

Abstract
We report investigations of the molecular structure of amyloid fibrils formed by residues 14–23 of the β-amyloid peptide associated with Alzheimer's disease (Aβ14-23), using solid-state nuclear magnetic resonance (NMR) techniques in conjunction with electron microscopy and atomic force microscopy. The NMR measurements, which include two-dimensional proton-mediated 13C-13C exchange and two-dimensional relayed proton-mediated 13C-13C exchange spectra, show that Aβ14-23 fibrils contain antiparallel β-sheets with a registry of backbone hydrogen bonds that aligns residue 17+k of each peptide molecule with residue 22−k of neighboring molecules in the same β-sheet. We compare these results, as well as previously reported experimental results for fibrils formed by other β-amyloid fragments, with theoretical predictions of molecular alignment based on databases of residue-specific alignments in antiparallel β-sheets in known protein structures. While the theoretical predictions are not in exact agreement with the experimental results, they facilitate the design of experiments by suggesting a small number of plausible alignments that are readily distinguished by solid-state NMR.
INTRODUCTION
Amyloid fibrils are filamentous aggregates formed by a large class of peptides and proteins with diverse amino-acid sequences. Current interest in amyloid fibrils arises from their involvement in amyloid diseases (including Alzheimer's disease, type 2 diabetes, dialysis-related amyloidosis, Parkinson's disease, transthyretin amyloidoses, transmissible spongiform encephalopathy, and others (1)), from the fairly recent realization that the propensity to form amyloid fibrils is not restricted to disease-associated peptides and proteins (but is instead a nearly generic property of polypeptides (2)), and from the possibility that amyloid fibrils may be a useful basis for development of self-assembled nanomaterials (3,4). Knowledge of the molecular-level details of amyloid fibril structures would contribute to our understanding of potential mechanisms by which amyloid fibrils contribute to or cause amyloid diseases, to the development of therapeutic agents (5–7), to our understanding of the intermolecular and intramolecular interactions that stabilize amyloid fibrils, and possibly to the development of amyloid-based nanomaterials. These molecular-level structural details are only recently becoming accessible, largely through the application of modern solid-state nuclear magnetic resonance (NMR) techniques such as multiple quantum NMR (8–10), dipolar recoupling (11–21), and various forms of multidimensional spectroscopy (10,18,19,22–26) in conjunction with magic-angle spinning (MAS). Solid-state NMR measurements have revealed that amyloid fibrils, which are known to be primarily β-sheet structures from x-ray fiber diffraction patterns (27–29), can contain either parallel or antiparallel β-sheets, depending on the amino-acid sequence. To date, antiparallel β-sheets have only been observed in fibrils formed by relatively short peptides that contain one β-strand segment (10,12,19). The precise registry of backbone hydrogen bonds (i.e., the alignment of neighboring β-strands within a single β-sheet) can be pH-dependent (19) and is not fully determined by the local amino acid sequence (e.g., by seven-residue segments). Not all short peptides form antiparallel β-sheets in amyloid fibrils (20,30), and the β-sheets in fibrils formed by short peptides can be switched from antiparallel to parallel by attachment of N-terminal alkyl chains (31).
Techniques other than solid-state NMR have also contributed greatly to our developing understanding of amyloid structures. These techniques include electron microscopy (32–36), x-ray crystallography (30,37), electron paramagnetic resonance (38–42), hydrogen/deuterium exchange (25,43–47), chemical cross-linking (13,48), limited proteolysis (49,50), and scanning mutagenesis (51–53). Results from these techniques are generally consistent with those from solid-state NMR, especially with regard to the types of β-sheets contained in amyloid fibrils.
In this article, we report the results of solid-state NMR measurements on fibrils formed by residues 14–23 of the full-length β-amyloid peptide associated with Alzheimer's disease (Aβ14-23, sequence Ac-HQKLVFFAED-NH2, with acetyl and amide capping groups at the N- and C-termini). We have chosen to study Aβ14-23 for the following reasons:
In earlier work, Tjernberg et al. (54) examined amyloid fibril formation by β-hairpin peptides containing the Aβ14-23 sequence on both sides of a type 1′ β-turn, constrained to align either residue 17+k with residue 20−k or residue 17+k with residue 21−k (54). Both β-hairpin peptides were found to form fibrils, raising the question of what alignment is preferred by the unconstrained Aβ14-23 peptide in amyloid fibrils (including the possibility of either antiparallel or parallel β-sheet structure).
Although the hexapeptide Ac-KLVFFAE-NH2 has been shown to form amyloid fibrils (10), other β-amyloid-derived peptides containing the LVFFA hydrophobic segment and a total of nine or fewer residues were found by Tjernberg et al. not to form fibrils (55), while all peptides examined that contained 11 or more residues did form fibrils (19,55). Residues 14–23 may therefore be considered the minimal segment of the β-amyloid sequence that is sufficient for fibril formation.
Given that our current understanding of amyloid structures and the interactions that stabilize these structures is relatively primitive (e.g., compared with our understanding of soluble, monomeric, globular protein structures), structural studies of model systems such as Aβ14-23 fibrils are expected to contribute important information about the variety and sequence dependence of amyloid structures and stabilizing interactions.
Model systems such as Aβ14-23 fibrils serve as test-beds for the development and demonstration of experimental methods for determining specific features of amyloid structures and theoretical methods for predicting these structural features.
The solid-state NMR measurements described below show that Aβ14-23 fibrils contain antiparallel β-sheets with hydrogen-bond registry that aligns residue 17+k with residue 22−k, for integral k (e.g., V18 of each Aβ14-23 molecule forms hydrogen bonds with A21 of a neighboring molecule in the same β-sheet). Moreover, the solid-state NMR data indicate a high level of order in the β-sheets, with no detectable defects in the 17+k ↔ 22−k hydrogen-bond registry. We compare these experimental results with theoretical predictions of β-strand alignment. In particular, we show that simple predictive tools based upon comparisons with known protein structures may be useful in guiding experimental design, although precise prediction of registry may not be possible.
METHODS
Peptide synthesis and fibril formation
Isotopically labeled amino acids were obtained from Cambridge Isotope Laboratories (Andover, MA). Aβ14-23 peptides containing labeled amino acids were synthesized on a Symphony/Multiplex solid-phase synthesizer (Protein Technologies, Tucson, AZ), using standard FMOC synthesis and cleavage protocols. Peptides were purified by high performance liquid chromatography using two mobile phases, water with 0.1% trifluoroacetic acid and acetonitrile with 0.1% trifluoroacetic acid, on a C18 reverse phase column (Grace Vydac, Hesperia, CA). Final purity was >95%, as confirmed by a Kompact MALDI TOF mass spectrometer (Kratos, Chestnut Ridge, NY). After lyophilization of the high performance liquid chromatography fraction containing these peptides, Aβ14-23 was dissolved at 5.3 mg/ml in 10 mM phosphate buffer, 0.01% NaN3, at pH 4.7. Fibrils formed within three weeks at room temperature. Aβ14-23 fibril samples were prepared with uniform 13C and 15N labeling of L17 and F20 (17,20-Aβ14-23), V18 and F20 (18,20-Aβ14-23), V18 and A21 (18,21-Aβ14-23), and L17 and A21 (17,21-Aβ14-23).
Electron microscopy and atomic force microscopy
Fibril formation was confirmed and fibril dimensions were determined using electron microscopy (EM) and atomic force microscopy (AFM). For EM measurements, 10 μl aliquots of incubated Aβ14-23 solutions were placed on specimen grids covered by a formvar/carbon support film. Excess fluid was wicked off after 2 min and the grids were negatively stained with 4 mg/ml uranyl acetate in water. The stained grids were then examined and photographed with a JEOL (Tokyo, Japan) JEM-100CXII transmission electron microscope.
For AFM, fibrils were diluted in 0.5% acetic acid (pH 3) to a peptide concentration of ∼0.5 mM. Both lyophilized and fully hydrated (i.e., never lyophilized or dried after incubation) fibrils were examined. A 50 μl aliquot was placed on freshly cleaved mica (1 cm2 area), allowed to adsorb for several minutes, and drained from the mica surface. The surface was washed twice with 100 μl of 0.5% acetic acid, then dried under a stream of nitrogen gas. AFM images were obtained in air with a MultiMode AFM system (Veeco Instruments, Santa Barbara, CA) in tapping mode, using microactuated probes with a nominal force constant of 3 N/m and a nominal tip radius of curvature of 10 nm. Approximately 100 images of 5 μm × 5 μm areas were recorded for both lyophilized and hydrated samples, with 1024 pixel resolution in each lateral dimension. AFM images in Fig. 1 b are portions of typical 5 μm × 5 μm areas.
FIGURE 1.

(a) Transmission electron microscope image of Aβ14-23 fibrils, with negative staining. (b) Atomic force microscope images of Aβ14-23 fibrils with (left) and without (right) lyophilization before dilution in 0.5% acetic acid and deposition on mica. Grayscales indicate height.
Solid-state NMR
NMR measurements were carried out at a magnetic field of 14.1 T (150.7 MHz 13C NMR frequency) using Varian (Palo Alto, CA) Infinity and InfinityPlus spectrometer consoles and a Varian MAS probe with 3.2 mm MAS rotor diameters and 11 μl maximum sample volumes. Additional Teflon plugs were inserted in the rotors to restrict smaller samples to the center of the radio-frequency (rf) coil in the NMR probe. Aβ14-23 fibril samples were pelleted in a microfuge (18,000 × g for 15 min), resuspended in deionized H2O, and lyophilized before packing into MAS rotors. Sample quantities were in the 2–6 mg range. Lyophilization permitted the use of small sample volumes with concomitantly high MAS speeds, rf field amplitudes, and NMR sensitivity. As shown by AFM images (Fig. 1 b), lyophilization breaks Aβ14-23 fibrils into 100-500 nm segments but does not otherwise affect fibril morphology. For certain measurements (specified below), lyophilized samples were rehydrated by addition of 2–3 μl of deionized H2O.
Rf pulse sequences used in solid-state NMR measurements are shown in Fig. 2. One-dimensional 13C NMR spectra (Fig. 2 a) were recorded with cross-polarization (CP) from protons (56) and with two-pulse phase-modulated proton decoupling (57). Decoupling fields were 110 kHz. 13C rf fields were ∼50 kHz during CP, with tangent-shaped amplitude modulation. Two-dimensional proton-mediated 13C-13C NMR exchange (2D-PME) spectra were recorded as described previously (19,58), using 150 μs CP periods, 200 μs proton spin diffusion (SD) periods in the exchange period, and MAS frequencies of 20.0–21.4 kHz (Fig. 2 b). Under these conditions, strong crosspeaks are observed between the NMR lines of 13C pairs with directly-bonded protons for which the proton-proton distances are <3 Å. In particular, strong intermolecular crosspeaks between NMR lines of 13C-labeled α-carbons (Cα) are detected when the corresponding residues are aligned in antiparallel β-sheets, leading to intermolecular distances of ∼2.2 Å between α-protons (Hα). Thus, the presence or absence of particular Cα/Cα crosspeaks in 2D-PME spectra can be used to determine hydrogen-bond registry in antiparallel β-sheets (19,58).
FIGURE 2.

Radio-frequency pulse sequences for one-dimensional 13C MAS NMR measurements (a), 2D-PME measurements (b), and 2D-RPME measurements (c). Radio-frequency-driven recoupling periods consist of one 13C π pulse per MAS rotor period. Dephasing delays τ are 1 ms. Phase cycles are: φ1 = x, −x; φ2 = x, x, −x, −x; φ3 = x or y for real or imaginary signals in t1; φ4 = x, x, y, y, −x, −x, −y, −y; φ5 = x, x, x, x, y, y, y, y, −x, −x, −x, −x, −y, −y, −y, −y. Receiver phase cycles are x, −x, y, −y, −x, x, −y, y in panel a and x, −x, −x, x, y, −y, −y, y, −x, x, x, −x, −y, y, y, −y in panels b and c.
Cα chemical shifts for L17 and F20 were found to be similar (0.7 ppm difference), preventing the observation of Cα/Cα crosspeaks in 2D-PME spectra of 17,20-Aβ14-23 fibrils even if L17 and F20 were aligned in antiparallel β-sheets. Therefore, a new solid-state NMR technique was designed in which alignment of uniformly 13C-labeled residues could be detected as an intermolecular crosspeak between carbonyl (CO) NMR lines. In this technique, called two-dimensional relayed proton-mediated 13C-13C NMR exchange (2D-RPME) spectroscopy (Fig. 2 c), 13C spin polarization is transferred between CO sites during the exchange period in five steps:
A short 13C-13C dipolar recoupling period for intraresidue CO→Cα transfer.
A short CP period for one-bond Cα→Hα transfer.
A short proton SD period for intermolecular Hα→Hα transfer.
A second short CP period for one-bond Hα→Cα transfer.
A second short 13C-13C dipolar recoupling period for intraresidue Cα→CO transfer. CP and SD conditions are the same as in 2D-PME measurements.
13C-13C dipolar recoupling periods employed the radio-frequency-driven recoupling pulse sequence (59,60), with one 8.0 μs 13C π-pulse per MAS rotation period for a total of 16 rotation periods (1.067 ms at a 15.0 kHz MAS frequency). 13C π-pulse phases followed the XY-16 pattern (61).
RESULTS
Electron microscopy and atomic force microscopy
The EM image in Fig. 1 a shows that Aβ14-23 forms fibrils with an apparently flat, ribbonlike morphology, with fibril widths of ∼30 nm. Fibril widths exceed the 3.5 nm length of a single Aβ14-23 molecule in a fully extended β-strand conformation, suggesting that each fibril contains many finer filaments with cross-β structures.
The AFM images in Fig. 1 b show that lyophilization breaks long Aβ14-23 fibrils into shorter fragments, which tend to coalesce into clumps under the conditions of AFM measurements, but otherwise has no detectable effect on fibril morphology. Apparent fibril heights in AFM images of both lyophilized and hydrated fibrils are 2.5 ± 0.5 nm. The fibril heights may correspond to the thickness of between two and four β-sheets in a laminated cross-β structure.
Solid-state NMR
Fig. 3 shows one-dimensional 13C MAS NMR spectra of the four Aβ14-23 fibril samples examined in this work. 13C chemical shift assignments, summarized in Table 1, are based on the known chemical shift ranges for individual carbon sites in amino acids, and are confirmed by the 2D-NMR spectra described below. Chemical shifts for CO, Cα, and β-carbon (Cβ) sites are consistent with a β-strand conformation for residues 17–21 in Aβ14-23 fibrils (i.e., upfield secondary shifts relative to random coil values (62) for CO and Cα, downfield secondary shifts for Cβ). Only the Cα line for L17 does not show a strong secondary shift. In the dry, lyophilized state, 13C MAS NMR line-widths for resolved single sites are 1.5 to 2.1 ppm (full width at half-maximum). In the rehydrated state (Fig. 3, a and b, bottom spectra), line-widths are 1.0 ppm. The reduction of 13C MAS NMR line-widths upon rehydration is attributable to increased molecular motion, which partially averages out the inhomogenous broadening that arises from structural variations within these noncrystalline fibril samples. Structural variations that may contribute to the observed line-widths include variations in backbone torsion angles within the β-strand segments by ∼±10°, variations in side-chain conformations, disorder at the extreme N- and C termini of Aβ14-23, variations in contacts between the fine filaments that presumably comprise the fibrils shown in Fig. 1, and variations in the number of or contacts between β-sheet layers within these filaments. When compared with the 2–3 ppm line-widths observed in 13C MAS NMR spectra of other noncrystalline systems, including peptide/antibody complexes (63,64) and helical proteins (65–67) in frozen solutions as well as other amyloid fibrils (17,21,22), the line-widths in Fig. 3 indicate a high degree of structural order, but without true crystallinity.
FIGURE 3.

One-dimensional 13C MAS NMR spectra of Aβ14-23 fibrils with uniform 13C and 15N labeling of L17 and F20 (a), V18 and A21 (b), V18 and F20 (c), and L17 and A21 (d). Spectra were recorded at a 13C NMR frequency of 150.7 MHz, with MAS frequencies of ∼20 kHz and between 64 and 256 scans.
TABLE 1.
13C NMR chemical shifts in Aβ14-23 fibrils
| Residue | CO | Cα | Cβ | Cγ | Cδ |
|---|---|---|---|---|---|
| L17 | 172.8 (175.9) | 53.3 (53.4) | 44.5 (40.7) | 24.7 (25.2) | 23.8 (23.2, 21.6) |
| V18 | 169.8 (174.6) | 59.2 (60.5) | 34.1 (31.2) | 19.6 (19.4, 18.6) | |
| F20 | 169.5 (174.1) | 54.0 (56.0) | 43.0 (37.9) | 136.5 (137.2) | |
| A21 | 172.5 (176.1) | 48.6 (50.8) | 21.4 (17.4) |
Values are in ppm relative to tetramethylsilane, based on an external reference of 177.95 ppm for the carboxylate line of polycrystalline L-alanine. Values in parentheses are random coil chemical shifts, taken from Wishart et al. (62) and adjusted to the tetramethylsilane reference by subtraction of 1.7 ppm.
In solid-state MAS NMR studies of microcrystalline proteins, 13C NMR line-widths <1 ppm are commonly observed at moderate temperatures (68–70). The line-widths increase substantially at low temperatures, where solvent within the crystal is immobilized and protein motions are quenched (71), although crystalline order is not lost. We infer from the 13C NMR line-widths that the level of molecular conformational order in Aβ14-23 fibrils is similar to, but not as high, as in microcrystalline proteins because 13C NMR lines for hydrated Aβ14-23 fibrils are not as narrow as 13C NMR lines for microcrystalline proteins. Hydrated amyloid fibrils formed by the HET-s protein of Podospora anserina have been shown by Siemer et al. to exhibit NMR line-widths on the order of 0.1 ppm (26), possibly because HET-s fibrils are responsible for an evolved biological function (namely, heterokaryon incompatibility (72)) and therefore have a highly homogeneous molecular structure.
Fig. 4 shows 2D-PME spectra of the four Aβ14-23 fibril samples. In the 2D-PME spectrum of 18,21-Aβ14-23 fibrils, strong crosspeaks (25% of diagonal peaks) are observed between Cα NMR lines of V18 and A21. Cα/Cα crosspeaks are not observed above the noise level in any of the other 2D-PME spectra. This result implies that Aβ14-23 fibrils contain antiparallel β-sheets in which V18 aligns with A21, i.e., antiparallel β-sheets with 17+k ↔ 22–k hydrogen-bond registry. Cα/Cα crosspeak intensities for 18,21-Aβ14-23 fibrils, relative to diagonal peak intensities, are approximately the same as in previously reported 2D-PME spectra of Aβ16-22 and Aβ11-25 fibrils for the Cα pairs that are aligned in antiparallel β-sheets in these fibrils (19,58). For example, under quite similar experimental conditions, Cα/Cα crosspeaks for V18/F20 and L17/A21 pairs in 2D-PME spectra of Aβ16-22 fibrils (which have 17+k ↔ 21–k registry (10)) were found to have 28% of the volume of V18 and A21 Cα diagonal peaks (58). The observed crosspeak intensities for 18,21-Aβ14-23 fibrils indicate that all V18 and A21 residues in Aβ14-23 fibrils participate in the 17+k↔22−k registry. Any putative alternations in registry (e.g., as previously suggested for Aβ34-42 fibrils (12)) or alternations between antiparallel and parallel β-sheet alignments would reduce the V18/A21 crosspeak intensities by at least a factor of two. Structures with alternating registry or alignment would also necessarily contain two or more inequivalent environments for Aβ14-23 molecules (i.e., more than one molecule in the asymmetric unit), potentially splitting each 13C NMR line into two or more components. No splittings are observed in the one- or two-dimensional spectra of Aβ14-23 fibrils.
FIGURE 4.

(a) Aliphatic regions of 2D-PME spectra of lyophilized Aβ14-23 fibrils with the indicated labeled residues. Single-headed arrows indicate assignments of certain intraresidue crosspeaks. Double-headed arrow indicates an interresidue crosspeak. (b) One-dimensional slices at the indicated Cα chemical shifts. Double-headed arrows indicate the only interresidue crosspeaks observed in these measurements. Spectra were recorded in 24–72 h, using recycle delays of 2 s and maximum t1 periods of 3.8 ms.
The absence of detectable Cα/Cα crosspeaks in the 2D-PME spectra of 18,20-Aβ14-23 and 17,21-Aβ14-23 fibrils places a constraint on the levels of certain defects in the antiparallel β-sheets. In particular, defects that produce 17+k ↔ 21−k alignments cannot be present at levels above 10%. This upper limit on defect concentration is dictated by the signal/noise ratio in the 2D-PME spectra.
Fig. 5 shows 2D-RPME spectra of 17,20-Aβ14-23 and 18,21-Aβ14-23 fibrils. As explained above, the 2D-RPME technique allows hydrogen-bond registry to be investigated in cases where the Cα NMR lines are not resolved. The 2D-RPME spectrum of 17,20-Aβ14-23 shows only intraresidue crosspeaks, including crosspeaks between the CO line of L17 (172.8 ppm) and aliphatic carbon lines of L17 (53.3, 44.5, and 24.7 ppm) and crosspeaks between the CO line of F20 (169.5 ppm) and aliphatic lines of F20 (54.0 and 43.0 ppm). In contrast, the 2D-RPME spectrum of 18,21-Aβ14-23 fibrils shows both intraresidue and interresidue crosspeaks, including crosspeaks between the CO line of V18 and the CO line of A21, crosspeaks between the CO line of V18 and aliphatic lines of A21, and crosspeaks between the CO line of A21 and aliphatic lines of V18. Although the signal/noise ratio is higher in the 2D-RPME spectrum of 18,21-Aβ14-23 fibrils (due to a larger sample quantity), the signal/noise ratio in the 2D-RPME spectrum of 17,20-Aβ14-23 fibrils is high enough to permit the observation of interresidue crosspeaks if they were present. The data in Fig. 5 confirm the 17+k↔22−k hydrogen-bond registry in Aβ14-23 fibrils. In addition, these data demonstrate the utility of the 2D-RPME technique in structural investigations of amyloid fibrils.
FIGURE 5.

(a) 2D-RPME spectra of rehydrated Aβ14-23 fibrils with the indicated labeled residues. (b) One-dimensional slices at the indicated CO chemical shifts. Spectra were recorded in 48–72 h, using recycle delays of 1.6 s and maximum t1 periods of 2.3 ms.
Comparison of the 2D-PME and 2D-RPME data for 18,21-Aβ14-23 (Figs. 4 and 5) indicates that the total 13C NMR signal amplitude in the 2D-RPME spectrum (i.e., the integrated signal in the first one-dimensional spectrum, at t1 = 0) is ∼25% greater in the 2D-RPME measurement for the same number of scans. Each interresidue crosspeak in the 2D-RPME spectrum has ∼15–30% of the volume of the interresidue Cα/Cα crosspeaks in the 2D-PME spectrum.
DISCUSSION
Summary of experimental findings
EM and AFM images in Fig. 1 demonstrate that Aβ14-23 forms amyloid fibrils, as observed previously for many other fragments of the Alzheimer's β-amyloid peptide (10,12,13,19). As in the case of fibrils formed by other short fragments (10,19), the fibril widths exceed the maximum length of a single peptide chain, suggesting that the observed fibrils are comprised of multiple finer filaments that cannot be resolved in the images. One-dimensional 13C NMR spectra of isotopically labeled Aβ14-23 fibrils in Fig. 3 indicate a high degree of structural order at the molecular level. 13C NMR chemical shifts indicate that residues 17–21 form a continuous β-strand. The 2D-PME and 2D-RPME spectra in Figs. 4 and 5 show that the β-sheets are of the antiparallel type, with a registry that produces a distance <3 Å between Hα sites of V18 and A21. Given the β-strand conformation of residues 17–21, this must be an intermolecular distance. Only 17+k ↔ 22−k registry is consistent with the data. Any alternation in registry or alignment within the β-sheets would be inconsistent with the intensity of Cα-Cα crosspeaks in the 2D-PME spectrum of 18,21-Aβ14-23 fibrils and the absence of splittings of any 13C NMR lines. A molecular model for the antiparallel β-sheets in Aβ14-23 fibrils is shown in Fig. 6.
FIGURE 6.

(a) Molecular model for an antiparallel β-sheet in Aβ14-23 fibrils, with the registry of backbone hydrogen bonds indicated by the solid-state NMR data in Figs. 4 and 5. (b) One-letter-code representation, showing hydrogen-bonded (red lines) and non-hydrogen-bonded (yellow lines) interstrand alignments of amino-acid pairs. Two different sets of alignments (i.e., patterns of red and yellow lines) alternate along the hydrogen-bonding direction of the antiparallel β-sheet, making two distinct contributions to the calculated free energy.
Antiparallel β-sheets have been established by previous solid-state NMR measurements on Aβ11-25 (19), Aβ16-22 (10) and Aβ34-42 (12) fibrils. The registry in Aβ16-22 fibrils prepared at pH 7.4 is 17+k↔21−k, while the registry in Aβ11-25 fibrils is 17+k↔20−k at pH 7.4 and 17+k↔22−k at pH 2.4. Thus, the β-sheet structure in Aβ14-23 fibrils prepared at pH 4.35 is the same as in Aβ11-25 fibrils prepared at low pH.
As observed in solid-state NMR studies of amyloid fibrils formed by other peptides (19,20,73), 13C NMR chemical shifts in lyophilized and hydrated Aβ14-23 fibrils are indistinguishable, indicating that the molecular structure is not affected significantly by hydration. Hydration produces a reduction in 13C NMR line-widths, also as previously observed. From a practical standpoint, lyophilized samples have the advantages of permitting the use of high MAS frequencies, as required for certain solid-state NMR measurements (74–76), and small sample volumes, which in turn permit high rf fields and high sensitivity. Lyophilized samples are not prone to rf-induced heating and to NMR probe tuning instabilities. On the other hand, hydrated samples may be preferred in experiments where the highest possible spectral resolution is required. Similar line-widths have been observed in spectra of samples that are fully hydrated without prior lyophilization (19) and in spectra of samples that were lyophilized and subsequently rehydrated (20,73).
Theoretical predictions
We now compare the experimental results with theoretical predictions of hydrogen-bond registry in antiparallel β-sheets. These predictions were made in advance of the experiments and were used to select the isotopic labeling patterns and solid-state NMR strategies described above.
The relative probability of a given registry can be estimated from the probability that such an alignment occurs in known protein structures (54). This probability is calculated from the relative probability that individual pairs of residues align in an antiparallel β-sheet in known protein structures, which we take from the database of Wouters and Curmi (77,78). Specifically, Wouters and Curmi report pair-correlation values 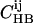 and
and 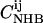 for all pairs of amino acids i and j, representing the ratio of the observed occurrence of i and j in positions of interstrand alignment (in a set of 253 nonredundant protein structures) to the predicted occurrence of i and j in positions of interstrand alignment if all amino acids in the antiparallel β-sheets were randomly distributed (77). Wouters and Curmi distinguish between hydrogen-bonded (HB) and non-hydrogen-bonded (NHB) alignments, which they find to have significantly different pair correlation values. We calculate the relative probability for a given registry of a given peptide sequence in antiparallel β-sheets in an amyloid fibril by multiplying the relevant
for all pairs of amino acids i and j, representing the ratio of the observed occurrence of i and j in positions of interstrand alignment (in a set of 253 nonredundant protein structures) to the predicted occurrence of i and j in positions of interstrand alignment if all amino acids in the antiparallel β-sheets were randomly distributed (77). Wouters and Curmi distinguish between hydrogen-bonded (HB) and non-hydrogen-bonded (NHB) alignments, which they find to have significantly different pair correlation values. We calculate the relative probability for a given registry of a given peptide sequence in antiparallel β-sheets in an amyloid fibril by multiplying the relevant 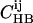 and
and 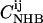 values for all aligned residue pairs in that registry. Residues that are unpaired in a given registry (i.e., dangling residues) are assigned a pair correlation value of 1. If the calculated probability is denoted by P, the relative free energy of binding for a given peptide registry is then given by ΔG = −RT ln(P), where T is the temperature and R is the gas constant.
values for all aligned residue pairs in that registry. Residues that are unpaired in a given registry (i.e., dangling residues) are assigned a pair correlation value of 1. If the calculated probability is denoted by P, the relative free energy of binding for a given peptide registry is then given by ΔG = −RT ln(P), where T is the temperature and R is the gas constant.
Note that each aligned residue pair occurs with both HB and NHB alignments in the antiparallel β-sheets under consideration here, because all β-strands have the same amino-acid sequence and because we consider only β-sheet structures with maximal symmetry. Therefore, we must evaluate two alignment probabilities (for the two combinations of HB and NHB alignments that alternate along the hydrogen-bonding direction of the β-sheet; see Fig. 6 b and accompanying caption for clarification) and two free energies. The final free energy is taken to be the average of the two.
Fig. 7 shows this free energy as a function of the offset N, defined by requiring that residue 17+k be aligned with residue N−k of neighboring peptide chains in the β-sheet. For Aβ14-23, the solid-state NMR results indicate that N = 22, so that V18 aligns with A21. Solid-state NMR data indicate that N = 21 for Aβ16-22 fibrils grown at pH 7.4, while for Aβ11-25 fibrils, N = 20 at pH 7.4 and N = 22 at pH 2.4. Calculated free energies predict N = 21 for all three peptides. In all three cases, the experimental offset is within a deep minimum of the predicted relative free energy. Thus, this simple predictive scheme may be useful to guide experimental design (for instance, by suggesting which residues should be isotopically labeled for solid-state NMR investigations), but does not capture all factors that determine the precise hydrogen-bond registries.
FIGURE 7.

Calculated free energies ΔG/RT as a function of the offset N in antiparallel β-sheets with 17+k ↔ N−k registry, plotted for the three peptides Aβ16-22, Aβ14-23, and Aβ11-25. See text for details of calculations.
It is interesting to note that the offset required for all residues to participate in an antiparallel β-sheet (i.e., to have no dangling residues) is 21 for Aβ16-22, but 20 for Aβ14-23 and 19 for Aβ11-25. Thus, if one were to attempt to improve the calculation by adding a free energy contribution biased against the number of dangling residues, one could improve the prediction in the case of Aβ11-25 fibrils at pH 7.4 but would worsen it in the other two cases.
We have also used the pair-information values Pi,j+m introduced by Steward and Thornton (79) to evaluate the relative probabilities of various registries, as previously described by Petkova et al. (19) The pair-information values take into account interstrand interactions between residues that are not directly aligned. For a given registry, we evaluate the sum of Pi,j+m values for m = −1, 0, and 1 (i.e., directly aligned residue pairs, and pairs that are shifted by one residue in either direction) and for i being residues 17, 18, 19, 20, and 21. Pi,j+m values for hydrogen-bonded and non-hydrogen-bonded pairs i and j are added together, because both types of pairing are present for each residue in the β-sheet structures under consideration. Total information scores are 450, 795, 1042, and 576 for N equal to 19, 20, 21, and 22, respectively. Replacing Asp by Asn and Glu by Gln to approximate the effects of low pH, the total scores become 450, 895, 1036, and 653. If we set Pi,j+m = 0, so that only directly aligned residues are considered, the scores are 80, 412, 631, and 204 at neutral pH, and 80, 412, 631, and 416 at low pH. Thus, the most likely registry according to the pair-information value treatment is always 17+k ↔ 21−k, in agreement with the free energy calculations in Fig. 7.
More sophisticated and accurate schemes could be formulated by including structural effects or energetics derived from other experimental techniques (80). The approaches described above do not take into account interactions between β-sheet layers, which have been elucidated experimentally in Aβ1-40 fibrils (81) and GNNQQNY fibrils (30), but not in the fibrils discussed above. In Aβ14-23 and Aβ11-25 fibrils, it is not yet known whether residues at the N- and C-termini are structurally ordered and participate in the antiparallel β-sheets. The 17+k ↔ 22−k registry observed in Aβ14-23 fibrils necessarily leaves H14 and Q15 unpaired and outside the β-sheets. Entropy associated with these residues may favor the observed registry over the predicted 17+k ↔ 21−k registry. In addition, the 17+k ↔ 22−k registry results in antiparallel β-sheets with two equivalent faces. Side chains of F19 and F20 create continuous rows of aromatic rings on each face, as shown in Fig. 6. These and other features may be advantageous from the standpoint of interactions between β-sheet layers.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, and by a grant to D.J.E.C. from the Alzheimer's Association. Development of solid-state NMR methodology was supported by a grant to R.T. from the Intramural AIDS Targeted Antiviral Program of the National Institutes of Health.
References
- 1.Sacchettini, J. C., and J. W. Kelly. 2002. Therapeutic strategies for human amyloid diseases. Nat. Rev. Drug Discov. 1:267–275. [DOI] [PubMed] [Google Scholar]
- 2.Dobson, C. M. 2003. Protein folding and misfolding. Nature. 426:884–890. [DOI] [PubMed] [Google Scholar]
- 3.Reches, M., and E. Gazit. 2003. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science. 300:625–627. [DOI] [PubMed] [Google Scholar]
- 4.Scheibel, T., R. Parthasarathy, G. Sawicki, X. M. Lin, H. Jaeger, and S. L. Lindquist. 2003. Conducting nanowires built by controlled self-assembly of amyloid fibers and selective metal deposition. Proc. Natl. Acad. Sci. USA. 100:4527–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wood, S. J., L. MacKenzie, B. Maleeff, M. R. Hurle, and R. Wetzel. 1996. Selective inhibition of Aβ fibril formation. J. Biol. Chem. 271:4086–4092. [DOI] [PubMed] [Google Scholar]
- 6.Cairo, C. W., A. Strzelec, R. M. Murphy, and L. L. Kiessling. 2002. Affinity-based inhibition of β-amyloid toxicity. Biochemistry. 41:8620–8629. [DOI] [PubMed] [Google Scholar]
- 7.Gordon, D. J., K. L. Sciarretta, and S. C. Meredith. 2001. Inhibition of β-amyloid(40) fibrillogenesis and disassembly of β-amyloid(40) fibrils by short β-amyloid congeners containing N-methyl amino acids at alternate residues. Biochemistry. 40:8237–8245. [DOI] [PubMed] [Google Scholar]
- 8.Antzutkin, O. N., R. D. Leapman, J. J. Balbach, and R. Tycko. 2002. Supramolecular structural constraints on Alzheimer's β-amyloid fibrils from electron microscopy and solid state nuclear magnetic resonance. Biochemistry. 41:15436–15450. [DOI] [PubMed] [Google Scholar]
- 9.Antzutkin, O. N., J. J. Balbach, R. D. Leapman, N. W. Rizzo, J. Reed, and R. Tycko. 2000. Multiple quantum solid state NMR indicates a parallel, not antiparallel, organization of β-sheets in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. USA. 97:13045–13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balbach, J. J., Y. Ishii, O. N. Antzutkin, R. D. Leapman, N. W. Rizzo, F. Dyda, J. Reed, and R. Tycko. 2000. Amyloid fibril formation by Aβ16–22, a seven-residue fragment of the Alzheimer's β-amyloid peptide, and structural characterization by solid state NMR. Biochemistry. 39:13748–13759. [DOI] [PubMed] [Google Scholar]
- 11.Griffiths, J. M., T. T. Ashburn, M. Auger, P. R. Costa, R. G. Griffin, and P. T. Lansbury. 1995. Rotational resonance solid state NMR elucidates a structural model of pancreatic amyloid. J. Am. Chem. Soc. 117:3539–3546. [Google Scholar]
- 12.Lansbury, P. T., P. R. Costa, J. M. Griffiths, E. J. Simon, M. Auger, K. J. Halverson, D. A. Kocisko, Z. S. Hendsch, T. T. Ashburn, R. G. S. Spencer, B. Tidor, and R. G. Griffin. 1995. Structural model for the β-amyloid fibril based on interstrand alignment of an antiparallel-sheet comprising a C-terminal peptide. Nat. Struct. Biol. 2:990–998. [DOI] [PubMed] [Google Scholar]
- 13.Benzinger, T. L. S., D. M. Gregory, T. S. Burkoth, H. Miller-Auer, D. G. Lynn, R. E. Botto, and S. C. Meredith. 1998. Propagating structure of Alzheimer's β-amyloid10–35 is parallel β-sheet with residues in exact register. Proc. Natl. Acad. Sci. USA. 95:13407–13412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gregory, D. M., T. L. S. Benzinger, T. S. Burkoth, H. Miller-Auer, D. G. Lynn, S. C. Meredith, and R. E. Botto. 1998. Dipolar recoupling NMR of biomolecular self-assemblies: determining inter- and intrastrand distances in fibrilized Alzheimer's β-amyloid peptide. Solid State Nucl. Magn. Reson. 13:149–166. [DOI] [PubMed] [Google Scholar]
- 15.Benzinger, T. L. S., D. M. Gregory, T. S. Burkoth, H. Miller-Auer, D. G. Lynn, R. E. Botto, and S. C. Meredith. 2000. Two-dimensional structure of β-amyloid10–35 fibrils. Biochemistry. 39:3491–3499. [DOI] [PubMed] [Google Scholar]
- 16.Burkoth, T. S., T. L. S. Benzinger, V. Urban, D. M. Morgan, D. M. Gregory, P. Thiyagarajan, R. E. Botto, S. C. Meredith, and D. G. Lynn. 2000. Structure of the β-amyloid10–35 fibril. J. Am. Chem. Soc. 122:7883–7889. [Google Scholar]
- 17.Balbach, J. J., A. T. Petkova, N. A. Oyler, O. N. Antzutkin, D. J. Gordon, S. C. Meredith, and R. Tycko. 2002. Supramolecular structure in full-length Alzheimer's β-amyloid fibrils: evidence for a parallel β-sheet organization from solid state nuclear magnetic resonance. Biophys. J. 83:1205–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antzutkin, O. N., J. J. Balbach, and R. Tycko. 2003. Site-specific identification of non-β-strand conformations in Alzheimer's β-amyloid fibrils by solid state NMR. Biophys. J. 84:3326–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petkova, A. T., G. Buntkowsky, F. Dyda, R. D. Leapman, W. M. Yau, and R. Tycko. 2004. Solid state NMR reveals a pH-dependent antiparallel β-sheet registry in fibrils formed by a β-amyloid peptide. J. Mol. Biol. 335:247–260. [DOI] [PubMed] [Google Scholar]
- 20.Chan, J. C. C., N. A. Oyler, W. M. Yau, and R. Tycko. 2005. Parallel β-sheets and polar zippers in amyloid fibrils formed by residues 10–39 of the yeast prion protein Ure2p. Biochemistry. 44:10669–10680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petkova, A. T., R. D. Leapman, Z. H. Guo, W. M. Yau, M. P. Mattson, and R. Tycko. 2005. Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils. Science. 307:262–265. [DOI] [PubMed] [Google Scholar]
- 22.Petkova, A. T., Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio, and R. Tycko. 2002. A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA. 99:16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaroniec, C. P., C. E. MacPhee, N. S. Astrof, C. M. Dobson, and R. G. Griffin. 2002. Molecular conformation of a peptide fragment of transthyretin in an amyloid fibril. Proc. Natl. Acad. Sci. USA. 99:16748–16753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaroniec, C. P., C. E. MacPhee, V. S. Bajaj, M. T. McMahon, C. M. Dobson, and R. G. Griffin. 2004. High-resolution molecular structure of a peptide in an amyloid fibril determined by magic-angle-spinning NMR spectroscopy. Proc. Natl. Acad. Sci. USA. 101:711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ritter, C., M. L. Maddelein, A. B. Siemer, T. Luhrs, M. Ernst, B. H. Meier, S. J. Saupe, and R. Riek. 2005. Correlation of structural elements and infectivity of the HET-s prion. Nature. 435:844–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siemer, A. B., C. Ritter, M. Ernst, R. Riek, and B. H. Meier. 2005. High-resolution solid state NMR spectroscopy of the prion protein HET-s in its amyloid conformation. Angew. Chem. Int. Ed. Engl. 44:2441–2444. [DOI] [PubMed] [Google Scholar]
- 27.Eanes, E. D., and G. G. Glenner. 1968. X-ray diffraction studies on amyloid filaments. J. Histochem. Cytochem. 16:673–677. [DOI] [PubMed] [Google Scholar]
- 28.Inouye, H., P. E. Fraser, and D. A. Kirschner. 1993. Structure of β-crystallite assemblies formed by Alzheimer β-amyloid protein analogs: analysis by x-ray diffraction. Biophys. J. 64:502–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sunde, M., L. C. Serpell, M. Bartlam, P. E. Fraser, M. B. Pepys, and C. C. F. Blake. 1997. Common core structure of amyloid fibrils by synchrotron x-ray diffraction. J. Mol. Biol. 273:729–739. [DOI] [PubMed] [Google Scholar]
- 30.Nelson, R., M. R. Sawaya, M. Balbirnie, A. O. Madsen, C. Riekel, R. Grothe, and D. Eisenberg. 2005. Structure of the cross-β spine of amyloid-like fibrils. Nature. 435:773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gordon, D. J., J. J. Balbach, R. Tycko, and S. C. Meredith. 2004. Increasing the amphiphilicity of an amyloidogenic peptide changes the β-sheet structure in the fibrils from antiparallel to parallel. Biophys. J. 86:428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldsbury, C., K. Goldie, J. Pellaud, J. Seelig, P. Frey, S. A. Muller, J. Kistler, G. J. S. Cooper, and U. Aebi. 2000. Amyloid fibril formation from full-length and fragments of amylin. J. Struct. Biol. 130:352–362. [DOI] [PubMed] [Google Scholar]
- 33.Goldsbury, C. S., S. Wirtz, S. A. Muller, S. Sunderji, P. Wicki, U. Aebi, and P. Frey. 2000. Studies on the in vitro assembly of Aβ1–40: implications for the search for Aβ fibril formation inhibitors. J. Struct. Biol. 130:217–231. [DOI] [PubMed] [Google Scholar]
- 34.Jimenez, J. L., E. J. Nettleton, M. Bouchard, C. V. Robinson, C. M. Dobson, and H. R. Saibil. 2002. The protofilament structure of insulin amyloid fibrils. Proc. Natl. Acad. Sci. USA. 99:9196–9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jimenez, J. L., J. L. Guijarro, E. Orlova, J. Zurdo, C. M. Dobson, M. Sunde, and H. R. Saibil. 1999. Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. EMBO J. 18:815–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Serpell, L. C., and J. M. Smith. 2000. Direct visualisation of the β-sheet structure of synthetic Alzheimer's amyloid. J. Mol. Biol. 299:225–231. [DOI] [PubMed] [Google Scholar]
- 37.Makin, O. S., E. Atkins, P. Sikorski, J. Johansson, and L. C. Serpell. 2005. Molecular basis for amyloid fibril formation and stability. Proc. Natl. Acad. Sci. USA. 102:315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Serag, A. A., C. Altenbach, M. Gingery, W. L. Hubbell, and T. O. Yeates. 2002. Arrangement of subunits and ordering of β-strands in an amyloid sheet. Nat. Struct. Biol. 9:734–739. [DOI] [PubMed] [Google Scholar]
- 39.Jayasinghe, S. A., and R. Langen. 2004. Identifying structural features of fibrillar islet amyloid polypeptide using site-directed spin labeling. J. Biol. Chem. 279:48420–48425. [DOI] [PubMed] [Google Scholar]
- 40.Der-Sarkissian, A., C. C. Jao, J. Chen, and R. Langen. 2003. Structural organization of α-synuclein fibrils studied by site-directed spin labeling. J. Biol. Chem. 278:37530–37535. [DOI] [PubMed] [Google Scholar]
- 41.Torok, M., S. Milton, R. Kayed, P. Wu, T. McIntire, C. G. Glabe, and R. Langen. 2002. Structural and dynamic features of Alzheimer's Aβ peptide in amyloid fibrils studied by site-directed spin labeling. J. Biol. Chem. 277:40810–40815. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka, M., P. Chien, N. Naber, R. Cooke, and J. S. Weissman. 2004. Conformational variations in an infectious protein determine prion strain differences. Nature. 428:323–328. [DOI] [PubMed] [Google Scholar]
- 43.Kheterpal, I., S. Zhou, K. D. Cook, and R. Wetzel. 2000. Aβ amyloid fibrils possess a core structure highly resistant to hydrogen exchange. Proc. Natl. Acad. Sci. USA. 97:13597–13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoshino, M., H. Katou, Y. Hagihara, K. Hasegawa, H. Naiki, and Y. Goto. 2002. Mapping the core of the β2-microglobulin amyloid fibril by H/D exchange. Nat. Struct. Biol. 9:332–336. [DOI] [PubMed] [Google Scholar]
- 45.Ippel, J. H., A. Olofsson, J. Schleucher, E. Lundgren, and S. S. Wijmenga. 2002. Probing solvent accessibility of amyloid fibrils by solution NMR spectroscopy. Proc. Natl. Acad. Sci. USA. 99:8648–8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whittemore, N. A., R. Mishra, I. Kheterpal, A. D. Williams, R. Wetzel, and E. H. Serpersu. 2005. Hydrogen-deuterium (H/D) exchange mapping of Aβ1–40 amyloid fibril secondary structure using nuclear magnetic resonance spectroscopy. Biochemistry. 44:4434–4441. [DOI] [PubMed] [Google Scholar]
- 47.Olofsson, A., A. E. Sauer-Eriksson, and A. Ohman. 2006. The solvent protection of Alzheimer amyloid-β1–42 fibrils as determined by solution NMR spectroscopy. J. Biol. Chem. 281:477–483. [DOI] [PubMed] [Google Scholar]
- 48.Shivaprasad, S., and R. Wetzel. 2004. An intersheet packing interaction in Aβ fibrils mapped by disulfide cross-linking. Biochemistry. 43:15310–15317. [DOI] [PubMed] [Google Scholar]
- 49.Kheterpal, I., A. Williams, C. Murphy, B. Bledsoe, and R. Wetzel. 2001. Structural features of the Aβ amyloid fibril elucidated by limited proteolysis. Biochemistry. 40:11757–11767. [DOI] [PubMed] [Google Scholar]
- 50.Baxa, U., K. L. Taylor, J. S. Wall, M. N. Simon, N. Q. Cheng, R. B. Wickner, and A. C. Steven. 2003. Architecture of Ure2p prion filaments: the N-terminal domains form a central core fiber. J. Biol. Chem. 278:43717–43727. [DOI] [PubMed] [Google Scholar]
- 51.Williams, A. D., E. Portelius, I. Kheterpal, J. T. Guo, K. D. Cook, Y. Xu, and R. Wetzel. 2004. Mapping Aβ amyloid fibril secondary structure using scanning proline mutagenesis. J. Mol. Biol. 335:833–842. [DOI] [PubMed] [Google Scholar]
- 52.Williams, A. D., S. Shivaprasad, and R. Wetzel. 2006. Alanine scanning mutagenesis of Aβ1–40 amyloid fibril stability. J. Mol. Biol. 357:1283–1294. [DOI] [PubMed] [Google Scholar]
- 53.Shivaprasad, S., and R. Wetzel. 2006. Scanning cysteine mutagenesis analysis of Aβ1–40 amyloid fibrils. J. Biol. Chem. 281:993–1000. [DOI] [PubMed] [Google Scholar]
- 54.Tjernberg, L. O., A. Tjernberg, N. Bark, Y. Shi, B. P. Ruzsicska, Z. M. Bu, J. Thyberg, and D. J. E. Callaway. 2002. Assembling amyloid fibrils from designed structures containing a significant amyloid β-peptide fragment. Biochem. J. 366:343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tjernberg, L. O., D. J. E. Callaway, A. Tjernberg, S. Hahne, C. Lilliehook, L. Terenius, J. Thyberg, and C. Nordstedt. 1999. A molecular model of Alzheimer amyloid β-peptide fibril formation. J. Biol. Chem. 274:12619–12625. [DOI] [PubMed] [Google Scholar]
- 56.Pines, A., M. G. Gibby, and J. S. Waugh. 1973. Proton-enhanced NMR of dilute spins in solids. J. Chem. Phys. 59:569–590. [Google Scholar]
- 57.Bennett, A. E., C. M. Rienstra, M. Auger, K. V. Lakshmi, and R. G. Griffin. 1995. Heteronuclear decoupling in rotating solids. J. Chem. Phys. 103:6951–6958. [Google Scholar]
- 58.Tycko, R., and Y. Ishii. 2003. Constraints on supramolecular structure in amyloid fibrils from two-dimensional solid state NMR spectroscopy with uniform isotopic labeling. J. Am. Chem. Soc. 125:6606–6607. [DOI] [PubMed] [Google Scholar]
- 59.Bennett, A. E., C. M. Rienstra, J. M. Griffiths, W. G. Zhen, P. T. Lansbury, and R. G. Griffin. 1998. Homonuclear radio-frequency-driven recoupling in rotating solids. J. Chem. Phys. 108:9463–9479. [Google Scholar]
- 60.Gullion, T., and S. Vega. 1992. A simple magic-angle spinning NMR experiment for the dephasing of rotational echoes of dipolar coupled homonuclear spin pairs. Chem. Phys. Lett. 194:423–428. [Google Scholar]
- 61.Gullion, T., D. B. Baker, and M. S. Conradi. 1990. New, compensated Carr-Purcell sequences. J. Magn. Reson. 89:479–484. [Google Scholar]
- 62.Wishart, D. S., C. G. Bigam, A. Holm, R. S. Hodges, and B. D. Sykes. 1995. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. 1. Investigations of nearest-neighbor effects. J. Biomol. NMR. 5:67–81. [DOI] [PubMed] [Google Scholar]
- 63.Sharpe, S., N. Kessler, J. A. Anglister, W. M. Yau, and R. Tycko. 2004. Solid state NMR yields structural constraints on the V3 loop from HIV-1 gp120 bound to the 447–52d antibody Fv fragment. J. Am. Chem. Soc. 126:4979–4990. [DOI] [PubMed] [Google Scholar]
- 64.Weliky, D. P., A. E. Bennett, A. Zvi, J. Anglister, P. J. Steinbach, and R. Tycko. 1999. Solid state NMR evidence for an antibody-dependent conformation of the V3 loop of HIV-1 gp120. Nat. Struct. Biol. 6:141–145. [DOI] [PubMed] [Google Scholar]
- 65.Blanco, F. J., S. Hess, L. K. Pannell, N. W. Rizzo, and R. Tycko. 2001. Solid state NMR data support a helix-loop-helix structural model for the N-terminal half of HIV-1 Rev in fibrillar form. J. Mol. Biol. 313:845–859. [DOI] [PubMed] [Google Scholar]
- 66.Long, H. W., and R. Tycko. 1998. Biopolymer conformational distributions from solid state NMR: α-helix and 310-helix contents of a helical peptide. J. Am. Chem. Soc. 120:7039–7048. [Google Scholar]
- 67.Havlin, R. H., and R. Tycko. 2005. Probing site-specific conformational distributions in protein folding with solid state NMR. Proc. Natl. Acad. Sci. USA. 102:3284–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Castellani, F., B. J. van Rossum, A. Diehl, K. Rehbein, and H. Oschkinat. 2003. Determination of solid state NMR structures of proteins by means of three-dimensional 15N-13C-13C dipolar correlation spectroscopy and chemical shift analysis. Biochemistry. 42:11476–11483. [DOI] [PubMed] [Google Scholar]
- 69.Franks, W. T., D. H. Zhou, B. J. Wylie, B. G. Money, D. T. Graesser, H. L. Frericks, G. Sahota, and C. M. Rienstra. 2005. Magic-angle spinning solid state NMR spectroscopy of the β1 immunoglobulin binding domain of protein G (GB1): 15N and 13C chemical shift assignments and conformational analysis. J. Am. Chem. Soc. 127:12291–12305. [DOI] [PubMed] [Google Scholar]
- 70.Igumenova, T. I., A. E. McDermott, K. W. Zilm, R. W. Martin, E. K. Paulson, and A. J. Wand. 2004. Assignments of carbon NMR resonances for microcrystalline ubiquitin. J. Am. Chem. Soc. 126:6720–6727. [DOI] [PubMed] [Google Scholar]
- 71.Martin, R. W., and K. W. Zilm. 2003. Preparation of protein nanocrystals and their characterization by solid state NMR. J. Magn. Reson. 165:162–174. [DOI] [PubMed] [Google Scholar]
- 72.Coustou, V., C. Deleu, S. Saupe, and J. Begueret. 1997. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc. Natl. Acad. Sci. USA. 94:9773–9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paravastu, A. K., A. T. Petkova, and R. Tycko. 2006. Polymorphic fibril formation by residues 10–40 of the Alzheimer's β-amyloid peptide. Biophys. J. 90:4618–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ishii, Y., J. J. Balbach, and R. Tycko. 2001. Measurement of dipole-coupled lineshapes in a many-spin system by constant-time two-dimensional solid state NMR with high-speed magic-angle spinning. Chem. Phys. 266:231–236. [Google Scholar]
- 75.Ishii, Y., and R. Tycko. 2000. Sensitivity enhancement in solid state 15N NMR by indirect detection with high-speed magic-angle spinning. J. Magn. Reson. 142:199–204. [DOI] [PubMed] [Google Scholar]
- 76.Ishii, Y., J. P. Yesinowski, and R. Tycko. 2001. Sensitivity enhancement in solid state 13C NMR of synthetic polymers and biopolymers by 1H NMR detection with high-speed magic-angle spinning. J. Am. Chem. Soc. 123:2921–2922. [DOI] [PubMed] [Google Scholar]
- 77.Wouters, M. A., and P. M. G. Curmi. 1995. An analysis of side-chain interactions and pair correlations within antiparallel β-sheets: the differences between backbone hydrogen-bonded and non-hydrogen-bonded residue pairs. Proteins. 22:119–131. [DOI] [PubMed] [Google Scholar]
- 78.Chu, C. K., L. L. Feng, and M. A. Wouters. 2005. Comparison of sequence and structure-based datasets for nonredundant structural data mining. Proteins. 60:577–583. [DOI] [PubMed] [Google Scholar]
- 79.Steward, R. E., and J. M. Thornton. 2002. Prediction of strand pairing in antiparallel and parallel β-sheets using information theory. Proteins. 48:178–191. [DOI] [PubMed] [Google Scholar]
- 80.Shi, Y., P. F. W. Stouten, N. Pillalamarri, L. Barile, R. V. Rosal, S. Teichberg, Z. M. Bu, and D. J. E. Callaway. 2006. Quantitative determination of the topological propensities of amyloidogenic peptides. Biophys. Chem. 120:55–61. [DOI] [PubMed] [Google Scholar]
- 81.Petkova, A. T., W. M. Yau, and R. Tycko. 2006. Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry. 45:498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]