

Abstract
Aminoacylase I (ACYI) catalyzes the stereospecific hydrolysis of L-acylamino acids and is generally assumed to be involved in the final step of the degradation of intracellular N-acetylated proteins. Apart from its crucial functions in intracellular amino acid metabolism, ACYI also has substantial commercial importance for the optical resolution of N-acylated DL-amino acids. As a zinc-dependent enzyme, ACYI is quite stable against heat-induced denaturation and can be regarded as a thermostable enzyme with an optimal temperature for activity of ∼65°C. In this research, the sequential events in ACYI thermal denaturation were investigated by a combination of spectroscopic methods and related resolution-enhancing techniques. Interestingly, the results from fluorescence and infrared (IR) spectroscopy clearly indicated that a pretransitional stage existed at temperatures from 50°C to 66°C. The thermal unfolding of ACYI might be a three-state process involving an aggregation-prone intermediate appearing at ∼68°C. The pretransitional structural changes involved the partial unfolding of the solvent-exposed β-sheet structures and the transformation of about half of the Class I Trp fluorophores to Class II. Our results also suggested that the usage of resolution-enhancing techniques could provide valuable information of the step-wise unfolding of proteins.
INTRODUCTION
Aminoacylase, a family of metalloenzymes found in many mammalian tissues and in microorganisms, catalyzes the stereospecific hydrolysis of acylamino acids to yield the corresponding organic acid and amino acid (1–3). Aminoacylase I (N-acyl-L-amino acid amido hydrolase, EC 3.5.1.14), also known as Acylase I (ACYI), is a homodimeric enzyme containing one zinc ion per subunit (4–6). ACYI, which was discovered in 1881 (7), is especially abundant in mammalian kidney and liver (8,9). The large amounts of ACYI in these tissues suggest that it might play an important role in intracellular metabolism. It is generally assumed that two distinct enzymes are involved in the intracellular degradation of N-acetylated proteins: the N-acetylated peptide hydrolase catalyzes the hydrolysis of the N-terminal amino acid residues from the N-acetylated proteins causing the release of acetylamino acids, which are finally hydrolyzed by ACYI (10,11). Due to its important physiological roles in intracellular amino acid metabolism, ACYI deficiency caused by genetic mutation has recently been related to inborn metabolism diseases (12,13). Apart from its crucial functions in intracellular metabolisms, ACYI also has substantial commercial importance for the optical resolution of N-acylated DL-amino acids. ACYI has been successfully applied in the industry to produce L-amino acids (14,15).
Unlike the thoroughly studied biochemical properties of ACYI, no high-resolution crystal structure of ACYI is presently available. Recently, the crystal structure of a D-aminoacylase from Alcaligenes faecalis was determined (16). However, the low homology among the amino acid sequences of D- and L-aminoacylases available suggests that they may have different fold and catalytic mechanisms (2). Based on mass spectrometric results, a structural model of ACYI was proposed, and it was suggested that each ACYI subunit might be composed of two domains with α/β folds: the catalytic domain and the dimerization domain (17). The folding of ACYI has been characterized to be a three-state process involving an inactive dimeric intermediate with molten globule-like characteristics (18,19). This suggested that the two domains of ACYI might fold hierarchically and the folding of the catalytic domain is the rate-limiting step. Despite the large amounts of ACYI that exist in mammalian kidney and liver, the reactivation of ACYI is particularly difficult in vitro (19–21), which suggests that the folding of ACYI in vivo might be assisted by the possible intracellular molecular chaperones and/or osmolytes. Previous studies have indicated that the zinc ion is essential for the stabilization of the active site conformation as well as the protein structure (18,22–24).
As a zinc-dependent enzyme, ACYI is quite stable against denaturation induced by heat stress. It can retain its activity when subjected to heat treatment at 60°C for 60 min (25), whereas it is irreversibly unfolded when heated at temperatures above 65°C. The optimal temperature for ACYI activity is ∼60°C (25,26), which indicates that it can be regarded as a thermostable enzyme. In general, the thermal denaturation of proteins can be demonstrated by a two-state thermodynamic mechanism. However, considerable structural changes were observed for ACYI when heated to 60°C (26). In this research, the sequential events in ACYI thermal unfolding were investigated by a combination of spectroscopic methods and the related resolution-enhancing techniques. Interestingly, the results from fluorescence and infrared (IR) spectroscopy clearly indicated that pretransitional conformational changes occurred at temperatures from 50°C to 66°C during ACYI thermal denaturation. The thermal unfolding of ACYI might be a three-state process involving an intermediate appearing at ∼68°C. Considering the commercial importance of aminoacylase, the results herein might facilitate the optimization of the conditions for the industrial use of ACYI. Moreover, the strategies used in this research might also facilitate the characterization of the potential pretransitional changes in the thermal transitions of other thermostable or thermophilic proteins.
MATERIALS AND METHODS
Materials
Sodium dodecyl sulfate (SDS) was purchased from Sigma (St. Louis, MO). Deuterated samples were from Cambridge Isotope Laboratories (Andover, MA). All other reagents were local products of analytical grade. ACYI was prepared from pig kidney according to the published procedures (4–6,18). The quality of the final products was characterized to be homogeneous on size exclusion chromatography and polyacrylamide gel electrophoresis in the presence and absence of SDS, and the purity of the protein was estimated to be above 98%. The enzyme concentration was determined by measuring the absorbance at 280 nm and using the absorption coefficient 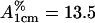 (5).
(5).
Aminoacylase activity assay
The enzyme was dissolved in 30 mM Tris-HCl, pH 7.5. The enzymatic activity was determined by measuring the absorbance at 238 nm accompanied with hydrolysis of the substrate and using the molar absorption coefficient ɛ238 = 185 M−1cm−1 as reported by Kordel and Schneider (5,6), except that chloroacetyl-L-leucine was used instead of pure-L-enantiomorph. The final enzyme concentration for the activity assay was 2 μM (∼0.18 mg/ml). The thermal dependence of ACYI activity was measured by incubating the enzyme solutions at given temperatures for 30 min, and then the activity assay was performed by mixing the enzyme solutions and reaction buffers preheated at the same temperatures.
Calorimetric measurements
Calorimetric measurements were carried out using a Setaram Micro DSC III differential scanning calorimeter (DSC) with a 0.8-ml cell (Setaram Scientific and Industrial Equipment, Caluire, France). The DSC curves were obtained using a scanning rate of 0.5 or 1.0 K/min from 20°C to 90°C. The enzymes were dissolved in 30 mM Tris-HCl, pH 7.5, with a final concentration of 1.0 or 1.5 mg/ml. Reversibility of the thermal transition was examined by reheating the samples after cooling from the first scan.
Infrared spectroscopy
The IR samples were prepared by dissolving ACYI in 100 mM deuterated phosphate saline buffer at a concentration of 50 mg/ml. The fully deuterated samples were obtained by incubating the protein solutions in a water bath at 50°C for 15 min, and then the samples were cooled to room temperature and lyophilized. The proteins were redissolved in D2O and centrifuged at 6000 × g for 10 min before use with a final pD of 8.0 adjusted using DCl or NaOD. The pD values were read directly from an Orion pH meter (Orion Research, Beverly, MA) and no corrections were made for isotope effects.
The Fourier transform infrared (FTIR) spectra were obtained using the published procedures (27). In brief, all FTIR experiments were performed on a Perkin-Elmer Spectrum 2000 spectrometer (Wellesley, MA) equipped with a dTGS detector. About 30 μl protein samples were placed between a pair of CaF2 windows separated by a 50-μm Teflon spacer. Spectra were collected from 30°C to 98°C at intervals of 2°C with a spectral resolution of 4 cm−1 and 256 scans. Baseline correction was performed before the further analysis of the IR data. Fourier self-deconvolution (FSD) was performed using the software Spectrum v3.02 (Perkin-Elmer) with a γ factor of 2.5 and Bessel smoothing of 70%, and second derivative was carried out using the algorithm in the software with a nine-point Savitzky-Golay smoothing. The transition curves of the amide I′ bands were obtained according to a method described previously (28). The amide I′ bands were assigned according to previous publications (24).
Two-dimensional infrared correlation (2D IR) analysis was carried out using SDIAPP software developed in-house (27) according to the generalized two-dimensional (2D) correlation algorithm (29). The synchronous and asynchronous correlation plots were constructed from nonnormalized FSD spectra. The averaged spectrum was used as a reference. Similar plots with relatively lower resolution could be obtained when using the original spectra for the 2D correlation analysis. The 2D IR plots were presented as contour maps produced by drawing the contour lines every 10% off from the maximum intensity of the whole correlation map. The sequence of the events was characterized by analyzing the signs of the peaks in the 2D IR correlation plots using rules proposed by Noda (29). The attempts to avoid artifacts in the 2D IR plots have been described in detail elsewhere (30).
Intrinsic fluorescence spectroscopy
The samples for the fluorescence measurements were prepared by dissolved ACYI in 30 mM Tris-HCl (pH 7.5) with a final concentration of 1 mg/ml. To evaluate the effect of the protein concentration on the fluorescence spectra, a diluted sample solution with a final concentration of 0.2 mg/ml was also prepared. Details with regard to the intrinsic fluorescence experiments were the same as those described before (30). In brief, the intrinsic Trp fluorescence emission spectra were measured using a Hitachi F-2500 spectrofluorometer (Tokyo, Japan) using 1-cm path-length cuvettes with an excitation wavelength of 295 nm. The spectra were measured after the samples had been equilibrated for 2 min at given temperatures controlled with a circulating water bath. Parameter A, which reflects the spectral shape of the intrinsic Trp fluorescence (31), was obtained by dividing the fluorescence intensity at 320 nm by the intensity at 365 nm.
The fitting of the fluorescence spectra was performed using the discrete states model of Trp residues in proteins and was calculated by a program developed in-house (30) based on the SIMS algorithms of decomposition (32). The fluorescence spectra were normalized before calculation. Since no crystal structure of ACYI is available, all four of the components (A and S, I, II, and III) of Trp fluorescence defined previously (33,34) were included in the curve-fitting process. The best fitting results were obtained according to the least root mean square criterion.
Phase diagram analysis of IR and fluorescence data
The phase diagram analysis, which is a sensitive tool to detect folding intermediates, was carried out as described previously (35). The original IR or fluorescence data were normalized by the corresponding intensity of the spectra recorded at 30°C. The phase diagram was constructed by the IR intensity at 1637 cm−1 vs. 1628 cm−1 or the fluorescence intensity at 320 nm vs. that at 365 nm at different temperatures. Each straight line in the phase diagram reflects an “all-or-none” process, and the joint position of two lines for a transition process indicates that an intermediate appeared at the corresponding temperature.
RESULTS AND DISCUSSION
The thermal dependence of ACYI activity was determined by measuring the catalytic activity of the samples equilibrated at given temperatures for 30 min. As shown in Fig. 1, the optimal temperature of ACYI activity was ∼65°C, which is consistent with the previous observations (25,26) when considering that different heat treatments were used. In the Arrhenius plot (Fig. 1B), the data yielded a straight line between 30°C and 65°C, from the slope of which the activation energy was calculated to be ∼31 kJ mol−1. The activity decreased rapidly at higher temperatures (>65°C) due to inactivation of the enzyme. These results suggested that the active site of the enzyme might remain intact at temperatures below 65°C under our conditions, whereas the loss of activity at temperatures above 65°C might be due to heat-induced unfolding.
FIGURE 1.

Thermal dependence of ACYI activity. The enzyme was dissolved in 30 mM Tris-HCl, pH 7.5, with a final concentration of 2 μM. The activity was measured by incubating the enzyme solutions at given temperatures for 30 min, and then the activity assay was performed by mixing the enzyme solutions and reaction buffers preheated at the same temperatures. (A) The data were normalized by taking the activity of the sample incubated at 25°C as 100%. (B) The Arrhenius plot for the ACYI activity in the temperature range 20°C–75°C. Activation energy calculated from the slope of the straight line (30–65°C) is 31 kJ mol−1.
DSC was used to investigate whether the thermal denaturation of ACYI was reversible or not. The heat flow versus temperature profile of ACYI contained an asymmetric positive peak arising from an exothermic transition (Fig. 2). The appearance of the unusual exothermic peak and the lack of endothermic peak suggested that the unfolding of ACYI was accompanied by serious aggregation, which might be a nucleation-dependent process (36). No significant difference was observed when the scanning rate was increased from 0.5 K/min to 1 K/min, whereas the exothermic peak moved to a relative low temperature when ACYI concentration was increased from 1 to 1.5 mg/ml. The concentration effect on DSC curves re-marked the appearance of the aggregation process. When the sample was heated up to 90°C, immediately cooled, and reheated, the reheating scan had no thermal effect over the temperature range of 30°C–90°C, which suggested that the thermal denaturation of ACYI was fully irreversible. These result confirmed the previous assumption that the irreversible inactivation of ACYI at high temperatures might be due to protein aggregation (25). Moreover, the thermal transition of ACYI could be explained by a two-state model, and no pretransitional changes could be identified from the DSC profile.
FIGURE 2.

Thermal dependence of the heat flow of ACYI thermal denaturation probed by DSC. The heating rates were 0.5 or 1.0 K/min, and the protein concentrations were 1.0 or 1.5 mg/ml. The dotted line represents the thermal transition examined by reheating the sample after cooling from the first scan (solid line). The positive peaks in the DSC profile are exothermic.
IR spectroscopy has the advantage that the measurements will not be interfered by the appearance of aggregates (37). Moreover, resolution-enhancing techniques, such as second derivative and 2D IR, have been shown to be sensitive tools to monitor the sequential events in protein conformational changes by FTIR (27,28,30,38–42). Thus IR experiments were performed to further investigate the sequential events during the thermal denaturation of ACYI. The amide I′ band of native ACYI in the original IR spectrum (Fig. 3 A) consisted of several overlapped components, which could be successfully distinguished by resolution-enhancing techniques, FSD (Fig. 3 B), and the second derivative spectra (Fig. 3 C). The amide I′ region of native ACYI was dominated by bands from the α-helix (1651 cm−1), α-helix and/or 310 helix (1660 cm−1), β-sheet (1629, 1637, and 1682 cm−1), β-turn (1668 and 1674 cm−1), and random coil (1644 cm−1). This assignment is consistent with the previous results (24) and the predicted α/β structure of ACYI (17). As the temperature was increased, the intensity of the bands from the native secondary structures decreased, and two new bands (1618 and 1682 cm−1) appeared at temperatures above 70°C. These two bands have been proposed to be the sign of the formation of intermolecular β-sheet in aggregates (42,43). These observations in the IR spectra coincided with the deduction based on DSC results that the thermal unfolding of ACYI is an irreversible process accompanied with protein aggregation.
FIGURE 3.

Thermal dependence of the original (A), FSD (B), and second derivative (C) IR spectra of ACYI. For clarity, only the typical spectra are presented. The arrows indicate the direction of intensity changes with the temperature increasing from 30°C to 40°C, 50°C, 60°C, 70°C, 80°C, 90°C, and 98°C, respectively. FSD was performed using the software Spectrum v3.02 with a γ-factor of 2.5 and a Bessel smoothing of 70%, and the second derivative was carried out using the algorithm in the software with a nine-point Savitzky-Golay smoothing. (A) The arrows indicate the bands at 1682, 1642, and 1618 cm−1, from left to right, respectively. (B) and (C) The arrows show the bands around 1682, 1660, 1651, 1645, 1637, 1630, and 1618 cm−1, from left to right, respectively.
The IR difference absorption spectra were calculated by subtracting the spectra at given temperatures from the spectrum recorded at 30°C (Fig. 4 A). The two positive peaks located at 1618 and 1682 cm−1 confirmed the appearance of the aggregates observed in Fig. 3. More importantly, the position of the negative peak, which reflects the unfolding of the native structures, was centered at around 1628 cm−1 in the temperature range 30–66°C, then shifted to around 1639 cm−1 when the temperature increased from 66°C to 80°C, and finally remained at this wavenumber upon heating. This suggested that the thermal unfolding of ACYI might involve step-wise transitions and the change of the band at 1628 cm−1 preceded those of the other bands. A similar conclusion could be deduced from the intensity changes of the amide I′ components shown in Fig. 4 B. The intensity of the 1630 cm−1 band slightly decreased when the temperature was increased to 68°C, whereas the intensity of other bands kept unchanged in this range. At temperatures above 70°C, all the bands except those from the β-turn (1668 and 1674 cm−1) possessed similar melting curves, which indicated that a major conformational change occurred in this temperature range. It is worth noting that the peak centered around 1639 cm−1 at temperatures above 80°C in the difference absorption spectra (Fig. 4 A) was the apparent peak consisting of the changes of most native bands in the amide I′ region, whereas the peak shift from 66°C to 80°C resulted from the transition from pretransitional to the dominant structural changes.
FIGURE 4.

The IR difference spectra (A) and the thermal melting curves (B) of ACYI. (A) The difference absorption spectra, shown at 4°C intervals, were obtained by subtracting the spectrum recorded at 30°C. The solid arrows indicate the directions of the intensity changes of bands (1682 and 1618 cm−1) from the cross-β structures in aggregates as the temperature increased. The dotted arrows show the directions of the intensity changes and positions of bands from the native structures upon heating. (B) The melting curves of the amide I′ bands were at 1674 (▪), 1651 (□), 1637 (○), 1630 (▵), and 1618 cm−1 (•), respectively. The data were calculated from the spectra in Fig. 3 C using the published procedures (28). The dashed line indicates the temperature at which aggregates were observed.
The results in Fig. 4 strongly supported the conclusion that pretransitional conformation changes occurred at temperatures where the enzyme retained high activity (see Fig. 1). To better visualize the sequential events occurring during ACYI thermal unfolding, 2D IR synchronous and asynchronous plots were constructed. In general, the autopeaks appearing on the diagonal in synchronous plots indicate that the intensity of the corresponding bands change with perturbation, whereas the crosspeaks show that the intensity changes of the two correlated bands are in the same (positive) or opposite (negative) direction. The appearance of a crosspeak in the symmetrical asynchronous plot indicates that the changes of the two correlated bands are out of phase. The sequence of events can be analyzed by comparing the signs of the corresponding crosspeaks in the synchronous and asynchronous plots according to the rules proposed by Noda (29). As shown in Fig. 5 A, when 2D plots were constructed over the whole temperature range, two dominant autopeaks centered at 1617 and 1638 cm−1 could be found in the synchronous plot, and negative crosspeaks developed between these two bands. Meanwhile, small autopeaks could be identified at 1649 and 1658 cm−1, and a minor autopeak centered at 1682 cm−1 was below the cutoff of the plot and was not presented. The dominant crosspeak in the asynchronous plot was a positive peak centered at (1629 cm−1, 1617 cm−1), whereas minor positive peaks could be found at (1648 cm−1, 1617 cm−1) and (1658 cm−1, 1617 cm−1). These results suggested that during ACYI thermal denaturation, the dominant events were the unfolding of the β-sheet (1638 cm−1) and the formation of the intermolecular β-sheet in aggregates (1617 cm−1). The order of events could be characterized as the intensity decrease of the band at 1629 cm−1 > the intensity increase of the band at 1617 cm−1 > the intensity decrease of the band at 1649 and 1658 cm−1. Since the frequency of the solvent-exposed secondary structures is smaller than the buried ones (44,45), the above sequential events could be interpreted as follows: the unfolding of the solvent-exposed β-sheet structures is the earliest event, followed by the formation of aggregates, and finally the unfolding of the other native structures.
FIGURE 5.

The 2D correlation analysis of the IR spectra of ACYI thermal denaturation. Synchronous (Φ) and asynchronous (Ψ) plots were constructed from the corresponding FSD spectra recorded in the temperature range of 30–98°C (A), 30–66°C (B), and 74–98°C (C). The spectra recorded at temperatures from 68°C to 72°C was not included in the 2D IR analysis in panel C to avoid possible artifacts caused by band shift accompanied with slight intensity changes (see also Fig. 4 A). The plots are presented as contour maps produced by drawing the contour lines every 10% off from the maximum intensity of the corresponding map. Clear and dark peaks represent positive and negative, respectively.
The reason that the autopeak at 1629 cm−1 did not appear might have been due to the overlapping of the adjacent intense peak at 1638 cm−1. To verify the results obtained in Fig. 5 A, 2D IR plots were constructed for the temperature ranges of 30°C–66°C and 74°C–98°C. The spectra recorded at temperatures from 68°C to 72°C were not included in the 2D IR analysis in Fig. 5 C to avoid the possible artifacts caused by large band shifts accompanied with slight intensity changes in this temperature range (see also Fig. 4 A) (46). The synchronous plot in Fig. 5 B clearly indicates that the change of the band at 1628 cm−1 was the dominant event at temperatures below 66°C, whereas the asynchronous plot was dominated by noise and no crosspeaks could be identified at a similar position in the synchronous plot. The synchronous plot in Fig. 5 C is similar to the one in Fig. 5 A, which coincided with the fact that major conformational changes occurred in this temperature range. The asynchronous plot was different from that in Fig. 5 A. Positive crosspeaks could be found at (1649 cm−1, 1636 cm−1), and negative crosspeaks were located at (1649 cm−1, 1617 cm−1) and (1658 cm−1, 1617 cm−1). However, the order of events from Fig. 5 C was not reliable due to the lack of some spectra, as mentioned above. Nevertheless, the 2D IR analysis also suggested that the pretransitional conformational changes involved the unfolding of the solvent-exposed β-sheet structures and aggregates appearing at a temperature at which the protein had not fully unfolded.
ACYI contains eight Trp residues in each of its two subunits (47,48), and the eight Trp residues are distributed in both the peptidase and dimerization domain (13,17). Thus the intrinsic Trp fluorescence was used to investigate the changes of the tertiary structure of ACYI during thermal denaturation. To ensure the accuracy of the curve fitting, two samples with final protein concentrations of 0.2 and 1 mg/ml were prepared and used for the Trp fluorescence analysis. The curves from the diluted solution (data not shown) were similar to those presented in Figs. 6 and 7 but with greater errors due to the relatively low signal/noise ratio and intactness of the thermal transition curve. Thus the sample with a concentration of 1 mg/ml was used for further analysis. Consistent with previous observations (26), the intensity of the intrinsic Trp fluorescence decreased with the increase of temperature, and meanwhile, the emission maximum wavelength red shifted from ∼333 nm to 340 nm (Fig. 6). At temperatures above 80°C, the fluorescence was significantly affected by the appearance of serious aggregation. The change of the intensity at 330 nm could not be fitted to a simple two-state model, and a platform could be found at the temperature range of 52°C–60°C (Fig. 6 B). Parameter A, which is a sensitive tool to reflect the position and shape of the Trp fluorescence spectrum, slightly decreased when the temperature increased to 64°C, and then an abrupt decrease was observed. To better understand the step-wise changes of the different Trp fluorophores, the spectrum recorded at each temperature was fitted using the discrete states model of Trp residues in proteins (30,32,34). Since no high-resolution structure of ACYI is available, all of the Trp fluorophores (Classes A and S, I, II, and III) defined previously (34) were included in the fitting procedure. The native ACYI contained Class I (∼60%) and II (∼40%) fluorophores, but did not contain Classes A, S, or III (Fig. 7 A). This indicated that most of the Trp residues in ACYI were buried in the interior (Class I) or exposed to bonded water (Class II), and no Trp residues were in a highly hydrophobic (Class A and S) or fully solvent-exposed (Class III) microenvironment. This result is, to some extent, consistent with the predicted ACYI structure (17). The intensity changes of the various fluorophores upon heating could be classified into three distinct stages. From 30°C to 50°C, no significant changes were observed for the four components. The content of the Class I component began to decrease when heated to ∼50°C, and meanwhile the Class II component began to increase and reached a level of ∼60% at 66°C. The other components remained at the same level as at 30°C in the temperature range of 50–66°C. At temperatures above 66°C, the content of the Class I fluorophore decreased continuously, whereas the Class III component increased. At 80°C, ACYI contains ∼10% Class I, 60% Class II, and 30% Class III fluorophores, which suggested that most Trp residues in the protein were accessible by solvent. It is noteworthy that the same results could be obtained for the diluted sample with a concentration (0.2 mg/ml) similar to that used for the activity assay, except that the transitions occurred at a temperature ∼2°C higher than the 1 mg/ml sample.
FIGURE 6.

Thermal dependence of the intrinsic Trp fluorescence spectra (A) and the melting curves (B) of ACYI. The protein concentration was 1 mg/ml. The spectra were obtained with an excitation wavelength of 295 nm to avoid the contributions of the Tyr residues. The abnormal change of the parameters above 80°C was due to serious aggregation and was not used in further analysis. (A) The dotted arrows indicate the directions of the intensity changes and positions of the emission maximum wavelength of ACYI as the temperature increased. The spectrum recorded at 82°C is represented by a dotted line. (B) The intensity change of the intrinsic fluorescence at 330 nm (○) and the change of Parameter A (•) are shown. Parameter A, which is a sensitive tool to reflect the shape and position of the Trp fluorescence spectrum (31), was obtained by dividing the intensity at 320 nm by that at 365 nm.
FIGURE 7.

Fitting of the experimental fluorescence spectra by the theoretical model of discrete states of Trp residues in proteins. (A–D) The fitting results of the spectra recorded at 30°C (A), 48°C (B), 64°C (C), and 78°C (D). The fitted spectra (dashed lines) are the sum of the four spectral components (solid lines), Classes A and S, I, II, and III fluorophores. The experimental data are represented by dotted lines. (E) The thermal dependence of the maximum intensity of Classes A and S (□), I (○), II (•), and III fluorophores (▪). The dashed lines indicate the temperature range of the pretransitional stage.
The above results from IR and intrinsic fluorescence spectroscopy strongly suggested that the thermal unfolding of ACYI was a step-wise process. The phase diagram analysis, which is a sensitive tool to detect folding intermediates (35), was performed to characterize whether an intermediate was involved in ACYI thermal unfolding. Although phase diagrams have usually been used in the analysis of Trp fluorescence data in previous publications, the theoretical analysis of the phase diagram is also expected to be true for IR data. To avoid misleading results from the heavy overlapping of the original IR amide I′ band, FSD data, which maintained the intensity of the original data, were used for the phase diagram analysis. The phase diagram was constructed by the FSD IR intensity at 1637 cm−1 versus that at 1628 cm−1 (Fig. 8 A) or the fluorescence intensity at 320 nm versus that at 365 nm (Fig. 8 B) at different temperatures. As can be seen in Fig. 8, both plots revealed that there are two linear segments that deviated at a temperature of around 68°C, and each straight line reflected a two-state process. The joint position of the changes in the two adjacent lines was around 68°C, suggesting the appearance of an intermediate at ∼68°C.
FIGURE 8.

Phase diagram analysis of the original IR (A) and intrinsic fluorescence (B) data. The phase diagram analysis was carried out as described previously (35). The FSD IR or fluorescence data were normalized by the corresponding intensity of the spectra recorded at 30°C. The phase diagram was constructed by the IR intensity at 1637 cm−1 vs. 1628 cm−1 (A) or the fluorescence intensity at 320 nm versus that at 365 nm (B). Each straight line in the phase diagram reflects a two-state process, and the joint position of two adjacent lines indicates that an intermediate appeared at the corresponding temperature.
Although the thermal unfolding of many proteins can successfully be illustrated by a two-state model, step-wise transitions have been observed in several proteins regardless of whether the process was reversible or irreversible (30,38,39,49–54). ACYI is, to some extent, a thermostable enzyme with an optimal temperature for activity at ∼65°C. Interestingly, considerable conformational changes were observed before the enzyme was fully inactivated by high temperature. The IR and fluorescence results strongly suggested that the thermal unfolding of ACYI could be illustrated as a two-stage process involving an intermediate: native state (N) → intermediate state (I) → aggregates (A). The I→A transition, which was referred to as the dominant unfolding process above, began at around 70°C and the midpoint of the transition (T1/2) occurred at ∼84°C (see Table 1). A significant intensity decrease was observed for most IR bands from native secondary structures, accompanied with the exposure of most Trp residues to water and the formation of aggregates during the I→A transition. The N→I transition, which was referred to as the pretransitional change, ended at ∼68°C and the T1/2 occurred at ∼60°C (see Table 1). The structural changes in this stage involved the partial unfolding of the solvent-exposed β-sheet structures and the transformation of about half of the Class I Trp fluorophores to Class II (see Fig. 7). The decrease in the content of Class I Trp fluorophores suggested that the intermediate had a loose tertiary structure, which allowed the penetration of water molecules into the hydrophobic interior. This two-stage scheme is similar to that from the chemical unfolding/refolding studies of ACYI (18,19), which also indicated that there existed a dimeric, aggregation-prone folding intermediate. It was difficult to distinguish in this research whether the pretransitional changes occurred in the catalytic domain or the dimerization domain of ACYI. Both domains contain α-helix and β-sheet structures, and both contain several Trp residues. It has been proposed that the catalytic domain contains a β-sheet sandwiched between α-helices and a second β-sheet located on the surface, whereas the dimerization domain consists of a β-sheet flanked on one side by two α-helices (17). Since ACYI can maintain its dimeric structure at a high GdnHCl concentration of 2 M (18), it seems that the dimerization domain was quite stable against denaturation. Thus the β-sheet on the surface of the catalytic domain, which is exposed to water in native ACYI, might unfold first. The unfolding of this β-sheet allows the water molecules to penetrate into the interior of the catalytic domain, which results in the transformation of some Class I fluorophores to Class II and a further inactivation of the enzyme. These pretransitional conformational changes also resulted in an intermediate with hydrophobic exposure, which could lead to serious aggregation in both chemical denaturants and heat-treatment conditions.
TABLE 1.
Thermal parameters of ACYI
| Method | T1/2* (°C) | Assignment |
|---|---|---|
| Activity assay | 65 | |
| IR bands (cm−1)† | ||
| 1674 | – | β-turn |
| 1651 | 89 ± 6 | α-helix |
| 1637 | 82.1 ± 0.9 | β-sheet |
| 1630 | 84 ± 1 | β-sheet |
| 1618 | 83.2 ± 0.4 | Intermolecular β-sheet in aggregates |
| Trp Fluorophore‡ | ||
| Class A and S | – | highly hydrophobic |
| Class I | 65 ± 2 | buried and inaccessible to water |
| Class II | 59 ± 2 | exposed to bonded water |
| Class III | >80 | highly exposed to solvent |
T1/2 is the midpoint of the major thermal transition or the temperature where the activity reached its maximum. The symbol “–” indicates no significant change was observed.
Only T1/2 values and assignments of the typical bands are shown.
Details regarding the correlation between Trp fluorophores and the microenvironmental parameters can be found in Reshetnyak et al. (34).
Acknowledgments
This investigation was supported by Grant 30500084 from the National Natural Science Foundation of China, and Grant 101023 from the Fok Ying Tong Education Foundation.
References
- 1.Anders, M. W., and W. Dekant. 1994. Aminoacylases. Adv. Pharmacol. 27:431–448. [DOI] [PubMed] [Google Scholar]
- 2.Hernick, M., and C. A. Fierke. 2005. Zinc hydrolases: the mechanisms of zinc-dependent deacetylases. Arch. Biochem. Biophys. 433:71–84. [DOI] [PubMed] [Google Scholar]
- 3.Seibert, C. M., and F. M. Raushel. 2005. Structural and catalytic diversity within the amidohydrolase superfamily. Biochemistry. 44:6383–6391. [DOI] [PubMed] [Google Scholar]
- 4.Birnbaum, S. M. 1955. Aminoacylase, amino acid acylase I and II from hog kidney. Methods Enzymol. 2:115–119. [Google Scholar]
- 5.Kordel, W., and F. Schneider. 1976. Chemical investigation on pig kidney aminoacylase. Biochim. Biophys. Acta. 445:446–457. [DOI] [PubMed] [Google Scholar]
- 6.Kordel, W., and F. Schneider. 1976. Chemical modification of two tryptophan residues abolishes the catalytic activity of aminoacylase. Hoppe Seylers Z. Physiol. Chem. 357:1109–1115. [DOI] [PubMed] [Google Scholar]
- 7.Schmiedeberg, O. 1881. Über Spaltungen und Synthesen im Thierkörper. Naunyn-Schmiedebergs Arch. Exp. Path. Pharma kol. 14:379–392. [Google Scholar]
- 8.Endo, Y. 1978. N-Acyl-L-aromatic amino acid deacylase in animal tissues. Biochim. Biophys. Acta. 523:207–214. [DOI] [PubMed] [Google Scholar]
- 9.Miller, Y. E., and B. Kao. 1989. Monoclonal antibody based immunoassay for human aminoacylase-1. J. Immunoassay. 10:129–152. [DOI] [PubMed] [Google Scholar]
- 10.Gade, W., and J. Brown. 1981. Purification, characterization and possible function of α-N-acylpeptide hydrolase from bovine liver. Biochim. Biophys. Acta. 662:86–93. [DOI] [PubMed] [Google Scholar]
- 11.Perrier, J., A. Durand, T. Giardina, and A. Puigserver. 2005. Catabolism of intracellular N-terminal acetylated proteins: involvement of acylpeptide hydrolase and acylase. Biochimie. 87:673–685. [DOI] [PubMed] [Google Scholar]
- 12.Van Coster, R. N., E. A. Gerlo, T. G. Giardina, U. F. Engelke, J. E. Smet, C. M. De Praeter, V. A. Meersschaut, L. J. De Meirleir, S. H. Seneca, B. Devreese, J. G. Leroy, S. Herga, J. P. Perrier, R. A. Wevers, and W. Lissens. 2005. Aminoacylase I deficiency: a novel inborn error of metabolism. Biochem. Biophys. Res. Commun. 338:1322–1326. [DOI] [PubMed] [Google Scholar]
- 13.Sass, J. O., V. Mohr, H. Olbrich, U. Engelke, J. Horvath, M. Fliegauf, N. T. Loges, S. Schweitzer-Krantz, R. Moebus, P. Weiler, A. Kispert, A. Superti-Furga, R. A. Wevers, and H. Omran. 2006. Mutations in ACY1, the gene encoding aminoacylase 1, cause a novel inborn error of metabolism. Am. J. Hum. Genet. 78:401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chenault, H. K., J. Dahmer, and G. M. Whitesides. 1989. Kinetic resolution of unnatural and rarely occurring amino acids: enantioselective hydrolysis of N-acyl amino acids catalyzed by acylase I. J. Am. Chem. Soc. 111:6354–6364. [Google Scholar]
- 15.Wang, H. J., J. H. Bai, D. S. Liu, T. Zhang, and H. M. Zhou. 1999. Preparation and properties of immobilized pig kidney aminoacylase and optical resolution of N-acyl-DL-alanine. Appl. Biochem. Biotechnol. 76:183–191. [DOI] [PubMed] [Google Scholar]
- 16.Liaw, S.-H., S.-J. Chen, T.-P. Ko, C.-S. Hsu, C.-J. Chen, A. H.-J. Wang, and Y.-C. Tsai. 2003. Crystal structure of D-aminoacylase from Alcaligenes faecalis DA1. A novel subset of amidohydrolases and insights into the enzyme mechanism. J. Biol. Chem. 278: 4957–4962. [DOI] [PubMed] [Google Scholar]
- 17.D'Ambrosio, C., F. Talamo, R. M. Vitale, P. Amodeo, G. Tell, L. Ferrara, and A. Scaloni. 2003. Probing the dimeric structure of porcine aminoacylase 1 by mass spectrometric and modeling procedures. Biochemistry. 42:4430–4443. [DOI] [PubMed] [Google Scholar]
- 18.Bai, J.-H., D. Xu, H.-R. Wang, S.-Y. Zheng, and H.-M. Zhou. 1999. Evidence for the existence of an unfolding intermediate state for aminoacylase during denaturation in guanidine solutions. Biochim. Biophys. Acta. 1430:39–45. [DOI] [PubMed] [Google Scholar]
- 19.Xie, Q., and H.-M. Zhou. 2004. Refolding intermediate of guanidine hydrochloride denatured aminoacylase. Int. J. Biochem. Cell Biol. 36:1332–1340. [DOI] [PubMed] [Google Scholar]
- 20.Kim, S.-H., Y.-B. Yan, and H.-M. Zhou. 2006. Role of osmolytes as chemical chaperones during the refolding of aminoacylase. Biochem. Cell Biol. 84:30–38. [DOI] [PubMed] [Google Scholar]
- 21.Kim, S.-H., J. Zhang, Y. Jiang, H.-M. Zhou, and Y.-B. Yan. 2006. Assisting the reactivation of guanidine hydrochloride-denatured aminoacylase by hydroxypropyl cyclodextrins. Biophys. J. 91:686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heese, D., S. Berger, and K. H. Röhm. 1990. Nuclear magnetic relaxation studies of the role of the metal ion in Mn2+-substituted aminoacylase I. Eur. J. Biochem. 188:175–180. [DOI] [PubMed] [Google Scholar]
- 23.Wang, Z.-X., H.-B. Wu, X.-C. Wang, H.-M. Zhou, and C.-L. Tsou. 1992. Kinetics of the course of inactivation of aminoacylase by 1,10-phenanthroline. Biochem. J. 281:285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang, Y., P.-R. Chen, B. He, and H.-M. Zhou. 1994. Secondary structure of holo-enzyme and apoenzyme of aminoacylase using CD and FTIR spectroscopy. Sci. China B. 37:1471–1478. [Google Scholar]
- 25.He, B., M. Yan, T. Zhang, and H.-M. Zhou. 1994. Unfolding and inactivation during thermal denaturation of aminoacylase. Chin. Sci. Bull. 39:1122–1127. [Google Scholar]
- 26.Xie, Q., F. G. Meng, and H. M. Zhou. 2004. Low temperature induced conformational changes of aminoacylase. Tsinghua Sci. Technol. 9:76–80. [Google Scholar]
- 27.Yan, Y.-B., Q. Wang, H.-W. He, X.-Y. Hu, R.-Q. Zhang, and H.-M. Zhou. 2003. Two-dimensional infrared correlation spectroscopy study of the heat induced unfolding and aggregation process of Myoglobin. Biophys. J. 85:1959–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang, J., and Y.-B. Yan. 2005. Probing conformational changes of proteins by quantitative second derivative infrared spectroscopy. Anal. Biochem. 340:89–98. [DOI] [PubMed] [Google Scholar]
- 29.Noda, I. 1993. Generalized two-dimensional correlation method applications to infrared, Raman, and other types of spectroscopy. Appl. Spectrosc. 47:1329–1336. [Google Scholar]
- 30.He, H.-W., J. Zhang, H.-M. Zhou, and Y.-B. Yan. 2005. Conformational change in the C-terminal domain is responsible for the initiation of creatine kinase thermal aggregation. Biophys. J. 89:2650–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turoverov, K. K., S. Y. Haitlina, and G. P. Pinaev. 1976. Ultra-violet fluorescence of actin. Determination of actin content in actin preparations. FEBS Lett. 62:4–6. [DOI] [PubMed] [Google Scholar]
- 32.Burstein, E. A., S. M. Abornev, and Y. K. Reshetnyak. 2001. Decomposition of protein tryptophan fluorescence spectra into log-normal components. I. Decomposition algorithms. Biophys. J. 81:1699–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reshetnyak, Y. K., and E. A. Burstein. 2001. Decomposition of protein tryptophan fluorescence spectra into log-normal components. II. The statistical proof of discreteness of tryptophan classes in proteins. Biophys. J. 81:1710–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reshetnyak, Y. K., Y. Koshevnik, and E. A. Burstein. 2001. Decomposition of protein tryptophan fluorescence spectra into log-normal components. III. Correlation between fluorescence and microenvironment parameters of individual tryptophan residues. Biophys. J. 81:1735–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bushmarina, N. A., I. M. Kuznetsova, A. G. Biktashev, K. K. Turoverov, and V. N. Uversky. 2001. Partially folded conformations in the folding pathway of bovine carbonic anhydrase II: a fluorescence spectroscopic analysis. ChemBioChem. 2:819–821. [DOI] [PubMed] [Google Scholar]
- 36.Berkowitz, S. A., G. Velicelebi, J. W. H. Sutherland, and J. M. Sturtevant. 1980. Observation of an exothermic process associated with the in vitro polymerization of brain tubulin. Proc. Natl. Acad. Sci. USA. 77:4425–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pelton, J. T., and L. R. McLean. 2002. Spectroscopic methods for analysis of protein secondary structure. Anal. Biochem. 277:167–176. [DOI] [PubMed] [Google Scholar]
- 38.Fabian, H., H. H. Mantsch, and C. P. Schultz. 1999. Two dimensional IR correlation spectroscopy: sequential events in the unfolding process of the λ Cro-V55C repressor protein. Proc. Natl. Acad. Sci. USA. 96:13153–13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dong, A., T. W. Randolph, and J. F. Carpenter. 2000. Entrapping intermediates of thermal aggregation in α-helical proteins with low concentration of guanidine hydrochloride. J. Biol. Chem. 275:27689–27693. [DOI] [PubMed] [Google Scholar]
- 40.Paquet, M.-J., M. Laviolette, M. Pézolet, and M. Auger. 2001. Two-dimensional infrared correlation spectroscopy study of the aggregation of cytochrome c in the presence of dimyristoylphosphatidylglycerol. Biophys. J. 81:305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noda, I. 2004. Advances in two-dimensional correlation spectroscopy. Vib. Spectrosc. 36:143–165. [Google Scholar]
- 42.Yan, Y.-B., J. Zhang, H.-W. He, and H.-M. Zhou. 2006. Oligomerization and aggregation of bovine pancreatic ribonuclease A: characteristic events observed by FTIR spectroscopy. Biophys. J. 90:2525–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jackson, M., and H. H. Mantsch. 1995. The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit. Rev. Biochem. Mol. Biol. 30:95–120. [DOI] [PubMed] [Google Scholar]
- 44.Manas, E. S., Z. Getahun, W. W. Wright, W. F. DeGrado, and J. M. Vanderkooi. 2000. Infrared spectra of amide groups in α-helical proteins: evidence for hydrogen bonding between helices and water. J. Am. Chem. Soc. 122:9883–9890. [Google Scholar]
- 45.Walsh, S. S. T., R. P. Cheng, W. W. Wright, D. O. Alonso, V. Daggett, J. M. Vanderkooi, and W. F. DeGrado. 2003. The hydration of amides in helices; a comprehensive picture from molecular dynamics, IR, and NMR. Protein Sci. 12:520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Czarnecki, M. A. 2000. Two-dimensional correlation sSpectroscopy: effect of band position, width, and intensity changes on correlation intensities. Appl. Spectrosc. 54:986–993. [Google Scholar]
- 47.Mitta, M., H. Ohnogi, A. Yamamoto, I. Kato, F. Sakiyama, and S. Tsunasawa. 1992. The primary structure of porcine aminoacylase 1 deduced from cDNA sequence. J. Biochem. (Tokyo). 112:737–742. [DOI] [PubMed] [Google Scholar]
- 48.Jakob, M., Y. E. Miller, and K. H. Rohm. 1992. Cloning and sequence analyses of cDNAs encoding aminoacylase I from porcine kidney. Biol. Chem. Hoppe Seyler. 373:1227–1231. [DOI] [PubMed] [Google Scholar]
- 49.Burgess, A. W., and H. A. Scheraga. 1975. A hypothesis for the pathway of the thermally-induced unfolding of bovine pancreatic ribonuclease. J. Theor. Biol. 53:403–420. [DOI] [PubMed] [Google Scholar]
- 50.Matheson, R. R., and H. A. Scheraga. 1979. Steps in the pathway of the thermal unfolding of ribonuclease A. A nonspecific photochemical surface-labeling study. Biochemistry. 18:2437–2445. [DOI] [PubMed] [Google Scholar]
- 51.Yan, Y.-B., Q. Wang, H.-W. He, and H.-M. Zhou. 2004. Protein thermal aggregation involves distinct regions: sequential events in the heat-induced unfolding and aggregation of hemoglobin. Biophys. J. 86:1682–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vermeer, A. W. P., and W. Norde. 2000. The thermal stability of immunoglobulin: unfolding and aggregation of a multi-domain protein. Biophys. J. 78:394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stelea, S. D., P. Pancoska, A. S. Benight, and T. A. Keiderling. 2001. Thermal unfolding of ribonuclease A in phosphate at neutral pH: deviations from two-state model. Protein Sci. 10:970–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stelea, S. D., and T. A. Keiderling. 2002. Pretransitional structural changes in the thermal denaturation of ribonuclease S and S protein. Biophys. J. 83:2259–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]