final concentration of 10 μM) was added at various time points using a reverse-time protocol to prevent radioligand re-association. Determination of nonspecific binding, termination of the reaction and determination of radioactivity were performed as described above.
Competition-binding assays
In all, 50 μg ml−1 (no pretreatment or ADA pretreated) or 20 μg ml−1 (urea pretreated) of A1 CHO cell membranes was incubated for 90 min at either 25 or 37°C (as indicated in the Results) in a 1-ml total volume of Tris(HCl) buffer containing 0.1 U ml−1 ADA±100 μM Gpp(NH)p (ADA-pretreated membranes only), 1 nM of [3H]DPCPX and a range of concentrations of one of the adenosine A1 receptor agonists R-PIA (0.01 nM–100 μM) or N6-cyclohexyladenosine (CHA) (0.01 nM–30 μM). In additional experiments, A1 CHO cell membranes were incubated for 90 min at 37°C in a 1-ml total volume of Tris(HCl) buffer containing 0.1 U ml−1 of ADA and 100 μM Gpp(NH)p (ADA-pretreated membranes only), 1 nM of [3H]DPCPX and a range of concentrations of adenosine (3 nM–3 mM). Nonspecific binding was defined using 100 μM R-PIA. Termination of the reaction and determination of radioactivity were performed as described above.
Data analysis
In all instances, the amount of radioligand bound was less than 10% of the total amount added; data were thus analysed using models that do not assume radioligand depletion. Data sets of total and nonspecific binding obtained from each saturation-binding assay were globally fitted to the following equation using Prism 4.0 (GraphPad Software, San Diego, CA, U.S.A.):
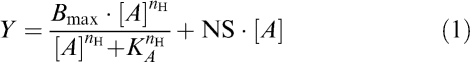 |
where Y represents radioligand binding, [A] denotes the concentration of radioligand, Bmax denotes the maximal density of binding sites, KA is the radioligand equilibrium dissociation constant, nH is an empirical slope factor and NS is the fraction of nonspecific binding. The hyperbolic term in this equation was not used when fitting the nonspecific binding data, whereas the parameter, NS, was shared between both total and nonspecific binding data sets (Motulsky & Christopoulos, 2004). An extra-sum-of-squares (F-test) determined that allowing the nH value to differ from 1 did not result in a significant improvement (P>0.05) in the fit; thus, the data were re-fitted with this value constrained to 1.
Data sets of percentage-specific binding obtained from each association kinetic assay were analysed according to the following equation for two-phase exponential association using Prism 4.0:
where Bt represents specific binding at time t, Amp1 represents the amplitude of the binding phase governed by the apparent association rate constant, k1, and Amp2 represents the amplitude of the binding phase governed by the apparent association rate constant, k2.
Dissociation kinetic data were fitted to the following monoexponential decay equation using Prism 4.0:
where Bt denotes the specific binding of radioligand at time t, B0 represents the specific binding of radioligand at equilibrium (time=0) and koff denotes the radioligand dissociation rate constant. An extra-sum-of-squares (F-test) determined that the data were adequately described by this model rather than a more complex two-phase dissociation model.
With the exception of the [3H]DPCPX/adenosine competition data in ADA-pretreated membranes, all competition-binding data were empirically fitted to either a one- (equation (4)) or two-site (equation (5)) inhibition mass-action curve using Prism 4.0:
where Top is the specific binding of the radioligand in the absence of any competing ligand, Bottom is the specific binding of the radioligand equal to nonspecific binding, IC50 is the concentration of competing ligand that produces radioligand binding half-way between the Top and Bottom, and X is the logarithm of the concentration of the competing ligand.
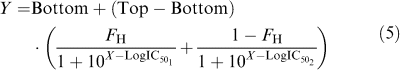 |
where Y, Top, Bottom and X are as defined above for equation (4); FH denotes the fraction of receptors inhibiting radioligand binding with a potency described by IC501, while IC502 representing the inhibitory potency of the remaining fraction of receptors. In all instances, an extra-sum-of-squares (F-test) was used to determine whether the data were better described by a one- versus a two-site model. For each agonist where a two-site fit was preferred over a one-site fit, the entire family of curves (representing different treatment conditions) were re-fitted to equation (5) using ‘global nonlinear regression analysis' (Motulsky & Christopoulos, 2004), whereby all curves from a single experiment were simultaneously fitted to the one model with selected parameters either allowed to float between individual curves or constrained to be shared by all curves (see Results for further details). The asymptotic values of Top and Bottom were always constrained to be shared across data sets. In all instances, an F-test was used to select the minimum global model that could describe the entire family of curves. Subsequently, the resulting IC50 values for the high- versus low-affinity states were converted to apparent (empirical) dissociation constants (KH or KL values, as appropriate) using the Cheng & Prusoff (1973) equation and the average KA value determined for [3H]DPCPX at either 25 or 37°C, as appropriate (see Results). The concentration of radioligand was measured separately for each individual experiment and was also used in the Cheng Prusoff correction.
All data represent the mean±s.e.m. for the number of experiments indicated. For the nonlinear regression procedures, a relative weighting scheme was applied to the datapoints (weight=1/Y2; Motulsky & Christopoulos, 2004). All affinity/potency estimates were determined as logarithms. Statistical analysis involved performing Student's t-test, F-test or one-way ANOVA (with Bonferroni post-test), as appropriate, and a probability (P) value of 0.05 was taken to indicate statistical significance.
Results
Impact of endogenous adenosine on [3H]DPCPX binding
The profound effect that endogenous adenosine has on [3H]DPCPX binding to human A1 receptors is demonstrated in Figure 1a. In comparison to CHO cell membranes that had not undergone any pretreatment, the addition of 0.1 U ml−1 ADA approximately doubled the specific binding of [3H]DPCPX. The addition of 100 μM Gpp(NH)p also restored [3H]DPCPX binding to a similar level as seen in the presence of 0.1 U ml−1 ADA. The combination of 0.1 U ml−1 ADA and 100 μM Gpp(NH)p did not increase [3H]DPCPX any further than either agent used alone. Figure 1b illustrates the results of an alternative approach whereby CHO cell homogenates had been pretreated with ADA during membrane preparation prior to washout and radioligand binding. Under these conditions, the subsequent addition of 0.1 U ml−1 ADA, 100 μM Gpp(NH)p or a combination of the two did not have a significant effect (P>0.05) on specific radioligand binding over and above that observed in the absence of added ADA and/or Gpp(NH)p.
Figure 1.

Effects of various treatments on the binding of [3H]DPCPX (1 nM) to human A1 receptors stably expressed in CHO cell membranes. (a) Membranes had no pretreatment during preparation prior to addition of the indicated treatment. [3H]DPCPX-specific binding in the absence of any treatment was 188±22 fmol mg−1 protein, in the presence of 0.1 U ml−1 ADA was 396±28 fmol mg−1 protein, in the presence of 100 μM Gpp(NH)p was 380±26 fmol mg−1 protein and in the presence of both 0.1 U ml−1 ADA and 100 μM Gpp(NH)p was 423±30 fmol ml−1 protein. **P<0.01; ***P<0.001; one-way ANOVA with Bonferroni post-test. (b) Membranes were pretreated with 10 U ml−1 ADA during preparation (as indicated in the Methods) prior to addition of the indicated treatment. [3H]DPCPX-specific binding in the absence of any treatment was 269±16 fmol mg−1 protein, in the presence of 0.1 U ml−1 ADA was 291±12 fmol mg−1 protein, in the presence of 100 μM Gpp(NH)p was 309±15 fmol mg−1 protein and in the presence of both 0.1 U ml−1 ADA and 100 μM Gpp(NH)p was 311±20 fmol mg−1 protein. In all cases, incubation was for 90 min at 25°C in 50 mM Tris(HCl) buffer. Nonspecific binding was defined using 10 μM R-PIA. Data points represent the mean±s.e.m. obtained from five separate experiments performed in triplicate. There were no significant differences (P>0.05) between groups.
Effect of urea pretreatment of [3H]DPCPX binding
An alternative method of removing the influence of endogenous adenosine explored in the present study was the use of the chaotropic agent, urea, to denature nonintegral membrane proteins; it has previously been established that pretreatment of cell homogenates with 7 M urea is required to obtain functional inactivation of G protein subunits (Lim & Neubig, 2001). As shown in the inset to Figure 2, chaotropic extraction of membrane proteins with urea led to 38% protein recovery compared to the procedure used in the ADA-pretreatment protocol. Also shown is the effect of mock extraction with buffer (see Methods), which itself yielded approx. 68% recovery (i.e., mock extraction in the absence of urea removes approx. 32% of nonintegral membrane proteins in its own right). Taken together, these findings indicate that urea is able to remove approx. 56% of membrane proteins. The subsequent addition of 0.1 U ml−1 ADA, 100 μM Gpp(NH)p or a combination of the two to membranes that had been pretreated with 7 M urea did not significantly increase (P>0.05) the [3H]DPCPX-specific binding in comparison to control (urea pretreatment only) homogenates (Figure 2, main panel). This indicates that urea pretreatment was sufficient to abolish the influence of any endogenous adenosine on the binding of radioligand.
Figure 2.

Inset: Protein recovery after membrane pretreatment with ADA (1350±125 μg ml−1; n=3), mock urea pretreatment (925±63 μg ml−1; n=3) or urea pretreatment (520±38 μg ml−1; n=3), as outlined in the Methods. *P<0.05; **P<0.01; one-way ANOVA with Bonferroni post-test. Main panel: Effects of various treatments on the binding of [3H]DPCPX (1 nM) to human A1 receptors stably expressed in CHO cell membranes that were pretreated with 7 M urea during preparation (as indicated in Methods). [3H]DPCPX-specific binding in the absence of any treatment was 474±93 fmol mg−1 protein, in the presence of 0.1 U ml−1 ADA was 539±109 fmol mg−1 protein, in the presence of 100 μM Gpp(NH)p was 550±114 fmol mg−1 protein and in the presence of both 0.1 U ml−1 ADA and 100 μM Gpp(NH)p was 494±115 fmol mg−1 protein. There were no significant differences (P>0.05) between groups. All other details as for Figure 1.
Comparison of the binding kinetics of [3H]DPCPX under different treatment conditions
Figure 3a and b show the association and dissociation kinetics of [3H]DPCPX at 25°C for ADA- and urea-pretreated membranes, respectively. For both sets of membranes, the dissociation of [3H]DPCPX from the receptor was monophasic and essentially complete by 30 min. The dissociation rate constant of [3H]DPCPX from the human adenosine A1 receptor in ADA-pretreated membranes was estimated as 0.27±0.03 min−1 (n=3), which was not significantly different from the value of 0.36±0.08 min−1 (n=3) determined using urea-pretreated membranes. In contrast, [3H]DPCPX displayed complex association kinetics, characterized by at least two phases. For ADA-pretreated membranes, nonlinear regression yielded apparent rate constants of k1=0.003 ±0.130 min−1 and k2=0.14±0.03 min−1, and amplitude estimates of Amp1=26±9% and Amp2=74±16% (n=3). Similar results were obtained in urea-pretreated membranes: k1=0.004±0.100 min−1 and k2=0.14±0.05 min−1, Amp1=20±10% and Amp2=80±5% (n=3). The large errors in the estimates of the k1 values reflect the poorly defined value of the slow rate constant.
Figure 3.

[3H]DPCPX association and dissociation kinetics at 25°C to human A1 receptors stably expressed in CHO cell membranes. (a) Membranes were pretreated with 10 U ml−1 ADA during preparation (as indicated in the Methods) and the assays were subsequently performed in the presence of 0.1 U ml−1 ADA. (b) Membranes were pretreated with 7 M urea during preparation (as indicated in Methods). [3H]DPCPX concentrations were 0.02 nM for association and 1 nM for dissociation experiments, in 50 mM Tris(HCl) buffer. Nonspecific binding was defined using 10 μM R-PIA. For the dissociation kinetic experiments, membranes were initially equilibrated with radioligand for 90 min prior to addition of 10 μM R-PIA at various time points. Data points represent the mean±s.e.m. obtained from three separate experiments conducted in triplicate.
[3H]DPCPX saturation binding
[3H]DPCPX saturation binding was determined using both ADA- and urea-pretreated membranes at two different temperatures. Figure 4 shows the results obtained at 25°C, which is a common temperature for many A1 receptor-binding assays reported in the literature. The negative logarithm of the radioligand dissociation constant (pKA value) in ADA-pretreated membranes was 8.94±0.03 (n=5) and the Bmax was 770±60 fmol mg−1 protein (Figure 4a). In urea-pretreated membranes, the [3H]DPCPX pKA was 8.66±0.08 (n=3) and the Bmax was 2110±280 fmol mg−1 protein (Figure 4b). There was no significant difference (P>0.05) between the two pKA values, but there was a significant enhancement (P<0.05) in Bmax after urea pretreatment. As there was no significant difference between the two pKA values, an average value was calculated (8.80) for subsequent use in the analysis of the competition-binding experiments. [3H]DPCPX saturation binding was also performed at the more physiologically relevant temperature of 37°C (Figure 5). At this temperature, the pKA value for ADA-pretreated membranes was 8.51±0.08 (n=3) and the Bmax was 1600±100 fmol mg−1 protein (Figure 5a), whereas using urea-pretreated membranes the DPCPX pKA was 8.60±0.27 (n=3) and the Bmax was 2631±500 fmol ml−1 protein (Figure 5b). Again, there was no significant difference (P>0.05) between pKA values determined at this temperature, yielding an average value of 8.55, but there was a significant difference (P<0.05) between Bmax values.
Figure 4.

[3H]DPCPX saturation binding (90 min, 25°C in 50 mM Tris(HCl) buffer) to human A1 receptors stably expressed in CHO cell membranes. (a) Membranes were pretreated with 10 U ml−1 ADA during preparation (as indicated in Methods) and the assays were subsequently performed in the presence of 0.1 U ml−1 ADA. (b) Membranes were pretreated with 7 M urea during preparation (as indicated in Methods). Data points represent the mean+s.e.m. obtained from five (a) or three (b) separate experiments conducted in triplicate. The curve superimposed on the data represents the best fit of the hyperbolic term in equation (1) using the parametric values for KA and Bmax stated in Results. All other details as for Figure 1.
Figure 5.

[3H]DPCPX saturation binding (90 min, 37°C in 50 mM Tris(HCl) buffer) to human A1 receptors stably expressed in CHO cell membranes. (a) Membranes were pretreated with 10 U ml−1 ADA during preparation (as indicated in Methods) and the assays were subsequently performed in the presence of 0.1 U ml−1 ADA. (b) Membranes were pretreated with 7 M urea during preparation (as indicated in Methods). Data points represent the mean+s.e.m. obtained from three separate experiments conducted in triplicate. All other details as for Figure 4.
[3H]DPCPX competition-binding studies
Figure 6a shows the results of competition binding between [3H]DPCPX and the agonist, R-PIA, at 25°C. In ADA-pretreated membranes, the agonist competition-binding isotherm was characterized by a Hill slope significantly less than 1 (P<0.05; Table 1), and was distinctly biphasic. The addition of 100 μM Gpp(NH)p, or the use of urea-pretreated membranes, resulted in a steepening of the curve, although the value of the Hill slope remained significantly less than 1 (P<0.05; Table 1). Global nonlinear regression analysis of the entire family of curves, in conjunction with an extra-sum-of-squares (F-test), indicated that the data were minimally described by a two-state binding model, with the effect of Gpp(NH)p or urea pretreatment being a reduction in the apparent affinity of the high-affinity state (pKH), with no significant (P>0.05; F-test) effect on the fraction of high-affinity states (FH) or on the value of the apparent dissociation constant for the low-affinity state (pKL); thus, a single estimate could be obtained for FH and pKL that described all the three curves, with the only difference between curves being the value of the pKH parameter (Table 1). This analysis was statistically preferred (P<0.05; F-test) to constraining the high-affinity state parameter to be shared, while allowing the fraction of high-affinity states to vary. Although allowing all parameters to vary resulted in a slight improvement in the curve fit, evidenced by a reduction in the overall sum-of-squares; this improvement in fit was not significant compared to that where FH and pKL were shared across all data sets (P>0.05; F-test). The results of the analysis are shown in Table 1. Similar experiments were also performed using CHA as the competing agonist (Figure 6b). Again, the results were qualitatively similar to the R-PIA competition assays, with the use of Gpp(NH)p or urea pretreatment leading to significant reductions (P<0.05) in the estimated agonist pKH value (Table 1); in this instance, the experiments performed in urea-pretreated membranes resulted in a complete loss of the high-affinity state, yielding a monophasic isotherm with a Hill slope not significantly different (P>0.05) from unity (Figure 6b; Table 1).
Figure 6.

[3H]DPCPX (2 nM) versus R-PIA (a) or CHA (b) competition-binding assays (90 min, 25°C in 50 mM Tris(HCl) buffer) performed at human A1 receptors stably expressed in CHO cell membranes. Membranes had undergone either 10 U ml−1 ADA pretreatment during the preparation plus subsequent addition of 0.1 U ml−1 ADA in the assay in the absence or presence of 100 μM Gpp(NH)p, or membranes have undergone 7 M urea pretreatment during preparation. Data points represent the mean+s.e.m. obtained from three or four separate experiments conducted in triplicate. Curves superimposed on the data represent the best of equation (5) estimated using simultaneous global nonlinear regression with Prism. All other details as for Figure 1.
Table 1.
[3H]DPCPX competition-binding parameters against various agonists at human adenosine A1 receptor stably expressed in CHO cell membranes
| Temperature | Agonist | Treatment | pKHa | pKLb | FHc | nHd | ne |
|---|---|---|---|---|---|---|---|
| 25°C | R-PIA | ADAf | 8.82±0.08g | 6.42±0.13 | 0.68±0.03 | 0.54±0.03h | 3 |
| Gpp(NH)pi | 7.72±0.06g | 6.42±0.13 | 0.68±0.03 | 0.72±0.05h | 3 | ||
| Ureaj | 7.71±0.06 | 6.42±0.13 | 0.68±0.03 | 0.63±0.04h | 3 | ||
| CHA | ADA | 8.64±0.09 | 6.15±0.19 | 0.69±0.04 | 0.52±0.05h | 3 | |
| Gpp(NH)p | 7.21±0.08 | 6.15±0.19 | 0.69±0.04 | 0.77±0.08h | 3 | ||
| Urea | ND | 6.15±0.19 | NA | 1.17±0.13 | 3 | ||
| 37°C | R-PIA | ADA | 8.00±0.23 | 6.39±0.20 | 0.51±0.11 | 0.63±0.07h | 3 |
| Gpp(NH)p | 7.16±0.14 | 6.39±0.20 | 0.51±0.11 | 1.05±0.14 | 3 | ||
| Urea | ND | 6.39±0.20 | NA | 1.17±0.17 | 3 | ||
| CHA | ADA | 8.22±0.14 | 6.37±0.14 | 0.54±0.06 | 0.55±0.05h | 3 | |
| Gpp(NH)p | 7.08±0.12 | 6.37±0.14 | 0.54±0.06 | 0.80±0.09h | 3 | ||
| Urea | ND | 6.37±0.14 | NA | 0.95±0.06 | 3 | ||
| Adenosine | Urea | ND | 5.66±0.05 | NA | 0.98±0.09 | 3 |
Assays were performed for 90 min in 50 mM Tris(HCl) buffer at the indicated temperatures. Nonspecific binding was defined using 10 μM R-PIA.
Negative logarithm of the apparent dissociation constant for the high-affinity agonist-binding state, determined by global nonlinear regression analysis of all three treatment groups to a common binding model (see Methods).
Negative logarithm of the apparent dissociation constant for the low-affinity agonist-binding state, determined by global nonlinear regression analysis of all three treatment groups to a common binding model. An extra-sum-of squares (F-test) concluded that a single value of the parameter could describe all the curves.
Fraction of the high-affinity agonist-binding state, determined by global nonlinear regression analysis of all three treatment groups to a common binding model. An extra-sum-of squares (F-test) concluded that a single value of the parameter could describe all the curves where a high-affinity state was evident.
Hill slope, determined by nonlinear regression analysis to a logistic function.
Number of experiments.
Membranes were pretreated with 10 U ml−1 ADA during preparation, and assays subsequently performed in the presence of 0.1 U ml−1 ADA.
Significantly different (P<0.05; Student's t-test) from the corresponding value at 37°C.
Significantly different (P<0.05; F-test) from 1.
As in footnote f above, but with the addition of 100 μM Gpp(NH)p.
Membranes were pretreated with 7 M urea during preparation.
ND, not determined.
NA, not applicable.
The competition-binding experiments were also repeated at 37°C (Figure 7). As with the experiments conducted at the lower temperature, competition binding using ADA-pretreated membranes in the absence of Gpp(NH)p resulted in biphasic inhibition isotherms with either R-PIA (Figure 7a) or CHA (Figure 7b). For the R-PIA-binding data in the presence of Gpp(NH)p, a nonrandom distribution of the best-fit curve around the experimentally determined data points was noted, but this most likely reflects the constraints imposed by the application of a two-state model to accommodate all the three curves; increasing the complexity of the model by introducing additional affinity states would improve the goodness-of-fit, but would not yield additional mechanistic insights, as the model is an empirical approximation of a mechanism involving interconverting affinity states.
Figure 7.

[3H]DPCPX (2 nM) versus R-PIA (a) or CHA (b) competition-binding assays (90 min, 37°C in 50 mM Tris(HCl) buffer) performed at human A1 receptors stably expressed in CHO cell membranes. Membranes had undergone either 10 U ml−1 ADA pretreatment during the preparation plus subsequent addition of 0.1 U ml−1 ADA in the assay in the absence or presence of 100 μM Gpp(NH)p, or membranes have undergone 7 M urea pretreatment during preparation. All other details as for Figure 6.
Interestingly, each agonist displayed a lower apparent dissociation constant (pKH) for the high-affinity state at 37°C relative to 25°C; this difference was statistically significant for R-PIA (P<0.05). There was no difference, however (P>0.05), between the low-affinity state estimates between temperatures (Table 1). Irrespective of the addition of 100 μM Gpp(NH)p, or the use of urea-pretreated membranes, each resulted in a reduction of the high-affinity state for each agonist at 37°C, which was complete in the urea-pretreated membranes (Table 1).
The findings from the competition experiments indicated that urea pretreatment is sufficient to allow the assessment of the binding of commonly used A1 adenosine receptor agonists to the low-affinity state of the receptor, especially at the physiologically relevant temperature of 37°C. Figure 8 illustrates the utility of this approach for quantifying the binding of the native agonist, adenosine, to the low-affinity state of the receptor. The standard practice of adding ADA to the assay buffer, or pretreating membranes with ADA prior to the assay, precludes the use of adenosine in routine binding assays due to residual effects of the enzyme. This is evident in Figure 8 by the inability of up to 300 μM adenosine to affect [3H]DPCPX binding in the presence of 100 μM Gpp(NH)p in membranes that had been pretreated by ADA (open squares). In contrast, the use of urea-pretreated membranes does not suffer from this drawback; the competition binding isotherm for adenosine in urea-pretreated membranes (circles, Figure 8) resulted in a curve with a Hill slope not significantly different (P>0.05) from unity, and allowed for the estimation of the dissociation constant of adenosine for the low-affinity state of the receptor (Table 1).
Figure 8.

[3H]DPCPX (2 nM) versus adenosine competition-binding assays (90 min, 37°C in 50 mM Tris(HCl) buffer) performed at human A1 receptors stably expressed in CHO cell membranes. Adenosine was used as the competing ligand. Membranes had undergone either 10 U ml−1 ADA pretreatment during the preparation plus subsequent addition of 0.1 U ml−1 ADA in the assay in the presence of 100 μM Gpp(NH)p, or membranes have undergone 7 M urea pretreatment during preparation. Data points are from either one experiment representative of two, or represent the mean±s.e.m. obtained from three separate experiments conducted in triplicate. All other details as for Figure 6.
Discussion
This study has demonstrated that the chaotropic extraction/denaturing of nonintegral membrane proteins, such as G proteins, with urea represents a useful and inexpensive alternative to the use of ADA or guanine nucleotides for cancelling the impact of endogenously produced adenosine on the binding properties of human adenosine A1 receptors. Moreover, urea pretreatment of membranes can eliminate the detection of multiple affinity states caused by the presence of both G protein-coupled and -uncoupled populations of receptors, thus allowing for a system-independent determination of the affinity of A1 agonists for the uncoupled form of the receptor.
Adenosine is constantly produced by all cell types as a product of metabolic reactions and transported out of the cells, where it is able to act at its target receptors. Despite the disruption of cells that occurs during membrane preparation and the accompanying multiple wash steps, appreciable amounts of adenosine remain trapped within the membrane preparation. The problem that this presents for all studies of adenosine receptor binding is clearly illustrated in Figure 1a, where [3H]DPCPX binding was studied in the absence of any form of membrane pretreatment. Relative to control levels, the addition of ADA to the reaction mixture, or the use of ADA in the membrane preparation stage (Figure 1b), leads to a significant increase in specific radioligand binding. ADA acts to catalyse the deamination of adenosine into inosine (Linden, 1989), and the resulting increase in specific radioligand binding thus reflects the removal of endogenously produced adenosine that was competing with the [3H]DPCPX for binding to the receptor.
A previous study by Cohen et al. (1996a) estimated the amount of endogenous adenosine produced by CHO cells to be approximately 60 nM. Given that the dissociation constant of adenosine for the G protein-coupled form of the receptor is approx. 10 nM (Cohen et al., 1996a), 60 nM endogenously produced adenosine would thus result in a significant occupancy of A1 receptors that exist in this high-affinity state. In contrast, the affinity of adenosine for the free (G protein-uncoupled) form of the A1 receptor is in the micromolar range (Figure 8, Table 1; also Cohen et al., 1996a), and thus any endogenously produced adenosine would have minimal impact on the determination of ligand binding to this state of the A1 receptor. This fact provides the rationale for another common method of removing the influence of endogenous adenosine on the determination of ligand binding to A1 receptors, namely, the inclusion of guanine nucleotides in the incubation medium. Figure 1a demonstrates that uncoupling adenosine A1 receptors from their G proteins using the nonhydrolysable guanine nucleotide analogue, Gpp(NH)p, is able to restore [3H]DPCPX binding to approximately the same level as that observed in the presence of ADA. Furthermore, each of these treatments appears maximally effective, as the combination of the two does not cause any further enhancement in radioligand binding (Figure 1).
Our study now highlights an alternative method for eliminating the influence of endogenous adenosine during A1 binding assays. Urea is a chaotropic agent that is thought to cause denaturation and/or extraction of all nonintegral membrane proteins (Lim & Neubig, 2001). G proteins are nonintegral membrane proteins that associate with the cell membrane via post-translational modifications. For instance, the Gα subunits are anchored to the membrane by either a myristoyl and/or palmitoyl moiety. The Gγ subunit associates with the cell membrane via a prenyl group, while the Gβ subunit is localized to the cell membrane region through interactions with Gγ subunits (Obrdlik et al., 2000). A previous study has demonstrated that while the activity of Gα subunits was attenuated by 5 M urea, the Gβγ subunits required 7 M urea to be functionally inactivated (Lim & Neubig, 2001). That same study estimated that the total loss of membrane proteins after urea extraction was approximately 60%; this compares favourably with the estimate from our study of 56% (Figure 2). On the basis of these results, we chose to use 7 M urea for our experiments. Surprisingly, this approach has rarely been used for radioligand-binding assays, but we have shown that this is as effective in removing the influence of endogenous adenosine on [3H]DPCPX binding (Figure 2), as either ADA and/or Gpp(NH)p (Figure 1). Although the absolute levels of specific [3H]DPCPX binding varied between the experiments shown in Figure 1 relative to those in Figure 2, this is to be expected, since the normalization of binding data to fmol mg−1 of protein would result in an increase of the apparent binding capacity for urea-treated membranes due to the selective loss of nonintegral membrane protein content, with no concomitant loss of receptor protein content.
One possible concern when using such a high concentration of urea to uncouple the G proteins from the receptors is that the urea may itself modify the ligand-binding properties and/or the function of the receptor. With respect to receptor function, Lim & Neubig (2001) have demonstrated that while 7 M urea pretreatment of membranes resulted in functional inactivation of the endogenous G protein subunits, normal α2 adrenoceptor function was restored after re-constitution with exogenous G proteins. Similarly, Northup and colleagues have shown that, after protein extraction with 6 M urea, 5-HT2C receptors, CB1 receptors and gastrin-releasing peptide receptors retain appropriate rank orders of potency for agonists and antagonists after G protein subunit reconstitution (Hartman & Northup, 1996; Hellmich et al., 1997; Glass & Northup, 1999). In terms of effects on ligand binding, our study has used a number of different methods to demonstrate that the binding properties of the adenosine A1 receptor were not altered after urea pretreatment. For instance, when comparing ADA- and urea-pretreated membranes, we found that both the association and dissociation kinetics of the antagonist [3H]DPCPX from the adenosine A1 receptor were similar in each case (Figure 3); the complex association kinetics observed for the antagonist are consistent with previous findings (Cohen et al., 1996b). Similarly, saturation-binding experiments revealed no significant effect on [3H]DPCPX affinity between the two approaches (Figures 4 and 5), although the estimated Bmax was significantly increased after urea pretreatment (for reasons outlined above).
Finally, to determine the effect of the various treatments on A1 agonist-binding affinity, [3H]DPCPX competition-binding assays were used; agonist saturation experiments were not attempted as the affinity of the receptor for agonist in the presence of guanine nucleotide or after urea pretreatment would be too low to allow the reliable detection of specific radioligand binding. Global nonlinear regression analysis, whereby the entire family of agonist curves were simultaneously fitted to the one model and parameters shared across data sets, revealed that all agonist competition isotherms, in the absence or presence of guanine nucleotide or urea pretreatment, could be described by a common dissociation constant (pKL in Table 1) for the low-affinity state of the receptor for each agonist. This is consistent with the notion that the affinity of an agonist for the G protein uncoupled form of the receptor should remain the same across the various treatments, and further supports our contention that ligand-binding properties of the free A1 receptor are not modified as a result of the urea pretreatment. However, albeit not surprisingly, both Gpp(NH)p and urea pretreatment did have a striking effect on agonist binding to the high-affinity state of the receptor (Table 1; Figures 6 and 7), manifested as a reduction in the value of the dissociation constant for the high-affinity state (pKH in Table 1). This finding is consistent with both treatments reducing the affinity of the G protein for the receptor.
Another interesting observation arising from the competition experiments was the effect of temperature on the agonist competition-binding isotherms. R-PIA displayed a significantly higher affinity for the high-affinity binding state at 25°C compared to 37°C (Table 1). A similar trend was evident for CHA, although this was not statistically significant. The mechanism underlying this effect is currently unknown, but it should be noted that A1 agonist high-affinity binding has previously been shown to be temperature-sensitive (Lorenzen et al., 2000). In contrast, the affinity values for the low-affinity state were similar irrespective of temperature. Temperature also appeared to play a role in the effectiveness of receptor–G protein uncoupling measures. At 25°C neither Gpp(NH)p nor urea pretreatment could fully abolish high-affinity agonist binding for R-PIA, although urea was able to abolish high-affinity CHA binding (Table 1; Figure 6). The inability of Gpp(NH)p to completely remove high-affinity agonist binding to the adenosine A1 receptor, although not a consistent observation (Cohen et al., 1996a, 1996b; Klotz et al., 1998), has been reported previously and suggested to reflect a particularly tight coupling between the A1 receptor and its G proteins (Stiles, 1985; Finlayson et al., 2003); we hypothesize that the inability of urea to completely remove high-affinity agonist binding under the same conditions reflects a similar phenomenon. However, it is worth noting that urea pretreatment was able to abolish high-affinity agonist binding (for CHA) under conditions where Gpp(NH)p could not. This was even more evident at the physiologically relevant temperature of 37°C (Figure 7; Table 1), where urea pretreatment revealed only one-state (low-affinity) binding for both R-PIA and CHA.
Taken together, our findings suggest that the use of urea-pretreated membranes at 37°C represents an optimal approach for investigating the binding properties of adenosine A1 ligands to the G protein-uncoupled form of the receptor. This has an advantage over the use of Gpp(NH)p in that it is inexpensive and completely promotes the uncoupled state of the receptor. Urea pretreatment also has an advantage over the use of ADA in that it allows for binding experiments to be performed using exogenously added adenosine (Figure 8). Although chaotropic extraction of nonintegral membrane proteins with urea will impact the receptor function, we propose that this approach remains valid and useful for studies of adenosine A1 ligand binding.
Acknowledgments
We are extremely grateful to Dr Nigel Birdsall for helpful comments and critical review of the manuscript. This work was supported by Project Grant No. 251538 of the National Health and Medical Research Council (NHMRC) of Australia. Arthur Christopoulos and Patrick Sexton are Senior Research Fellows of the NHMRC. Lauren May is the recipient of a Melbourne Research Scholarship of the University of Melbourne.
Abbreviations
- ADA
adenosine deaminase
- AEBSF
4-(2-aminoethyl)benzenesulphonyl fluoride
- CHA
N6-cyclohexyladenosine
- CHO
Chinese hamster ovary
- DMEM
Dulbecco's modified Eagle medium
- EGTA
ethylene glycol-bis-aminoethylethertetraacetic acid
- FBS
foetal bovine serum
- GPCR
G protein-coupled receptor
- Gpp(NH)p
5′-guanylylimidodiphosphate
- [3H]DPCPX
[3H]cyclopentyl-1,3-dipropylxanthine 8 dipropyl-2,3
- MOPS
4-morpholinepropanesulphonic acid
- R-PIA
(−)-N6-(2-phenylisopropyl)-adenosine
References
- BARALDI P.G., ZAID A.N., LAMPRONTI I., FRUTTAROLO F., PAVANI M.G., TABRIZI M.A., SHRYOCK J.C., LEUNG E., ROMAGNOLI R. Synthesis and biological effects of a new series of 2-amino-3-benzoylthiophenes as allosteric enhancers of A1-adenosine receptor. Bioorg. Med. Chem. Lett. 2000;10:1953–1957. doi: 10.1016/s0960-894x(00)00379-6. [DOI] [PubMed] [Google Scholar]
- BÖHM M., GRABEL C., KIRCHMAYR R., LENSCHE H., ERDMANN E., GIERSCHIK P. C-terminal modifications of pertussis toxin-sensitive G-protein alpha-subunits differentially affect immunoreactivity. Evidence against endogenous ADP-ribosylation in human heart, lung, thrombocytes and adipose tissue. Biochem. Pharmacol. 1993;46:2145–2154. doi: 10.1016/0006-2952(93)90603-t. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- BRUNS R.F., FERGUS J.H. Allosteric enhancement of adenosine A1 receptor binding and function by 2-amino-3-benzoylthiophenes. Mol. Pharmacol. 1990;38:939–949. [PubMed] [Google Scholar]
- CHENG Y., PRUSOFF W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- COHEN F.R., LAZARENO S., BIRDSALL N.J. The affinity of adenosine for the high- and low-affinity states of the human adenosine A1 receptor. Eur. J. Pharmacol. 1996a;309:111–114. doi: 10.1016/0014-2999(96)00415-3. [DOI] [PubMed] [Google Scholar]
- COHEN F.R., LAZARENO S., BIRDSALL N.J. The effects of saponin on the binding and functional properties of the human adenosine A1 receptor. Br. J. Pharmacol. 1996b;117:1521–1529. doi: 10.1111/j.1476-5381.1996.tb15316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FINLAYSON K., MAEMOTO T., BUTCHER S.P., SHARKEY J., OLVERMAN H.J. Comparison of effects of MgCl2 and Gpp(NH)p on antagonist and agonist radioligand binding to adenosine A1 receptors. Acta Pharmacol. Sin. 2003;24:729–740. [PubMed] [Google Scholar]
- GLASS M., NORTHUP J.K. Agonist selective regulation of G proteins by cannabinoid CB(1) and CB(2) receptors. Mol. Pharmacol. 1999;56:1362–1369. doi: 10.1124/mol.56.6.1362. [DOI] [PubMed] [Google Scholar]
- HAAS H.L., SELBACH O. Functions of neuronal adenosine receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 2000;362:375–381. doi: 10.1007/s002100000314. [DOI] [PubMed] [Google Scholar]
- HARTMAN J.I., NORTHUP J.K. Functional reconstitution in situ of 5-hydroxytryptamine2c (5HT2c) receptors with alphaq and inverse agonism of 5HT2c receptor antagonists. J. Biol. Chem. 1996;271:22591–22597. doi: 10.1074/jbc.271.37.22591. [DOI] [PubMed] [Google Scholar]
- HELLMICH M.R., BATTEY J.F., NORTHUP J.K. Selective reconstitution of gastrin-releasing peptide receptor with G alpha q. Proc. Natl. Acad. Sci. U.S.A. 1997;94:751–756. doi: 10.1073/pnas.94.2.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLOTZ K.N., HESSLING J., HEGLER J., OWMAN C., KULL B., FREDHOLM B.B., LOHSE M.J. Comparative pharmacology of human adenosine receptor subtypes – characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;357:1–9. doi: 10.1007/pl00005131. [DOI] [PubMed] [Google Scholar]
- KOLLIAS-BAKER C., RUBLE J., DENNIS D., BRUNS R.F., LINDEN J., BELARDINELLI L. Allosteric enhancer PD 81,723 acts by novel mechanism to potentiate cardiac actions of adenosine. Circ. Res. 1994;75:961–971. doi: 10.1161/01.res.75.6.961. [DOI] [PubMed] [Google Scholar]
- KOUROUNAKIS A., VISSER C., DE GROOTE M., AP I.J. Differential effects of the allosteric enhancer (2-amino-4,5-dimethyl-trienyl)[3-trifluoromethyl) phenyl]methanone (PD81,723) on agonist and antagonist binding and function at the human wild-type and a mutant (T277A) adenosine A1 receptor. Biochem. Pharmacol. 2001;61:137–144. doi: 10.1016/s0006-2952(00)00536-0. [DOI] [PubMed] [Google Scholar]
- LI X., CONKLIN D., MA W., ZHU X., EISENACH J.C. Spinal noradrenergic activation mediates allodynia reduction from an allosteric adenosine modulator in a rat model of neuropathic pain. Pain. 2002;97:117–125. doi: 10.1016/s0304-3959(02)00011-8. [DOI] [PubMed] [Google Scholar]
- LI X., CONKLIN D., PAN H.L., EISENACH J.C. Allosteric adenosine receptor modulation reduces hypersensitivity following peripheral inflammation by a central mechanism. J. Pharmacol. Exp. Ther. 2003;305:950–955. doi: 10.1124/jpet.102.047951. [DOI] [PubMed] [Google Scholar]
- LIM W.K., NEUBIG R.R. Selective inactivation of guanine-nucleotide-binding regulatory protein (G-protein) alpha and betagamma subunits by urea. Biochem. J. 2001;354:337–344. doi: 10.1042/0264-6021:3540337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LINDEN J. Adenosine deaminase for removing adenosine: how much is enough. Trends Pharmacol. Sci. 1989;10:260–262. doi: 10.1016/0165-6147(89)90021-7. [DOI] [PubMed] [Google Scholar]
- LORENZEN A., GUERRA L., CAMPI F., LANG H., SCHWABE U., BOREA P.A. Thermodynamically distinct high and low affinity states of the A1 adenosine receptor induced by G protein coupling and guanine nucleotide ligation states of G proteins. Br. J. Pharmacol. 2000;130:595–604. doi: 10.1038/sj.bjp.0703339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIZUMURA T., AUCHAMPACH J.A., LINDEN J., BRUNS R.F., GROSS G.J. PD 81,723, an allosteric enhancer of the A1 adenosine receptor, lowers the threshold for ischemic preconditioning in dogs. Circ. Res. 1996;79:415–423. doi: 10.1161/01.res.79.3.415. [DOI] [PubMed] [Google Scholar]
- MOTULSKY H., CHRISTOPOULOS A. Fitting Models to Biological Data Using Linear and Nonlinear Regression. Oxford: Oxford University Press; 2004. [Google Scholar]
- OBRDLIK P., NEUHAUS G., MERKLE T. Plant heterotrimeric G protein beta subunit is associated with membranes via protein interactions involving coiled-coil formation. FEBS Lett. 2000;476:208–212. doi: 10.1016/s0014-5793(00)01706-3. [DOI] [PubMed] [Google Scholar]
- PAN H.L., XU Z., LEUNG E., EISENACH J.C. Allosteric adenosine modulation to reduce allodynia. Anesthesiology. 2001;95:416–420. doi: 10.1097/00000542-200108000-00025. [DOI] [PubMed] [Google Scholar]
- STILES G.L. The A1 adenosine receptor. Solubilization and characterization of a guanine nucleotide-sensitive form of the receptor. J. Biol. Chem. 1985;260:6728–6732. [PubMed] [Google Scholar]
- TRANBERG C.E., ZICKGRAF A., GIUNTA B.N., LUETJENS H., FIGLER H., MURPHREE L.J., FALKE R., FLEISCHER H., LINDEN J., SCAMMELLS P.J., OLSSON R.A. 2-Amino-3-aroyl-4,5-alkylthiophenes: agonist allosteric enhancers at human A(1) adenosine receptors. J. Med. Chem. 2002;45:382–389. doi: 10.1021/jm010081p. [DOI] [PubMed] [Google Scholar]
- WILLARDSON B.M., POU B., YOSHIDA T., BITENSKY M.W. Cooperative binding of the retinal rod G-protein, transducin, to light-activated rhodopsin. J. Biol. Chem. 1993;268:6371–6382. [PubMed] [Google Scholar]