

Abstract
Exposure of cultured cortical neurons to elevated extracellular K+ concentrations (25 mM) induces membrane depolarization and an increase in action-potential firing.
Long-term high K+ treatment was associated with an increased neuronal cell death.
In surviving neurons, multiple changes occurred in the proportion of individual NMDA receptor subunit 1 (NR1) splice variant mRNA expression, whereas the overall expression of NR1, NR2A and NR2B transcripts remained unaffected. The high K+-induced changes in NR1 splice variant expression were virtually abolished upon a concurrent administration of tetrodotoxin (TTX; 3 μM).
In voltage-clamp recordings performed on neurons resistant to high K+ treatment, inward currents induced by NMDA (1–1000 μM) were reduced.
In K+-resistant cells, the activity of calpain but not of caspase-3 was diminished compared with controls kept in regular medium.
NR function as well as calpain activity was not affected in cultures concomitantly treated with high K+ and either TTX or a NR antagonist (CGS19755 (selfotel) or memantine).
In conclusion, the present data indicate adaptive changes in NR1 splice variant expression and a decrease in NR function upon a sustained increase in neurotransmission. Accordingly, alternative splicing could be an endogenous mechanism to counteract cellular damage due to overactivation of excitatory NRs and may be associated with an impairment of necrotic mechanisms.
Keywords: Neuroprotection, NMDA receptors, receptor adaptation, calpain, caspase-3, cultured cortical neurons
Introduction
Native (N-methyl-D-aspartate) (NMDA) receptors are multimeric ligand-gated ion channels consisting of at least one member of the NMDA receptor (NR)1 subunit family and at least one of four different subunits of the NR2 family (NR2A–D). The NR1 family comprises eight splice variants generated by alternative splicing of the NR1 subunit at exon 5, encoding a 21 amino-acid sequence in the extracellular N-terminal domain (designated as the N1 cassette), and at adjacent exons 21 and 22, encoding a 37 (C1 cassette) and a 38 amino-acid sequence (C2 cassette), respectively, in the intracellular C-terminal domain (Laurie & Seeburg, 1994; Zukin & Bennett, 1995). The composition and stoichiometry of NRs appear to vary within different brain regions (Dunah et al., 1998). However, NR1 expression is presumed to be obligatory for NR function.
Among the glutamate receptors, NRs are unique in exhibiting a high Ca2+/Na+ permeability ratio and a requirement for the coagonist glycine (Dingledine et al., 1999). The glycine binding site is expressed on NR1, whereas the glutamate-binding site is localized on an extracellular region of the NR2 subunits (Hirai et al., 1996; Laube et al., 1997). NR-mediated increases in intracellular Ca2+ levels may acutely affect receptor and channel activities, but also nuclear gene transcription. NRs participate in neuronal differentiation and maturation, learning and memory, and are implicated in neuronal survival and neurotoxicity associated with neuronal disorders like stroke, epilepsy and Parkinson's disease (Parsons et al., 1998, Vallano, 1998; Cull-Candy et al., 2001).
The pharmacological properties of NRs are largely dependent on their subunit composition. NR-cytoskeletal linkage – via the C1 cassette of NR1 and α-actinin-2 – is essential for synaptic localization and receptor activity (Wyszynski et al., 1997; Chandler et al., 1998). Phosphorylation of serine residues within the C1 cassette by protein kinase C dispersed NR1 clusters in transfected QT6 fibroblasts (Ehlers et al., 1995). Furthermore, Ca2+-calmodulin binding to the C1 cassette subsequent to Ca2+ entry disrupts the receptor–cytoskeletal interaction, and reduces channel activity (Ehlers et al., 1996). In addition to these modulatory mechanisms, expression of NR1 splice variants lacking the C1 cassette may be involved in long-term regulation of receptor clustering and function (Chandler et al., 1998). Since excessive Ca2+ influx through NRs can result in neuronal death, negative feedback mechanisms such as phosphorylation of C1 cassette residues or alternative splicing could be crucial protective mechanisms to prevent excitotoxic cell death. The present study investigated the effects of long-term high K+ treatment on NR1 subunit expression and NR function, as well as on calpain and caspase-3 activation in rat cultured cortical neurons.
Methods
Cell cultures
Primary cultures of cortical neurons were prepared from the frontal/parietal cortex of Wistar rats (gestational day 16) as described previously (Scheibler et al., 1999). The tissue sections were pooled in ice-cold (Hanks balanced salt solution (HBSS); Gibco BRL, Gaithersburg, MD, U.S.A.), and then dissociated by treatment with 0.25% trypsin (Gibco BRL) in the presence of DNase (≈200 U) and by careful trituration. Subsequent to centrifugation, the supernatant was discarded and the pellet was dispersed in a 1 : 1 mixture of DMEM/F12 supplemented with 20% fetal calf serum (FCS; Seromed), 2 mM L-glutamine, 50 μg ml−1 gentamycin (Gibco BRL; 50 mg ml−1) and 36 mM D-glucose (Sigma, St Louis, MO, U.S.A.). The cell suspension was plated on poly-L-lysine-coated (25 μg ml−1; Sigma) culture dishes with a density of 150,000 cells cm−2. Cells were maintained in culture up to 15 days at 37°C in an atmosphere of 5% CO2/95% air. The culture medium was replaced every 5–7 days. To prevent proliferation of non-neuronal cells, 10 μM cytosine-arabinoside (cytosine-β-arabinofuranoside; Sigma) was added for 24 h on DIV6, and FCS was reduced to 10%, thereafter. Immediately after plating, cortical neurons were grown in medium containing either 5 or 25 mM K+. TTX (3 μM; Tocris, Avonmouth, U.K.) was added either up to DIV10 (immunocytochemical studies) or from DIV10 to 15 (mRNA expression and electrophysiological studies). The NR antagonists memantine (Tocris) and CGS19755 (Tocris) were present from DIV10 to 15 as indicated.
Immunocytochemistry
Cortical neurons (DIV10) were stained with a monoclonal mouse antibody directed against microtubule-associated protein-2 (MAP-2; Sigma). NR1 immunoreactivity was detected with a polyclonal anti-NR1 (NMDA zeta 1, C-20; Santa Cruz) antibody raised in goat against a peptide mapping at C22 of rat NR1 terminus (identical to the corresponding mouse and human sequences); NR2A and NR2B were stained using a polyclonal anti-NR2A (Santa Cruz Sc-1468) and a polyclonal anti-NR2B (Santa Cruz Sc-1469) antibody also raised in goat. Astrocytes were detected with a monoclonal mouse antibody directed against glial fibrillary acidic protein (GFAP; Sigma). The cultures were fixed in methanol (−20°C) for 10 min and then washed twice for 5 min in Tris-buffered saline (TBS). For permeabilization and blocking, the cells were treated with 0.5% Triton X-100 and 5% FCS in TBS for 30 min at room temperature. Thereafter, the permeabilized cells were incubated with a mixture of MAP-2 antibody (1 : 2000) or GFAP antibody (1 : 1000) and one of the corresponding NR antibodies (1 : 200) in TBS containing 0.5% Triton X-100 for 24 h at 4°C. After washing, the cells were incubated in a mixture of secondary antibodies of Cy2-tagged rabbit anti-mouse IgG (1 : 600; Dianova, Hamburg, Germany) and Cy3-tagged rabbit anti-goat IgG (1 : 600; Dianova) for 2 h at room temperature. After rinsing, the specimens were dehydrated and mounted in glycerol gelatine (Sigma). Immunofluorescence was studied by means of a confocal laser-scanning microscope (LSM 510, Zeiss) with an excitation wavelength of 488 nm (Cy2) or of 543 nm (Cy3).
Counting of neurons
In control cultures (5 mM K+), cultures treated with high potassium (25 mM K+) and cultures treated with high K+ plus TTX (3 μM), the number of neurons at DIV10 was determined subsequent to staining of the neuronal nuclei with monoclonal mouse neuron-specific nuclear protein (NeuN; 1 : 200; Chemicon, Temecula, CA, U.S.A.) antibody and secondary Cy2-tagged rabbit anti-mouse IgG antibody (1 : 600; for experimental details, see above). For each preparation, immunofluorescence was recorded in 4–6 randomly selected fields of view (921.3 μm × 921.3 μm) using the LSM 510 (Plan-Neofluar × 10/0.30 W objective). In Adobe Photoshop 6.0, the LSM images (enlarged to 100% of size) were overlaid with a grid pattern consisting of 54 × 54 single squares and the neurons were counted manually.
Molecular biology
RNA preparation and reverse transcription
Total RNA from primary rat cortical cultures was extracted at DIV10 (control and high K+ group) and at DIV15 (control, high K+ and high K+ plus TTX group) using Total Quick RNA Cells and Tissues kit mini (Talent, Italy) according to the protocol of the manufacturer. Samples treated with RNase-free DNase (10 U μl−1; Roche, No. 776785) and subsequently repurified on silica resin were devoid of genomic DNA. First-strand cDNA was synthesized by reverse transcription of 1 μg total RNA with 100 U Superscript™-II reverse transcriptase (Life Technologies, Grand Island, NY, U.S.A.) in first-strand buffer containing 12.5 mM dithiothreitol, 500 μM of each dNTP, 5 μM pd(N)6 (Pharmacia Biotech) and 20 U. RNase block ribonuclease inhibitor (Stratagene) according to standard protocols. The reaction was stopped by heating at 70°C for 15 min.
Real-time fluorescence PCR Generation of cDNA calibration fragments specific for NR subunits
PCR amplification of cDNA calibration fragments specific for NR subunits NR1, NR2A and NR2B, respectively, was performed on a PTC-200 Thermocycler (MJ Research) in a final volume of 25 μl PCR buffer (10 mM Tris-HCl, pH 8.3, 50 mM KCl) containing 1 μl cDNA, 1 U Ampli-Taq DNA Polymerase (Applied Biosystems, Foster City, CA, U.S.A.), 200 nM (anti)sense primer, 200 μM of each dNTP and 1 mM MgCl2. The primers used are given in Table 1a. The amplification conditions were as follows: 94°C for 5 min followed by 35 cycles of denaturation (94°C for 30 s), annealing (NR1: 58°C, NR2A: 56°C, NR2B: 56.3°C for 30 s, each), extension (72°C for 90 s) and a final extension step at 72°C for 7 min. Amplified products were separated by agarose gel electrophoresis (1.5%) and visualized by ethidium bromide fluorescence. cDNA was extracted from agarose gel by means of QIAquick Gel Extraction Kit (Qiagen) and subsequently used for the generation of cDNA reference curves for the respective NR subunits.
Table 1.
RT–PCR primers and probes
| (a) Primers used for amplification of specific cDNA calibration fragments | ||||
|---|---|---|---|---|
| Subunit | Sequences of sense (s) and antisense (as) primer (5′ → 3′) | Position (nt) | Fragment seize (bp) | Accession code (NCBI) |
| NR1 | s GGA GTG GAA CGG AAT GAT GG | 1753–1772; exon 13 | 787 | X63255 |
| as AAA GCC TGA GCG GAA GAA CA | 2520–2539; exon 18 | |||
| NR2A | s ATG GCG GGC GTG TTC TAC AT | 2671–2690; exon 13 | 1030 | M91561 |
| as TGC GGA AGT TCT CAT TGT GA | 3681–3700; exon 14 | |||
| NR2B | s CTT TGC TTC TAC CGG CTA TG | 2325–2344; exon 11 | 376 | M91562 |
| as TTG GCG CTC CTC TAT GGC TA | 2681–2700; exon 13 | |||
| (b) Real-time RT–PCR primers and probes | ||||
|---|---|---|---|---|
| Subunit | Sequences (5′ → 3′) of sense (s) primer, antisense (as) primer and probes | Position (nt) | ||
| NR1 | s CGC ATC ACG GGC ATC A | 2252–2267; exon 16 | ||
| as GCT TTA CAG TTG CGT AGA TGA ACT TG | 2321–2296; exon 17 | |||
| FAM-ACC CCA GGC TCA GAA ACC CCT CAG-TAMRA | 2271–2294; exon 17 | |||
| NR2A | s AGC TGC GCT TCT GCT TCA C | 2753–2771; exon 13 | ||
| as CCC CAT GGA TGC AAC TAT AGA TG | 2820–2842; exon 14 | |||
| FAM-CCC GGG CTG CTC TTC TCC ATC A-TAMRA | 2791–2812; exon 13 | |||
| NR2B | s CTA TCC TGC AGC TGT TTG GAG A | 2393–2415; exon 11 | ||
| as CTC ATT CTT CTC ATT GTG GCA AAT | 2455–2478; exon 12 | |||
| FAM-AGA TGG AAG AAC TGG AAG CTC TCT GGC TCA-TAMRA | 2420–2449; exon 12 | |||
FAM: 6-carboxyfluorescein; TAMRA: 6-carboxytetramethylrhodaminc.
Analysis of NR subunit gene transcript expression
NR1, NR2A and NR2B gene transcript expression was measured by a ready-to-use real-time quantitative fluorescence PCR assay. Briefly, conventional 96-well bases were loaded with a given number of sample tubes, eight reference tubes coated with eight different amounts of cDNA of the NR subunit under investigation and with another eight tubes coated with eight different cDNA amounts of GAPDH serving as internal standard in each sample. Aliquots (23 μl) of a master mix containing mRNA-specific sense and antisense primer (see Table 1b), the TaqMan probe (5′-labelled with the fluorescent reporter dye 6-carboxyfluorescein (FAM), and the common 3′-fluorescent quencher dye 6-carboxytetramethyl-rhodamin (TAMRA)), PCR buffer, passive fluorescence dye 6-carboxy-tetramethyl-rhodamin (ROX), MgCl2, dNTPs and AmpliTaq® ‘GOLD' (1.5 U; Applied Biosystems) were added to each tube (sample as well as reference and internal standard tubes) by means of a BIOMEK®2000 laboratory automation workstation (Beckman Instruments Inc.). A final reaction volume of 25 μl per tube was completed by the addition of 2 μl aliquots of sample cDNA and 2 μl of H2O to sample tubes and reference/internal standard tubes, respectively. PCR amplification and detection were performed with an ABI PRISM® 7000 Sequence Detection System (Applied Biosystems). Each sample was run in duplicate. Sample cDNA transcripts for the various NR subunits (NR1, N2A, and NR2B) were determined from the respective reference curves obtained with the simultaneously processed control strips and normalized by the number of GAPDH cDNA transcripts (in attomol) determined in the same sample (Taubert et al., 2000).
NR1 splice variant expression
NR1 splice variant expression was investigated by a semiquantitative approach described previously by Allgaier et al. (1999), using primer pairs flanking exon 5 and exons 21 and 22, respectively (see Figure 1). For C-terminal NR1 splice variant fragment amplification a common sense primer (sE21/E22) and different antisense primers (asE21; asE22) were used for exon 22-containing isoforms (NR1-1, NR1-2) and exon 22-deleted isoforms (NR1-3, NR1-4). The amplification conditions were as follows: 94°C for 5 min followed by 35 cycles of denaturation (94°C for 30 s), annealing (58°C for 30 s) and extension (72°C for 1 min), and a final extension step at 72°C for 10 min. N-terminal NR1 cDNA fragments were amplified using the specific primer pair sE5/asE5 (Figure 1) at an annealing temperature of 64°C (Allgaier et al., 1999). For further details on PCR amplification conditions see above (Generation of cDNA calibration fragments). Amplified products were separated by agarose gel electrophoresis (2%) and visualized by ethidium bromide fluorescence. C-terminal amplification products were semiquantitatively analysed by Southern hybridization with a digoxygenin-labelled DNA probe and chemiluminescence detection (CSPD using a Diana II Digital Image Analyzer (raytest) (Allgaier et al., 1999).
Figure 1.

(a) Cartoon on NR1 splice variant terminology and nucleotide (nt) sequences of primer pairs used for PCR amplification of NR1 splice variant cDNA fragments. Alternative splicing at exons 5, 21 and 22 encoding amino-acid cassettes N1, C1 and C2, respectively, generates a total number of eight different splice variants. (b) Primer sequences and product size (bp).
Western blotting
Cultured cortical neurons were rinsed on ice with phosphate-buffered saline (PBS). Then the samples were scraped off the culture dishes with 0.5 ml of lysis buffer (5 mM Tris-HCl, 1 mM EDTA, 0.1 M PMSF, 1 M OVNa, 2 mg ml−1 aprotinin; pH 7.4) and homogenized in a Teflon homogenizer. After 10 strokes, the resulting suspension was centrifuged at 4°C for 30 min in a centrifuge (Sigma; centrifuge 3K15) at 13,500 r.p.m. The supernatant was removed and the pellet was resuspended in buffer (50 mM Tris-HCl, 0.1 M PMSF, 1 M OVNa, 2 mg ml−1 aprotinin). Protein concentration was determined by the Bradford method using BSA as a standard (BioRad). The protein samples were stored at −80°C until used for electrophoresis. Cell extracts containing equivalent amounts of protein were boiled for 5 min in sample loading buffer (0.5 M Tris-HCl, pH 6.8, 10% glycerol, 2% w v−1 SDS, 5% v v−1 2-β-mercaptoethanol, 0.05% bromophenol blue). In all, 20 μg of protein was loaded onto a 7.5% SDS–polyacrylamide gel and subsequently transferred to a polyvinylidene difluoride membrane (PVDF) (Immobilon™-P; Millipore Corp., Bedford, MA, U.S.A.) using a transblot apparatus (BioRad). Nonspecific binding was blocked with 5% nonfat dry milk dissolved in TBS-T buffer (50 mM Tris; NaCl 1.5%; 0.05% Tween 20, pH 7.5) for 1 h. Blots were incubated with a primary polyclonal antimyosin antibody (1 : 3000) (Paesel+Lorei, Hanau, Germany) and a primary monoclonal anti-α-spectrin antibody (1 : 1000) (Chemicon, Temecula, CA, U.S.A.) as a marker of necrosis (related to calpain activation) and apoptosis (related to caspase-3 activation) in TBS-Tween with 5% nonfat milk at room temperature for 1 h. Then, the blots were washed twice in TBS-Tween buffer, incubated with horseradish peroxidase-conjugated secondary mouse monoclonal antibody (1 : 2000; Cell Signalling) in TBS-Tween for 30 min and finally washed three times for 5 min in TBS-Tween buffer. Immunoreactive protein was visualized using an enhanced chemiluminescence kit (ECL; BioRad). Protein loading was routinely monitored by phenol red staining of the membrane. Images were acquired by chemilux programme (CSX-1400M Camera Controller).
Electrophysiology
Experiments were performed at 20–23°C on multipolar cortical neurons DIV15 incubated either under control conditions or in the presence of high K+, or in the presence of high K+ plus TTX (3 μM; from DIV10 to DIV15). In some experiments, memantine (50 μM; Tocris) or CGS19755 (50 μM; Tocris) were used instead of TTX. NMDA-induced currents were recorded at a holding potential of −70 mV in the whole-cell configuration of the patch-clamp technique. The bath solution contained (in mM): NaCl 162, KCl 2.4, CaCl2 1.2, HEPES 10, glycine 0.01 and glucose 11 (∼320 mosmol, pH 7.3 with NaOH). TTX (0.3 μM; Tocris), picrotoxin (100 μM; Sigma) and strychnine (2 μM; Sigma) were added to inhibit spontaneous activity as well as ligand-gated chloride channels. The pipette solution contained (in mM): CsCl 95, CaCl2 1, HEPES 10, MgCl2 6, EGTA 11, K-ATP 4, potassium creatine phosphate 20 and 50 U ml−1 phosphocreatine kinase (∼300 mosmol, pH 7.2 with CsOH). Whole-cell currents were recorded by means of an EPC-9 amplifier, filtered at 1 kHz, digitized at 3 kHz and analysed using Pulse software (HEKA). NMDA was applied by means of a pressurized fast-flow superfusion system (DAD-12, Adams and List). In order to asses concentration–response relationships for NMDA, increasing concentrations of the agonist (1–1000 μM) were applied every 120 s for 2 s each, and peak current amplitudes were measured. Curves were fitted to weighted (1/s.d.) mean data points using the equation:
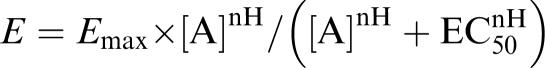 |
where E is the observed current, Emax the extrapolated maximal current, [A] the NMDA concentration (μM), EC50 the concentration of NMDA that produces 50% of Emax and nH the Hill coefficient (slope).
The blocking action of extracellular Mg2+ on NRs was investigated over a broad membrane potential range. To this end, consecutive voltage ramps (2 s from −100 to +40 mV) were applied from the holding potential of −70 mV, 20 s before application of NMDA, and around the peak currents evoked by pressure application of NMDA (30 μM), first in the absence and second 2 min later in the presence of 1 mM Mg2+. NR-mediated current–voltage relationships for both conditions (Mg2+-free, 1 mM Mg2+) were then generated by digitally subtracting the respective membrane currents obtained in the absence of NMDA from those obtained in its presence. Mg2+-induced changes were measured as differences in the corresponding current amplitudes at membrane potentials of −80, −60, −40 and −20 mV and expressed as percent inhibition.
The acute effect of elevated extracellular K+ on membrane potential was measured under current clamp on rat cortical neurons DIV15 incubated either under control conditions (5 mM K+) or in the presence of high K+ (25 mM K+). To this end, TTX, picrotoxin and strychnine were omitted from the standard bath solution and 1 mM MgCl2 was added. In the 25 mM K+ bath solution, 22.6 mM NaCl was replaced by the same amount of KCl. The pipette solution contained (mM): KCl 140, CaCl2 1, MgCl2 2.0, HEPES 10 and EGTA 11 (pH 7.2; ∼300 mosmol). High K+ solution was pressure-applied two times for 10 s to the neuron under investigation, first in the absence of TTX and second after 2 min of superfusion in the presence of TTX (0.3 μM).
Statistics
All data are given as arithmetic means±s.e.m. The significance of differences between the examined groups was determined by Student's t-test, or one-way ANOVA followed by Bonferroni's t-test in the case of multiple comparisons.
Results
Effect of extracellular high K+ on membrane potential
The membrane potential of cortical neurons grown either in 5 or 25 mM K+ and subsequently measured in the standard bath solution containing 2.4 mM K+ were −65±4 and −63±5 mV, respectively (n=6, each). Only one out of the 12 neurons (5 mM K+ culture), included in these experiments, fired spontaneous action potentials, albeit at low frequency (two action potentials during a 30-s observation period). Pressure application (10 s) of standard bath solution containing 5 mM K+ had no effect on the membrane potential in the 12 cells (data not shown). In contrast, acutely pressure-applied high K+ (25 mM; for 10 s) induced depolarization and action potential firing in both control (Figure 2a) and high K+-treated neurons. During the 10 s of high K+ application, 6.6±2.8 (range 2–16) and 7.2±1.7 (range 2–12) action potentials were elicited in neurons grown in 5 and 25 mM K+, respectively. Accordingly, these data indicate that neurons even after long-term K+ treatment exhibited regular responses to acutely elevated extracellular K+. TTX (0.3 μM) had no effect on its own on the membrane potential, but prevented action potentials in response to acutely pressure-applied high K+ (Figure 2b). When measured in the presence of TTX, a highly selective inhibitor of voltage-dependent Na+ channels, the high K+-induced depolarizations were 27±2 and 31±2 mV in cells grown in either 5 or 25 mM K+.
Figure 2.

Effects of high K+ (25 mM) on the membrane potential of rat cortical neurons. Membrane potential was measured under current clamp in the whole cell configuration of the patch-clamp technique. The figure shows responses in a representative control neuron grown in 5 mM K+, which, however, were qualitatively and quantitatively similar to the responses obtained in neurons grown in high K+ (25 mM; see Results). The high K+ (25 mM)-containing bath solution was pressure-applied two times for 10 s to the neurons under investigation, first in the absence of TTX (a) and second after 2 min of superfusion and in the continued presence of TTX (0.3 μM; b). Horizontal lines above membrane potential traces indicate times of high K+ and TTX application. Dotted lines indicate the membrane potential (−69 mV) of the cell. Fast vertical deflections are action potentials, which may not be resolved in their full height due to the low sampling frequency (3 kHz).
Effect of sustained elevated K+ levels on cell viability and NR1 expression
Using NeuN, a specific marker for neuronal nuclear protein, it was studied whether long-term high-potassium treatment was associated with a loss of cortical neurons. To this end, primary cultures of rat cortical neurons (DIV10) were kept either under control conditions (5 mM K+), in the presence of high K+ (25 mM) or in the presence of high K+ plus TTX (3 μM), and subsequently stained overnight with anti-NeuN primary antibody (1 : 200). A careful evaluation of the LSM images after visualization of the stained cortical neurons with Cy2-tagged secondary antibody revealed a significant decrease in the number of NeuN-positive neurons by about 30% by high potassium treatment (Figure 3a and b). This high potassium-induced neuronal loss was prevented upon concurrent administration with 3 μM TTX (Figure 3a and b).
Figure 3.

(A) Determination of the number of neurons in rat primary cortical cultures incubated under control conditions, in the presence of high K+ (25 mM) or in the presence of high K+ plus TTX (3 μM). Evaluated data of neurons (DIV10) stained with primary antibody against NeuN (1 : 200) and subsequently visualized with Cy2-tagged secondary antibody (green fluorescence; scale bar: 20 μm). (B) Means±s.e.m. (expressed in % of the number of neurons in controls) of the experiments described in (A) are given. In all, 4–6 images per group were evaluated in two preparations each. An average number of 1878±90 (n=21) neurons was determined in each field of view in controls. Significant differences vs control: *P<0.01. (C) Confocal images of rat cultured cortical neurons double-stained with primary antibodies against MAP-2 (1 : 2000) (image a), GFAP (1 : 1000) (image d) and NR1 (1 : 200) (images b, e). MAP-2 and GFAP immunoreactivity was visualized by Cy2-tagged (green fluorescence) secondary antibodies. NR1 immunoreactivity was revealed by Cy3-tagged (red fluorescence) secondary antibodies. Merged images are shown in images c, f. Scale bar: 20 μm. Note that NR1 immunoreactivity was detectable only in neurons but not in glial cells.
Double-staining experiments with anti-NR1 antibody and with a monoclonal antibody directed against MAP-2, a cytoskeletal protein expressed in nerve cell bodies and dendrites but virtually absent from most axons (Caceres et al., 1984; Matus et al., 1986), demonstrated a distinct NR1 immunoreactivity located mostly at the somatic cell membrane (Figure 3c). In contrast, no NR1 immunoreactivity could be detected in astrocytes double-stained with anti-NR1 antibody and with a monoclonal antibody directed against GFAP (Figure 3c). No differences were observed between cultures kept under control conditions and high potassium-treated cultures. There was no specific staining detectable in control experiments performed without primary anti-NR1 antibody or with primary antibody preincubated with antigen.
Effect of sustained elevated K+ levels on NR subunit mRNA expression
Quantitative RT–PCR revealed no differences in overall NR1, NR2A and NR2B expression in cultured cortical neurons, whether incubated under control conditions (5 mM K+) or in the presence of high K+ (25 mM K+) (Figure 4). However, a detailed semiquantitative analysis in NR1 splice variant expression revealed significant differences between control cultures and high potassium-treated cultures (Figure 5).
Figure 4.

Real-time RT–PCR analysis of NR subunit (NR1, NR2A, NR2B) expression in primary cultures (DIV10) of cortical neurons grown in medium containing 5 mM (control) or 25 mM K+ (high K+). Data are presented as mean±s.e.m. of six qRT–PCR determinations from three preparations. The number of NR1, NR2A and NR2B transcripts per attomol GAPDH is given, respectively.
Figure 5.

NR1 subunit mRNA expression in rat cortical neurons (DIV10, a–c; DIV15, d–f) cultured under control conditions or treated with 25 mM K+. In some instances, TTX (3 μM) was present in high K+ cultures from DIV10 to DIV15 (d–f). C-terminal splice variants were detected using a common sense primer flanking E21 and distinct antisense primers flanking E21 and E22, respectively. N-terminal splice variants were detected using primer pairs flanking E5. The ratios of the C-terminal (a, b, d, e) and N-terminal (c, f) splice forms were estimated after Southern hybridization and ethidium bromide staining, respectively. Relative percentages of each NR1 splice variant were calculated from the corresponding total expression values. See Methods for detailed experimental protocol. Means±s.e.m. of 9–15 determinations from 3–5 individual experiments are given. Significant differences vs control: *P<0.05; **P<0.01.
In control cultures, splice variants NR1-1 and NR1-4 predominated over NR1-2 and NR1-3 by 2–3-fold, respectively (Figure 5a, b, d and e). Expression of NR1-a mRNA largely exceeded that of NR1-b (Figure 5c and f). In contrast, cultures grown in medium containing 25 mM K+ exhibited multiple changes in mRNA expression of the various NR1 splice variants (Figure 5a–f). For example, at DIV10 the proportion of the expression of NR1-1, NR1-3 and NR1-b was decreased by high K+ treatment from 72.3±1.4 to 50.2±4.1%, from 23.5±3.8 to 7.8±2.3% and from 16.2±1.7 to 8.9±0.5%, respectively. Accordingly, mRNA expression of NR1-2, NR1-4 and NR1-a was increased from 27.7±1.4 to 49.8%, from 76.5±3.8 to 92.2±2.3% and from 83.8±1.7 to 91.1±0.5%, respectively (Figure 5a–c) (n=15 determinations from five individual experiments, each). There was a similar overall NR1 splice variant expression pattern also after DIV15 both for control and high K+-treated cultures.
After administration of TTX (3 μM) from DIV10 to 15, the K+-induced changes on mRNA expression were diminished or even abolished (Figure 5d–f). In contrast, TTX given to controls with regular K+ concentration had no effect on the NR1 subunit expression pattern (data not shown).
Effect of sustained elevated K+ levels on NMDA-induced membrane currents
In voltage-clamp recordings, NMDA (plus 10 μM glycine; holding potential: −70 mV) caused inward currents in all neurons challenged (n=77) in a concentration-dependent manner. Under control conditions (5 mM K+), the NMDA concentration–response curve levelled off at about 100 μM, and had a Hill slope of 1.36 and an EC50 of 15.7 μM. The extrapolated maximum was −831 pA (Figure 6b; n=7). In neurons grown in 25 mM K+, the NMDA concentration–response curve also reached a plateau at about 100 μM. Likewise, the Hill slope (1.30) as well as the EC50 (14.7 μM) were very similar to the values found under control conditions. However, the maximum current evoked by NMDA was halved to −415 pA (Figure 6b; n=7). Administration of TTX (3 μM), or the uncompetitive NR antagonist memantine (50 μM; Parsons et al., 1993), or the competitive NR antagonist CGS19755 (50 μM) (Lehmann et al., 1988) from DIV10 to 15 prevented the high K+-induced diminution of the NMDA 300 μM-induced responses (Figure 6c). To account for the possibility that high K+-pretreated neurons were smaller than control cells, and, hence, may simply bear fewer functional NRs, whole cell capacitance as a measure of the surface area was assessed in all cells. However, the membrane capacitance of control cells (18.5±1.4 pF; n=21) was not significantly different from that of cells incubated in high K+ alone (17.0±1.3 pF; n=21), high K+ plus TTX (19.0±0.9 pF; n=7) high K+ plus memantine (16.4±1.7 pF; n=7) or high K+ plus CGS19755 (17.9±1.9 pF; n=7).
Figure 6.

NMDA-induced transmembrane currents in rat cortical neurons cultured under control conditions and treated with 25 mM K+. (a) Representative inward currents in response to increasing concentrations of NMDA (3, 30 and 300 μM) at a holding potential of −70 mV in neurons treated either with 5 mM K+ (control, upper panel) or 25 mM K+ (lower panel). The dotted lines indicate zero current level; horizontal bars above the current traces indicate times of NMDA application. (b) Concentration–response curves of NMDA (1–1000 μM, plus 10 μM glycine) in control (○) and high K+-treated cultures (•). (c) NMDA (300 μM) induced current amplitudes obtained for controls (control), high K+ (K+), high K+ plus 3 μM TTX (K+-TTX), high K+ plus 50 μM memantine (K+-Memantine) and high K+ plus 50 μM CGS 19755 (K+-CGS 19755). The respective current densities (peak current amplitudes divided by whole-cell capacitance) are shown. Means±s.e.m. of n=7–21 experiments are given in (b) and (c). Significant differences vs control: *P<0.01.
NR-mediated current voltage relationships recorded in the absence of extracellular Mg2+ were linear both in control (Figure 7a) and in high K+-treated neurons. As to expect for currents mediated by nonselective cationic channels, the current reversal potential was close to 0 mV under each condition. In control neurons (no Mg2+) and in the presence of Mg2+ (1 mM), it was −1.2±3.5 and 1.2±3.6 mV, respectively (n=7). However, a region of negative conductance at hyperpolarized membrane potentials was induced by application of Mg2+ (Figure 7a). The inhibitory effect of Mg2+ on NR channels was absent at −20 mV (0.2±6.4% inhibition), pronounced at −40 mV (51.4±8.2% inhibition) and increased further in parallel with membrane hyperpolarization to an inhibition of 85.4±5.1% at −80 mV (Figure 7a and b). Neurons grown in high K+ had a reversal potential of −0.3±2.5 mV, which also remained unchanged upon addition of Mg2+ (1.3±2.9 mV; n=7). The inhibition by Mg2+ showed a similar strength and voltage dependence as found in control neurons: it amounted to 0.5±5.3, 49.9±4.2 and 84.8±4.3% at −20, −40 and −80 mV, respectively (P>0.05). Also in the absence of extracellular Mg2+ current amplitudes evoked by NMDA were significantly smaller in the high K+-treated cells than in control cells. Upon application of 30 μM NMDA (+10 μM glycine), current amplitudes measured at −80 mV were 727.4±79.8 and −390.6±45.5 pA in control and high K+-treated cells, respectively (P<0.05).
Figure 7.

Voltage-dependent block of NRs in rat cortical neurons by extracellular Mg2+. (a) NMDA (30 μM)-induced membrane currents recorded from a representative control (5 mM K+) neuron during 2 s voltage ramps from −100 to +40 mV in the absence (Mg2+-free) and presence of extracellular Mg2+ (1 mM). (b) Inhibition of NMDA-evoked currents by Mg2+ (1 mM) measured at different membrane potentials in neurons grown either in 5 mM K+ (control: open bars) or 25 mM K+ (K+: filled bars; n=7 each; P>0.05).
Effect of sustained elevated K+ levels on α-spectrin breakdown products (SBDP)
Upon calpain activation α-spectrin is cleaved into two characteristic fragments of 150 and 145 kDa, referred to as SBDP150 and SBDP145, respectively. Moreover, caspase-3 activation produces a distinct 120 kDa fragment (SBDP120) and may also contribute to SBDP150. In cortical neurons (DIV10) exposed to 5 or 25 mM of K+, generation of SBDP was studied by Western blot analysis. In neurons subjected to high K+ treatment, α-spectrin fragmentation showed a decrease in calpain activity indicated by a decrease in SBDP 150/145. In contrast, in high K+-treated neurons, SBDP120 expression specifically indicating caspase-3 activation was not different from controls (Figure 8). Spectrin degradation was not affected by treatment with TTX or memantine alone compared with control (data not shown). The high K+-induced decrease in calpain activity was at least partly related to sustained neuronal activity and NR activation as the decrease in enzyme activity was diminished by coadministration with either TTX or memantine.
Figure 8.

Western blot analysis of α-spectrin breakdown in cortical neurons. Total protein was extracted from cortical neurons (DIV10) exposed to 5 mM K+ (control), 25 mM K+, 25 mM K+ plus either TTX (3 μM) or memantine (50 μM); 20 μg of protein was loaded on each lane. (a) Full-lengh α-spectrin is represented by the 240 kDa band. Calpain activation is specifically indicated by formation of α-spectrin breakdown product SBDP145; caspase-3 activation is specifically indicated by the 120 kDa fragment SBDP120. Fragment SBDP150 indicates both calpain and caspase-3 activation. A representative blot is given. (b) Bar chart representing a semiquantitative densiometric band analysis of SBDP150/145 and SBDP120 in the same experimental conditions. The bands were normalized to myosin (200 kDa) levels and expressed as arbitrary units. Histograms represent mean±s.e.m. from 4–6 different cultures. Significant differences vs respective controls: *P<0.05.
Discussion
Although the functional effects of different NR2 subunits have received most attention (e.g., Flint et al., 1997; Allgaier, 2002), NR1 splice variant variations also strongly modify the properties of NRs (Vallano, 1998; Cull-Candy et al., 2001). The present study demonstrates that long-term exposure of cortical neurons to increased extracellular K+ (25 mM), which was associated with an enhancement in action-potential firing (Figure 2a) and, hence, neurotransmission, produced an increase in neuronal cell death (Figure 3a and b). However, in the surviving neurons high K+ treatment caused changes in the expression pattern of individual NR1 splice variants (Figure 5), whereas the overall NR1 mRNA expression remained unaffected (Figure 4).
Most prominently, mRNA levels of splice variants NR1-1 and NR1-3 (each containing exon 21) were reduced, whereas those of NR1-2 and NR1-4 (each lacking exon 21) were increased, accordingly. Since exon 21 encodes the C1 cassette involved in anchoring and clustering of NRs (Ehlers et al., 1995; 1998), the increase in subunits lacking the C1 cassette may be associated with a decrease in receptor function (Ehlers et al., 1996).
NR1-a mRNA expression (lacking exon 5), which in cortical neurons largely predominates over that of NR1-b mRNA (containing exon 5) (present data and, e.g., Laurie & Seeburg, 1994), was only slightly enhanced by high K+ treatment. The presence of exon 5 determines the sensitivity of NR to protons and polyamines: at physiological pH, splice variants that contain exon 5 are fully active, whereas those lacking exon 5 are partially blocked (Traynelis et al., 1995). Thus, the increase in NR1-a expression could principally contribute to the decrease in NR function observed. However, in view of the predominant NR1-a expression in controls, it remains open whether the further increase in NR1-a expression by high K+ was of functional significance. As no histochemical evidence was found for a glial NR1 expression (Figure 3c), the observed changes in NR1 splice variant expression occurred only in neurones and not in astrocytes.
NR2A and NR2B are the predominant NR2 subunits expressed in cortical as well as in hippocampal neurons (Zhong et al., 1994). NR2 subunit expression is regulated developmentally (Hoffmann et al., 2000; Allgaier, 2002; Law et al., 2003) and in response to various pathophysiological conditions such as spinal cord injury (Grossman et al., 2000). In cerebellar granule neurons, NR2A as well as NR2B expression was reduced upon prolonged blockade of the glutamate transporter (Cebers et al., 2001). In the present study, both NR2A and NR2B expression remained unchanged by high K+ treatment (Figure 4).
The inhibitory action of NRs by extracellular Mg2+ was also not affected by high K+ treatment (Figure 7). As is typical for NR2A- and/or NR2B-containing receptors, the voltage-dependent Mg2+ block appeared at relatively depolarized membrane potentials (negative to −20 mV), whereas in NR2C/NR2D-containing NRs, considerably stronger hyperpolarizations were required (negative to −45 mV; Monyer et al., 1994). Accordingly, from these functional data, it is rather unlikely that the high K+-induced decrease in NR function was due to a switch in the expression pattern from high-conductance NR2A/NR2B-containing to low-conductance NR2C/NR2D-containing receptors.
TTX, a blocker of voltage-dependent sodium channels, prevented high K+-induced neuronal cell death as well as changes in NR1 splice variant expression in the surviving neurons, suggesting that both events were related to enhanced neuronal activity. In high K+-treated neurons, the NMDA-induced cationic currents were reduced by half compared to untreated controls, whereas the agonist potency remained virtually unchanged. The inhibition of NR function could be prevented by administration of either TTX or NR antagonists, the latter acting either in a competitive manner at the glutamate-binding site (CGS19755) or as an open-channel blocker (memantine). Hence, these observations provide a direct functional link between the effects of a long-term increase in glutamate release and downregulation of the NR function. The K+-induced decrease in NR function could be explained by the changes in NR1 mRNA splice variant expression discussed above, all the more as TTX prevented both effects in parallel and the NR antagonists were able to prevent the K+-induced changes in NR function. Accordingly, the changes in NR1 splice variant expression and the decrease in receptor function observed upon long-term high K+ treatment are presumably related to a sustained action potential-induced glutamate release. A decrease in NR1-1 expression has already been suggested as a compensatory mechanism in knockout mice deficient in neuronal nitric oxide synthase α to make the brain less vulnerable to enhanced levels of glutamate (Putzke et al., 2000). In cultured spinal cord neurons, a downregulation (by 15–35%) in the number of distinct NR1 splice variant receptor clusters has been observed upon chronic treatment with low (10 μM) concentrations of NMDA (Pauly et al., 2005).
Other mechanisms, such as an ongoing elevation of intracellular calcium resulting from high K+-induced depolarization, could contribute to the decrease in NR function (Rosenmund et al., 1995). However, this possibility appears unlikely, since high K+ treatment was associated with less calpain activity indicated by a decrease in α-spectrin cleavage fragments SBDP150/145. As calpains that play a crucial role in mediating cellular adaptive changes are distinctly Ca2+-activated intracellular proteases (Goll et al., 2003; Nixon, 2003), this finding is in line with a decreased NR function that is associated with a diminution of Ca2+ influx (Dutta et al., 2002; Araujo et al., 2005). The high K+-induced decrease in calpain was prevented by coadministration of TTX or memantine. Accordingly, the present data demonstrate that under conditions of ongoing enhanced neurotransmission NR adaptive mechanisms were initiated, reducing the efficacy of necrotic mechanisms.
It is well accepted that inappropriate activation of excitatory NRs is implicated in the pathophysiology of several brain disorders such as stroke and epilepsy (Cull-Candy et al., 2001). For instance, neuronal hyperexcitability underlying epilepsy may – inter alia – result from an increase in extracellular K+ concentration caused by the glial inability to buffer excess extracellular K+, with the consequence of a constant neuronal depolarization (Janigro et al., 1997). Excessive calcium influx through NRs can initiate intracellular cascades that cause neuronal injury and, finally, cell death. Accordingly, NR antagonists were found to be neuroprotective in animal models of both stroke and seizure, at least when applied at an early stage (Lee et al., 1999). However, rather disappointingly, NR antagonists failed in human stroke and epileptic trials or could not be used because of adverse effects (Ikonomidou & Turski, 2002; Vajda, 2002; Wahlgren & Ahmed, 2004). The present data may suggest that a delayed postinjural application of NR antagonists could counteract neuronal survival-promoting signals associated with an ongoing ‘low' NR activation (Ikonomidou & Turski, 2002). In agreement with this hypothesis, using a mouse model of traumatic brain injury, it has been demonstrated that NR hyperactivation after injury is short-lived and is followed by a distinct and long-lasting loss of receptor function (Biegon et al., 2004). NR activation postinjury attenuated neurological deficits and subsequently improved cognition performance.
Acknowledgments
This work was supported by the Interdisziplinäre Zentrum für Klinische Forschung (IZKF) at the Faculty of Medicine of the University of Leipzig (Projects C20, CA, and C5; AR). We wish to express their gratitude to Dr J. Grosche (Paul-Flechsig-Institute of Brain Research, Leipzig) for LSM measurements, Dr E. Lobos for providing the myosin antibody, and to Helga Sobottka and Angelika Sommer for skilful technical assistance.
Abbreviations
- DIV
day(s) in vitro
- nt
nucleotide
- GFAP
glial fibrillary acidic protein
- MAP-2
microtubule-associated protein-2
- NeuN
neuron-specific nuclear protein
- NR
NMDA receptor subunit
- RT–PCR
reverse transcriptase–polymerase chain reaction
- TTX
tetrodotoxin
References
- ALLGAIER C. Ethanol sensitivity and NMDA receptors. Neurochem. Int. 2002;41:377–382. doi: 10.1016/s0197-0186(02)00046-3. [DOI] [PubMed] [Google Scholar]
- ALLGAIER C., SCHEIBLER P., MÜLLER D., FEUERSTEIN T.J., ILLES P. NMDA receptor characterization and subunit expression in rat cultured mesencephalic neurones. Br. J. Pharmacol. 1999;126:121–130. doi: 10.1038/sj.bjp.0702284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARAUJO I.M., XAPELLI S., GIL J.M., MOHAPEL P., PETERSEN A., PINHEIRO P.S., MALVA J.O., BAHR B.A., BRUNDIN P., CARVALHO C.M. Proteolysis of NR2B by calpain in the hippocampus of epileptic rats. Neuroreport. 2005;16:393–396. doi: 10.1097/00001756-200503150-00017. [DOI] [PubMed] [Google Scholar]
- BIEGON A., FRY P.A., PADEN C.M., ALEXANDROVICH A., TSENTER J., SHOHAMI E. Dynamic changes in N-methyl-D-aspartate receptors after closed head injury in mice: implications for treatment of neurological and cognitive deficits. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5117–5122. doi: 10.1073/pnas.0305741101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CACERES A., BANKER G., STEWARD O., BINDER L., PAYNE M. MAP2 is localized to the dendrites of hippocampal neurons which develop in culture. Brain Res. 1984;315:314–318. doi: 10.1016/0165-3806(84)90167-6. [DOI] [PubMed] [Google Scholar]
- CEBERS G., CEBERE A., KOVACS A.D., HOGBERG H., MOREIRA T., LILJEQUIST S. Increased ambient glutamate concentration alters the expression of NMDA receptor subunits in cerebellar granule neurons. Neurochem. Int. 2001;39:151–160. doi: 10.1016/s0197-0186(01)00014-6. [DOI] [PubMed] [Google Scholar]
- CHANDLER L.J., HARRIS R.A., CREWS F.T. Ethanol tolerance and synaptic plasticity. Trends Pharmacol. Sci. 1998;19:491–495. doi: 10.1016/s0165-6147(98)01268-1. [DOI] [PubMed] [Google Scholar]
- CULL-CANDY S., BRICKLEY S., FARRANT M. NMDA receptor subunits: diversity, development and disease. Curr. Opin. Neurobiol. 2001;11:327–335. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- DINGLEDINE R., BORGES K., BOWIE D., TRAYNELIS S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- DUNAH A.W., LUO J., WANG Y.-H., YASUDA R.P., WOLFE B.B. Subunit composition of N-methyl-D-aspartate receptors in the central nervous system that contain the NR2D subunit. Mol. Pharmacol. 1998;53:429–437. doi: 10.1124/mol.53.3.429. [DOI] [PubMed] [Google Scholar]
- DUTTA S., CHIU Y.C., PROBERT A.W., WANG K.K. Selective release of calpain produced alphalI-spectrin (alpha-fodrin) breakdownproducts by acute neuronal cell death. Biol. Chem. 2002;383:785–791. doi: 10.1515/BC.2002.082. [DOI] [PubMed] [Google Scholar]
- EHLERS M.D., FUNG E.T., O'BRIEN J., HUGANIR R.L. Splice variant-specific interaction of the NMDA receptor subunit NR1 with neuronal intermediate filaments. J. Neurosci. 1998;18:720–730. doi: 10.1523/JNEUROSCI.18-02-00720.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EHLERS M.D., TINGLEY W.G., HUGANIR R.L. Regulated subcellular distribution of the NR1 subunit of the NMDA receptor. Science. 1995;269:1734–1737. doi: 10.1126/science.7569904. [DOI] [PubMed] [Google Scholar]
- EHLERS M.D., ZHANG S., BERNHARDT J.P., HUGANIR R.L. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell. 1996;84:745–755. doi: 10.1016/s0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- FLINT A.C., MAISCH U.S., WEISHAUPT J.H., KRIEGSTEIN A.R., MONYER H. NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J. Neurosci. 1997;17:2469–2476. doi: 10.1523/JNEUROSCI.17-07-02469.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOLL D.E., THOMPSON V.F., LI H., WEI W., CONG J. The calpain system. Physiol. Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- GROSSMAN S.D., WOLFE B.B., YASUDA R.P., WRATHALL J.R. Changes in NMDA receptor subunit expression in response to contusive spinal cord injury. J. Neurochem. 2000;75:174–184. doi: 10.1046/j.1471-4159.2000.0750174.x. [DOI] [PubMed] [Google Scholar]
- HIRAI H., KIRSCH J., LAUBE B., BETZ H., KUHSE J. The glycine binding site of the N-methyl-D-aspartate receptor subunit NR1: identification of novel determinants of co-agonist potentiation in the extracellular M3–M4 loop region. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6031–6036. doi: 10.1073/pnas.93.12.6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOFFMANN H., GREMME T., HATT H., GOTTMANN K. Synaptic activity-dependent developemental regulation of NMDA receptor subunit expresssion in cultured neocortical neurons. J. Neurochem. 2000;75:1590–1599. doi: 10.1046/j.1471-4159.2000.0751590.x. [DOI] [PubMed] [Google Scholar]
- IKONOMIDOU C., TURSKI L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury. Lancet Neurol. 2002;1:383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- JANIGRO D., GASPARINI S., D'AMBROSIO R., MCKHANN G., DIFRANCESCO D. Reduction of K+ uptake in glia prevents long-term depression maintenance and causes epileptiform activity. J. Neurosci. 1997;17:2813–2824. doi: 10.1523/JNEUROSCI.17-08-02813.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAUBE B., HIRAI H., STURGESS M., BETZ H., KUHSE J. Molecular determinants of agonist discrimination by NMDA receptor subunits: analysis of the glutamate binding site on the NR2B subunit. Neuron. 1997;18:493–503. doi: 10.1016/s0896-6273(00)81249-0. [DOI] [PubMed] [Google Scholar]
- LAURIE D.J., SEEBURG P.H. Regional and developmental heterogeneity in splicing of the rat brain NMDAR1 mRNA. J. Neurosci. 1994;14:3180–3194. doi: 10.1523/JNEUROSCI.14-05-03180.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAW A.J., WEICKERT C.S., WEBSTER M.J., HERMAN M.M., KLEINMAN J.E., HARRISON P.J. Expression of NMDA receptor NR1, NR2A and NR2B subunit mRNAs during development of the human hippocampal formation. Eur. J. Neurosci. 2003;18:1197–1205. doi: 10.1046/j.1460-9568.2003.02850.x. [DOI] [PubMed] [Google Scholar]
- LEE J.M., ZIPFEL G.J., CHOI D.W. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- LEHMANN J., HUTCHISON A.J., MCPHERSON S.E., MONDADORI C., SCHMUTZ M., SINTON C.M., TSAI C., MURPHY D.E., STEEL D.J., WILLIAMS M., CHENEY D.L., WOOD P.L. CGS 19755, a selective and competitive N-methyl-D-aspartate-type excitatory amino acid receptor antagonist. J. Pharmacol. Exp. Ther. 1988;246:65–75. [PubMed] [Google Scholar]
- MATUS A., BERNHARDT R., BODMER R., ALAIMO D. Microtubule-associated protein 2 and tubulin are differently distributed in the dendrites of developing neurons. Neuroscience. 1986;17:371–389. doi: 10.1016/0306-4522(86)90253-8. [DOI] [PubMed] [Google Scholar]
- MONYER H., BURNASHEV N., LAURIE D.J., SAKMANN B., SEEBURG P.H. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- NIXON R.A. The calpains in aging and aging-related diseases. Ageing Res. Rev. 2003;2:407–418. doi: 10.1016/s1568-1637(03)00029-1. [DOI] [PubMed] [Google Scholar]
- PARSONS C.G., DANYSZ W., QUACK G. Glutamate in CNS disorders as a target for drug development: an update. Drug News Perspect. 1998;11:523–569. doi: 10.1358/dnp.1998.11.9.863689. [DOI] [PubMed] [Google Scholar]
- PARSONS C.G., GRUNER R., ROZENTAL J., MILLAR J., LODGE D. Patch clamp studies on the kinetics and selectivity of N-methyl-D-aspartate receptor antagonism by memantine (1-amino-3,5-dimethyladamantan) Neuropharmacology. 1993;32:1337–1350. doi: 10.1016/0028-3908(93)90029-3. [DOI] [PubMed] [Google Scholar]
- PAULY T., SCHLICKSUPP A., NEUGEBAUER R., KUHSE J. Synaptic targeting of N-methyl-D-aspartate receptor splice variants is regulated differentially by receptor activity. Neuroscience. 2005;131:99–111. doi: 10.1016/j.neuroscience.2004.10.039. [DOI] [PubMed] [Google Scholar]
- PUTZKE J., SEIDEL B., HUANG P.L., WOLF G. Differential expression of alternatively spliced isoforms of neuronal nitric oxide synthase (nNOS) and N-methyl-D-aspartate receptors (NMDAR) in knockout mice deficient in nNOS alpha (nNOS alpha(Delta/Delta) mice) Mol. Brain Res. 2000;85:13–23. doi: 10.1016/s0169-328x(00)00220-5. [DOI] [PubMed] [Google Scholar]
- ROSENMUND C., FELTZ A., WESTBROOK G.L. Calcium-dependent inactivation of synaptic NMDA receptors in hippocampal neurons. J. Neurophysiol. 1995;73:427–430. doi: 10.1152/jn.1995.73.1.427. [DOI] [PubMed] [Google Scholar]
- SCHEIBLER P., KRONFELD A., ILLES P., ALLGAIER C. Trichloroethanol impairs NMDA receptor function in rat mesencephalic and cortical neurones. Eur. J. Pharmacol. 1999;366:R1–R2. doi: 10.1016/s0014-2999(98)00924-8. [DOI] [PubMed] [Google Scholar]
- TAUBERT H., KOEHLER T., MEYE A., BARTEL F., LAUTENSCHLAGER C., BORCHERT S., BACHE M., SCHMIDT H., WURL P. mdm2 mRNA level is a prognostic factor in soft tissue sarcoma. Mol. Med. 2000;6:50–59. [PMC free article] [PubMed] [Google Scholar]
- TRAYNELIS S.F., HARTLEY M., HEINEMANN S.F. Control of proton sensitivity of the NMDA receptor by RNA splicing and polyamines. Science. 1995;268:873–876. doi: 10.1126/science.7754371. [DOI] [PubMed] [Google Scholar]
- VAJDA F.J. Neuroprotection and neurodegenerative disease. J. Clin. Neurosci. 2002;9:4–8. doi: 10.1054/jocn.2001.1027. [DOI] [PubMed] [Google Scholar]
- Vallano M.L. Developmental aspects of NMDA receptor function. Crit. Rev. Neurobiol. 1998;12:177–204. doi: 10.1615/critrevneurobiol.v12.i3.20. [DOI] [PubMed] [Google Scholar]
- WAHLGREN N.G., AHMED N. Neuroprotection in cerebral ischaemia: facts and fancies – the need for new approaches. Cerebrovasc. Dis. 2004;17:153–166. doi: 10.1159/000074808. [DOI] [PubMed] [Google Scholar]
- WYSZYNSKI M., LIN J., RAO A., NIGH E., BEGGS A.H., CRAIG A.M., SHENG M. Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- ZHONG J., RUSSELL S.L., PRITCHETT D.B., MOLINOFF P.B., WILLIAMS K. Expression of mRNAs encoding subunits of the N-methyl-D-aspartate receptor in cultured cortical neurons. Mol. Pharmacol. 1994;45:846–853. [PubMed] [Google Scholar]
- ZUKIN S., BENNETT M. Alternatively spliced isoforms of the NMDAR1 subunit expression. Trends Neurosci. 1995;18:306–313. doi: 10.1016/0166-2236(95)93920-s. [DOI] [PubMed] [Google Scholar]